
ABSTRACT
Introduction
Primary chylomicronemia is characterized by pathological accumulation of chylomicrons in the plasma causing severe hypertriglyceridemia, typically >10 mmol/L (>875 mg/dL). Patients with the ultra-rare familial chylomicronemia syndrome (FCS) subtype completely lack lipolytic capacity and respond minimally to traditional triglyceride-lowering therapies. The mainstay of treatment is a low-fat diet, which is difficult to follow and compromises quality of life. New therapies are being developed primarily to prevent episodes of life-threatening acute pancreatitis.
Areas covered
Antagonists of apolipoprotein (apo) C-III, such as the antisense oligonucleotide (ASO) volanesorsen, significantly reduce triglyceride levels in chylomicronemia. However, approval of and access to volanesorsen are restricted since a substantial proportion of treated FCS patients developed thrombocytopenia. Newer apo C-III antagonists, namely, the ASO olezarsen (formerly AKCEA-APOCIII-LRx) and short interfering RNA (siRNA) ARO-APOC3, appear to show efficacy with less risk of thrombocytopenia. Potential utility of antagonists of angiopoietin-like protein 3 (ANGPTL3) such as evinacumab and the siRNA ARO-ANG3 in subtypes of chylomicronemia remains to be defined.
Expert opinion
Emerging pharmacologic therapies for chylomicronemia show promise, particularly apo C-III antagonists. However, these treatments are still investigational. Further study of their efficacy and safety in patients with both rare FCS and more common multifactorial chylomicronemia is needed.
1. Introduction
Primary chylomicronemia refers to a group of conditions in which there is a pathological accumulation of chylomicrons in the plasma, resulting in severe hypertriglyceridemia (HTG) [Citation1]. There are two main types of chylomicronemia: familial chylomicronemia syndrome (FCS) and multifactorial chylomicronemia syndrome (MCS) [Citation1]. FCS is an ultra-rare autosomal recessive disorder that is present from childhood and is characterized by triglyceride (TG) levels >10 mmol/L (>875 mg/dL) and is associated with an elevated risk of acute pancreatitis [Citation1,Citation2]. MCS, the more common form of chylomicronemia, is a polygenic condition expressed later in life, resulting from the combination of many genetic variants and secondary factors causing HTG [Citation2,Citation3].
The treatment of chylomicronemia is challenging because the current pharmacologic options are minimally effective [Citation1–4]. Treatment aims to reduce plasma TG levels to minimize the risk of developing complications, most notably acute pancreatitis, which can be life-threatening [Citation1–4]. The mainstay of the treatment for chylomicronemia, particularly FCS, is a strict, low-fat diet [Citation5–7]. However, compliance can be difficult and can compromise the patient’s quality of life [Citation5–7]. Treatment of MCS also relies on lifestyle measures and addressing the patient’s secondary predisposing factors, such as obesity, diabetes, alcohol consumption, and TG-raising medications [Citation2,Citation4,Citation8]. Traditional TG-lowering therapies, such as fibrates and omega-3 fatty acids, while somewhat effective in MCS, are ineffective in FCS since these patients lack lipolytic capacity [Citation1,Citation2,Citation8]. Given the current paucity of effective medications for FCS, there is great interest in developing novel pharmacologic treatments. Recent advances in TG-lowering therapies for FCS patients include antagonists of apolipoprotein (apo) C-III and angiopoietin-like protein type 3 (ANGPTL3), although each has limitations [Citation9–13]. Here, we review the safety and efficacy of existing and novel pharmacologic treatments in patients with chylomicronemia, focusing on FCS. Because FCS is so challenging to manage clinically, safe and effective treatments for FCS might also help in the management of MCS.
We performed a literature search of MEDLINE, EMBASE, and Cochrane Central Register of Controlled Trials from 1 January 1990 to 30 April 2022 using the following keywords: ANGPTL3 antagonists, apo C-III antagonists, chylomicronemia, familial chylomicronemia syndrome, multifactorial chylomicronemia, hypertriglyceridemia, and acute pancreatitis.
2. Chylomicronemia
2.1. Definition and subtypes
Primary chylomicronemia refers to the pathological presence of chylomicrons in the plasma after a fasting period of 12–14 hours, associated with severe HTG (i.e. TG >10 mmol/L or >875 mg/dL) [Citation1]. FCS, formerly called Fredrickson type 1 hyperlipoproteinemia or lipoprotein lipase (LPL) deficiency, is an ultra-rare disorder with essentially absent LPL activity resulting from inheritance of two copies of a pathogenic DNA variant (i.e. bi-allelic inheritance), leading to accumulation of chylomicrons together with relative deficiencies of other lipoprotein species [Citation2,Citation14]. In contrast, MCS, formerly called type 5 hyperlipoproteinemia is about 100 times more common than FCS and has a complex etiology arising from the combination of multiple inherited common genetic variants, and/or a single rare pathogenic variant, often in the context of secondary aggravating factors that worsen HTG and precipitate chylomicronemia [Citation3,Citation14]. Other lipoprotein species, such as very-low density lipoproteins (VLDL), are markedly elevated in MCS, while they are low to normal in FCS. Primary chylomicronemia has an estimated prevalence of ~1 in 400 to 500 adults in the general population, with >95% of cases being MCS [Citation1,Citation7,Citation14]. In contrast, monogenic recessive FCS accounts for <5% of cases and has an estimated prevalence of 1 in 100,000 to 1,000,000 in the general population [Citation1,Citation7]. The prevalence of MCS is likely to rise in the future due to the increasing rates of obesity, and type 2 diabetes mellitus.
2.2. Genetics and biochemistry
FCS arises from biallelic (i.e. two copies of) pathogenic loss-of-function variants in one of the five canonical lipolysis genes: LPL, APOC2, APOA5, LMF1, and GPIHBP1 [Citation1,Citation14]. The product of each gene plays an important role in the catabolism of TG-rich lipoproteins (see ). The functions of the non-LPL genes are as follows: APOC2 encodes apo C-II, an essential co-factor of LPL; APOA5 encodes apo A-V, which stabilizes the LPL-apo C-II complex; LMF1 encodes lipase maturation factor 1 (LMF1), which aids in the maturation and transport of LPL across the endoplasmic reticulum; and GPIHBP1 encodes glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 (GPIHBP1), a protein that mediates the transport of LPL into the capillary endothelium and anchors it to the endothelial surface [Citation3,Citation14].
Figure 1. TG-rich lipoprotein metabolism and novel drug targets. TG-rich lipoproteins are assembled from FA in the liver or intestine. CM are made in the small intestine and released into circulation via the lymphatic system, whereas VLDL is made in the liver and released directly into the blood stream. Both types of TG-rich lipoproteins are hydrolyzed by circulating LPL into respective remnant particles. CM remnants provide substrate for assembly of VLDL precursors in the liver, so completely impaired LPL function as in familial chylomicronemia syndrome is actually associated with deficiency of VLDL. In contrast, partially impaired LPL activity in multifactorial chylomicronemia syndrome is associated with elevations in CM, VLDL and their respective remnants. Several proteins modulate LPL activity and are targets for novel pharmacologic therapies for chylomicronemia. Most notably, apo C-III inhibits LPL activity and is the target of apo C-III antagonists (e.g. volanesorsen, olezarsen and ARO-APOC3). ANGPTL3 also inhibits LPL, as well as endothelial lipase activity, and antagonists of ANGPTL3 (e.g. evinacumab, vupanorsen and ARO-ANG3) are in development for the treatment of primary chylomicronemia. ANGPTL4 is another antagonist of LPL that may be a potential target for chylomicronemia treatment. Apo C-II is a co-factor for LPL and apo A-V stabilizes the LPL-apo C-II complex.
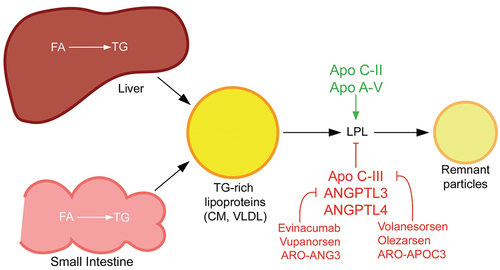
Pathogenic variants in LPL account for ~80–90% of FCS cases, while biallelic variants in one of the other four genes account for the remaining ~10–20% [Citation14,Citation15]. In patients with MCS, a different genetic pattern is seen: patients can have zero or just one copy (i.e. heterozygous) of a rare pathogenic variant in one of the five above canonical lipolysis genes, but more often there is a statistically significant accumulation of many common TG-raising DNA variants from throughout the genome. Each common variant has an individually small, incremental and reproducible TG-raising effect, but is not directly pathogenic for FCS, instead contributing to susceptibility to HTG [Citation6,Citation8,Citation14]. The degree of TG elevation in MCS can be just as severe as in FCS.
LPL encodes lipoprotein lipase (LPL), which is the main enzyme responsible for the lipolysis and uptake of TG into peripheral tissues [Citation16,Citation17]. LPL is synthesized in adipose tissue and skeletal and cardiac myocytes, and subsequently localizes to the endothelial surface [Citation16,Citation17]. When LPL is functioning normally, it hydrolyzes TG within chylomicrons, after which chylomicrons remnants are cleared from the plasma within 3–4 hours of eating [Citation17–19]. Rare pathogenic variants in the LPL gene that abolish lipolytic function result in increased levels of chylomicrons and persistent HTG even after fasting [Citation8,Citation16,Citation17]. The metabolic blockade is so severe that smaller lipoproteins normally formed by remodeling of chylomicrons are deficient [Citation3]. FCS due to biallelic variants in LPL versus non-LPL genes show approximately equivalent phenotypic severity, with some subtle differences [Citation15].
2.3. Clinical features
Chylomicronemia syndrome refers to the presence of >1 associated clinical feature in patients with primary chylomicronemia [Citation1]. Common clinical features include failure to thrive, nausea and vomiting, abdominal pain, eruptive xanthomas on the trunk and limbs, lipemia retinalis, and hepatosplenomegaly [Citation4,Citation19,Citation20]. Less common clinical features include anemia, intestinal bleeding, diarrhea, seizures, and encephalopathy [Citation7]. FCS develops in childhood, adolescence, or in early adulthood [Citation2]. In contrast, MCS tends to develop later in life, usually in adulthood, often in the context of a secondary medical condition or medication [Citation2]. Therefore, patients with MCS more often present with poorly controlled or undiagnosed diabetes mellitus, obesity, excessive alcohol intake, poor diet, chronic kidney disease, nephrotic syndrome, and hypothyroidism, all of which are associated with HTG [Citation2]. Clinical features of MCS are similar to those of FCS, but without the pediatric features such as failure to thrive [Citation2]. Moreover, patients with FCS tend to have normal or low weight, whereas patients with MCS have a high prevalence of overweight and obesity [Citation2].
2.4. Complications
The most serious complication of chylomicronemia is hyperlipidemic acute pancreatitis (HLAP), which can be recurrent and potentially fatal [Citation21]. HLAP is more likely to develop in FCS than MCS due to lifelong, sustained, and severe HTG [Citation2,Citation21]. The lifetime risk of HLAP in FCS is 60–80% compared to 10–20% in MCS [Citation3,Citation19]. Moreover, TG >20 mmol/L carry a 5–6% mortality associated with HLAP [Citation1].
HLAP is thought to occur in chylomicronemia when pancreatic lipase, which is normally an exocrine secretion, becomes mislocalized in pancreatic capillaries, leading to aberrant intravascular lipolysis and release of free fatty acids that activate trypsinogen causing autodigestion of the pancreas [Citation1]. The risk of HLAP has been shown to markedly decrease with the reduction of TG levels to <5.6 mmol/L (<500 mg/dl) [Citation22]. Complications of HLAP include recurrent or chronic pancreatitis, endocrine and exocrine insufficiency, pancreatic necrosis, pancreatic abscess, and pancreatic pseudocyst [Citation21,Citation23].
A further complication of MCS, but not FCS, is atherosclerotic cardiovascular disease (ASCVD) [Citation20,Citation24]. This occurs due to the accumulation of a much wider population of lipoprotein species, since metabolic blockade is only partial in MCS in contrast to FCS. Therefore, in addition to excess chylomicrons in MCS, there is also an accumulation of atherogenic species, such as remnant lipoproteins, which are small enough to penetrate the vessel wall leading to atherogenesis, and ultimately causing ASCVD [Citation20,Citation24]. In contrast, FCS patients develop an isolated chylomicronemia: chylomicrons are too large to penetrate the arterial wall. Furthermore, the low levels of smaller atherogenic lipoprotein species in FCS does not raise ASCVD risk [Citation20,Citation24]. Chylomicronemia is also associated with the development of nonalcoholic steatohepatitis with accumulation of intrahepatic TG, and also with susceptibility to insulin resistance [Citation2,Citation18].
2.5. Diagnosis
Chylomicronemia should be suspected in patients with a fasting plasma TG level >10 mmol/L [Citation25] and also with direct observation of opalescent, lactescent, or lipemic plasma. A thorough history and physical examination may yield evidence of clinical features of chylomicronemia syndrome and also of secondary factors linked to HTG, including diabetes mellitus, obesity, poor diet, excessive alcohol consumption, chronic kidney disease, and TG-raising medications [Citation1,Citation16,Citation25]. There have been several FCS diagnostic tools proposed; one such FCS scoring system was proposed by a panel of European experts, and includes features such as fasting TG >10 mmol/L, lack of secondary factors (except pregnancy and oral estrogens), a young age of onset, and a history of acute pancreatitis [Citation26]. A notable exception to the lack of secondary factors usually seen in FCS would be pregnancy and oral estrogens, as patients with HTG precipitated by these factors may still have FCS [Citation26]. This FCS scoring system has demonstrated a sensitivity of 88% and specificity of 85%, and is a useful clinical tool in differentiating FCS and MCS and identifying patients who are high-priority for diagnostic testing [Citation26]. The gold standard for diagnosing FCS is gene sequencing using a targeted panel [Citation1,Citation25]. Traditional biochemical assessment of lipolytic activity in plasma post-heparin infusion is not clinically available (only available as a research assay) and is often unreliable as the necessary skill and experience have been lost today compared to previous generations of clinical biochemists [Citation11].
3. Current treatment landscape and gaps for chylomicronemia patients
The treatment of chylomicronemia aims to lower TG levels primarily to prevent pancreatitis and secondarily to reduce the risk of ASCVD [Citation1,Citation27,Citation28]. Differentiating between FCS and MCS is important as the treatments are somewhat different. Treatment of MCS relies on a combination of lifestyle measures, addressing secondary factors exacerbating HTG, and standard TG-lowering therapies, such as fibrates and omega-3 fatty acids, often in combination with statin therapy to reduce ASCVD risk [Citation3,Citation4]. For FCS patients, the foundational treatment is dietary as these patients respond minimally to traditional TG-lowering therapies [Citation1,Citation4]. Also, new pharmacological therapies are currently in development for FCS, such as apo C-III and ANGPTL3 antagonists; these will be explored in detail below.
3.1. Treating secondary factors
Addressing treatable secondary factors is crucial for TG lowering in patients with chylomicronemia, particularly MCS [Citation2,Citation16,Citation29]. Clinical conditions associated with HTG include diabetes mellitus, obesity, chronic kidney disease, nephrotic syndrome, and hypothyroidism; optimizing these conditions will help to reduce TG levels [Citation2,Citation16,Citation29]. Medications associated with HTG include exogenous estrogen, beta-blockers, thiazide diuretics, second-generation antipsychotics, first-generation protease inhibitors, and corticosteroids [Citation18,Citation29]. If a patient with chylomicronemia happens to be on one of these medications, a less metabolically disruptive alternative should be sought [Citation18]. If there is no alternative, the lowest possible dose should be used.
3.2. Lifestyle measures
A strict, low-fat diet is the cornerstone of treatment for patients with chylomicronemia [Citation5]. It is recommended that patients with FCS limit fat intake to <15–20% of their daily calories (i.e. <30–50 g/day) [Citation6,Citation7]. This diet is recommended to reduce fatty acids in the body, which are constituents of triglycerides, and thus reduce the formation of chylomicrons [Citation6,Citation7]. Adhering to a low-fat diet is associated with improvement of clinical manifestations, including attenuation of hepatosplenomegaly and eruptive xanthomas, and decreased risk of pancreatitis [Citation1,Citation16]. However, the required extreme fat restriction can reduce quality of life and is challenging for patients to maintain over the long term. Medium-chain TG oil has been proposed as a supplement to increase the overall caloric intake; it is absorbed through the hepatic portal vein and metabolized through a chylomicron-independent pathway, so it does not contribute to chylomicron formation compared to longer-chain fatty acids [Citation5,Citation30,Citation31]. It is available primarily via prescription [Citation5,Citation30,Citation31]. Moreover, monitoring and supplementing fat-soluble vitamins (A, D, E, and K) is also recommended [Citation5].
Macronutrient composition also should be adjusted [Citation5,Citation30,Citation31]; patients with primary chylomicronemia should reduce their consumption of carbohydrates to <60% of their daily caloric intake since excess carbohydrates can promote increased production of hepatically derived VLDL [Citation5]. Complex carbohydrates are preferred, and simple, refined carbohydrates should be avoided [Citation5]. Furthermore, patients with chylomicronemia should limit their alcohol consumption both to reduce the production of TG-rich lipoproteins and to reduce pancreatitis risk [Citation5,Citation21,Citation32]. There are no specific physical activity guidelines for chylomicronemia, but the National Lipid Association suggests that patients with HTG should engage in 150 minutes of moderate-to-high intensity physical activity per week [Citation33].
3.3. Treatment of acute pancreatitis
The main complication of FCS is HLAP, which can be recurrent and life-threatening [Citation21]. The current treatment of HLAP is conservative: withholding oral intake (i.e. nil per os or NPO), intravenous fluid support, and analgesia if needed [Citation16,Citation19]. In patients with FCS, complete fasting cuts off any further dietary source of TG, allowing these patients’ compromised LPL to process the accumulated chylomicrons thus reducing TG levels [Citation34]. Intravenous insulin may also be of benefit in this setting, as it lowers TG levels by activating LPL and inhibiting hormone-sensitive lipase, particularly in patients with diabetes and uncontrolled hyperglycemia [Citation19,Citation35]. Therapeutic plasma exchange (TPE) has been suggested as a potential treatment for HLAP to rapidly lower TG levels. An observational study showed that TG levels were reduced by ~50% at 24 hours with conservative measures alone compared to ~70% with TPE [Citation36]. However, this additional ~20% reduction in TG levels was mostly lost after 48–72 hours with no benefit seen in acute clinical end points [Citation34,Citation36]. Therefore, the use of TPE is not currently recommended for the acute treatment of HLAP, nor is it recommended for chronic use since TG levels rebound without ongoing medical therapy [Citation34]. A possible exception might be chylomicronemia during pregnancy. TPE may also be considered in the refractory cases of HLAP, particularly in the small subset of FCS patients with apo-CII deficiency and normal LPL as the replacement of apo-CII would have longer-lasting benefits [Citation37].
3.4. Traditional pharmaceutical therapies for hypertriglyceridemia
Existing TG-lowering therapies are used to treat mild-to-moderate HTG but have minimal effect in primary chylomicronemia [Citation1,Citation3,Citation16]. Some of these therapies, such as fibrates, require a functional lipolytic pathway; thus, they may have some benefit in patients with MCS, but not in FCS, which is defined by essentially absent lipolysis [Citation1,Citation3,Citation16].
3.4.1. Statins
Statins inhibit 3-hydroxy-3-methylglutaryl-coenzyme A reductase, which results in the upregulation of hepatic low-density lipoprotein (LDL) receptors, acting to reduce LDL cholesterol levels [Citation33]. While they do not impact TG metabolism directly, statins are consistently associated with a 10–20% reduction in TG levels, which is thought to occur nonspecifically both via suppression of production and increased catabolism of specific receptor mediated uptake of TG-rich lipoprotein particles. Co-administration of a statin and ezetimibe has been shown to decrease the production of TG-rich lipoproteins, including chylomicrons, although the clinical impact is minor [Citation38]. Statins are sometimes given together with fibrates in patients with MCS, mainly for protection against ASCVD, but are not clinically useful in FCS [Citation16].
3.4.2. Fibrates
Fibrates are activators of peroxisome proliferator-activated receptors (PPARs), particularly the alpha isoform; their multiple effects include suppression of hepatic apo C-III production, which reduces the production of hepatic VLDL and disinhibits LPL activity [Citation39]. Fibrates can reduce TG levels by up to 50% in patients with a functional lipolytic pathway [Citation40], and via clinical experience are proven to be useful in patients with severe HTG due to MCS. However, in FCS patients with essentially no LPL activity, the response to fibrates is minimal [Citation1]. Available fibrates include gemfibrozil, fenofibrate, bezafibrate, and pemafibrate. The effect of fibrates on ASCVD reduction in statin-treated patients is currently unclear; there have been post-hoc analyses in fibrate trials suggesting ASCVD risk reduction in patients with HTG [Citation41]. The PROMINENT trial is the only study to specifically test this hypothesis, but the study was terminated early due to futility and results have not yet been published [Citation42]. Fibrates are still considered to have value in MCS patients with severe HTG where the clinical concern is acute pancreatitis, and possibly to treat nonalcoholic fatty liver disease.
3.4.3. Niacin
Niacin has been used clinically since the 1970s as a lipid lowering therapy and has multiple effects on the lipid profile. Although its mechanism of action is still uncertain, niacin is known to increase the expression of adipose tissue lipase, which improves tissue TG uptake and inhibits intracellular diacylglycerol O-acyltransferase 2, which in turn, decreases hepatic VLDL synthesis [Citation43]. Niacin can lower TG levels by 5–35% [Citation44]. Clinical experience indicates that in MCS patients who have HTG due to high levels of chylomicrons and VLDL, niacin lowers total TG primarily by lowering VLDL [Citation1]. Experience also indicates that effects in FCS patients are minimal. Niacin has several side effects, including insulin resistance, flushing, pruritis, and light-headedness [Citation44]. Furthermore, niacin failed to reduce ASCVD in clinical trials, leading to its withdrawal from many markets [Citation45].
3.4.4. Omega-3 fatty acids
Omega-3 fatty acids, specifically eicosapentaenoic acid (EPA) moderately reduce TG levels, possibly by inhibiting synthesis of hepatic VLDL and inhibiting apo C-III [Citation1,Citation46]. Some uncontrolled case series suggest the potential efficacy of omega-3 fatty acids in primary chylomicronemia, predominantly MCS [Citation46–48], but no controlled trials have been performed [Citation1]. In the past, some opinion leaders have advised against using additional oral fatty acids, including omega-3 fatty acids in patients with FCS, but clinical experience indicates that any impact on TG in this situation is likely neutral rather than deleterious.
Icosapent ethyl (IPE), a highly purified EPA ethyl ester, has shown benefit in ASCVD risk reduction in patients with HTG without chylomicronemia [27]. In the REDUCE-IT trial, the risk of ischemic events, including cardiovascular death, was 25% lower in the IPE group compared to placebo [Citation27]. The median reduction in TG was 19.7% greater in the IPE group, compared to placebo [Citation27]. Thus, this study demonstrates a potential association between TG-lowering and ASCVD risk reduction, which has not been previously established in the literature. Another study, the STRENGTH trial, demonstrated that among statin-treated patients with a high cardiovascular risk, a mixture of EPA and docosahexaenoic acid did not significantly reduce major cardiovascular events compared to corn oil [Citation28]. Combined, the results of the REDUCE-IT and STRENGTH trial suggest that pure IPE, but not other omega-3 fatty acid mixtures, is associated with ASCVD risk reduction, which has important implications for patients with MCS [Citation27,Citation28].
3.4.5. Lomitapide
Lomitapide is a microsomal TG transfer protein (MTTP) inhibitor that lowers TG levels by preventing the transfer of TG to nascent apo B-containing lipoproteins, including chylomicrons [Citation49–51]. It is approved in North America and Europe for the treatment of homozygous familial hypercholesterolemia (HoFH) but not FCS Citation1,Citation52]. A case study of a FCS patient with recurrent severe pancreatitis episodes annually for decades who was treated with lomitapide showed a reduction in TG levels by 60–70% with no further episodes of pancreatitis while on treatment [Citation50], although the patient eventually developed hepatic fibrosis. Common mechanism-related side effects of lomitapide include nausea, vomiting, and diarrhea, though these can be mitigated by gradual dose escalation [Citation49,Citation50,Citation52]. Patients taking lomitapide are instructed to follow a low-fat diet, which can help to reduce gastrointestinal side effects [Citation52]. A more serious side effect of lomitapide is a fatty liver with transaminase elevation, although this is reversible with dose de-escalation or cessation [Citation49,Citation50,Citation52]. Widespread use of lomitapide has been limited by its cost and adverse gastrointestinal and hepatic effects.
4. Novel pharmacologic therapies for chylomicronemia
Given the lack of efficacy of current pharmacologic therapies, and considering the complications and poor quality of life experienced by FCS patients in particular, there has been much interest in the development of novel therapies for these patients. Because of the overlap in phenotypic features, it may seem intuitive that novel agents that are effective for FCS patients will automatically be effective in MCS patients, and vice versa, though this is not necessarily the case. The most promising new agents are apo C-III antagonists, and others that are being evaluated include ANGPTL3 antagonists (see ).
Table 1. Emerging treatments for chylomicronemia
Apo C-III is a 79 amino acid protein produced in the liver and small intestine, encoded by the APOC3 gene on chromosome 11q23 [Citation11,Citation53–55]. Apo C-III is a component of hepatically and probably intestinally secreted TG-rich lipoproteins and acts to decrease LPL activity, leading to impaired lipolysis of TG-rich lipoproteins [Citation56,Citation57]. Apo C-III offsets the activity of apo C-II, which is a necessary cofactor for LPL activity. Blocking apo C-III removes hindrance upon apo C-II, increasing LPL activity and ultimately decreasing TG levels [Citation54]. Antagonizing apo C-III acts to reduce TG-rich lipoprotein particles, either at the level of production or perhaps by promoting non-catalytic receptor mediated removal [Citation54]. Genetic studies have consistently shown that naturally occurring loss-of-function variants of APOC3 are associated with reduced TG levels and reduced risk of ASCVD [Citation58–60]. This spurred the development of new therapies targeting apo C-III for treatment across a range of HTG disease states, including FCS. There are three novel therapies targeting apo C-III currently in development: volanesorsen, olezarsen (AKCEA-APOCIII-LRx), and ARO-APOC3.
4.1. Anti-apo C-III antisense oligonucleotide: volanesorsen
Volanesorsen (ISIS 304810; Akcea-Ionis Pharmaceuticals) is a 20-nucleotide partial 2’-O-methoxyethyl gapmer that functions by entering the nucleus, binding to its complementary mRNA and inhibiting the translation of APOC3 mRNA [Citation11,Citation54]. There have been two phase 3 trials studying volanesorsen. The first, the APPROACH trial (A Study of ISIS 304801 in Patients With Familial Chylomicronemia Syndrome, NCT02211209), was a phase 3, multi-center, randomized, double-blind, placebo-controlled trial investigating the safety and efficacy of volanesorsen in patients with FCS with fasting TG ≥8.4 mmol/L [Citation12]. This trial enrolled 66 FCS patients of whom about 80% had biallelic LPL loss-of-function variants: 33 of them received volanesorsen 300 mg subcutaneously once weekly and 33 received matching placebo. At 3 months, the study found that patients receiving volanesorsen had an 84% reduction in mean plasma apo C-III levels compared to a 6.1% increase in the placebo group (p < 0.001). Correspondingly, the study found a 77% reduction in mean plasma TG levels in the volanesorsen group compared to an 18% increase in the placebo group (p < 0.001). Furthermore, 77% of participants in the volanesorsen group achieved TG levels <8.5 mmol/L compared to 10% in the placebo group. Volanesorsen was also found to be associated with decreases in chylomicron TG (by 83%), apo B-48 (by 76%), non-HDL cholesterol (by 46%), and VLDL cholesterol (by 58%), and was associated with increases in HDL cholesterol (by 46%), apo A1 (by 14%), LDL cholesterol (by 136%), and total apo B (by 20%). The main adverse event of volanesorsen identified from this study was thrombocytopenia. Thrombocytopenia, with platelet levels <100,000/μL, occurred in 15 out of 33 participants in the volanesorsen group compared to none in the placebo group. Two participants in the volanesorsen group had platelets <25,000/μL, which recovered after cessation of volanesorsen.
Subsequent to the APPROACH trial, the open-label extension trial called ReFOCUS (Retrospective Findings and Observations Captured in Burden of Illness Survey in FCS Patients) was a retrospective, global, web-based survey aiming to evaluate the quality of life of patients who had received volanesorsen for more than 3 months [Citation61]. The study found that patients who received volanesorsen had reduced symptoms, including reduced steatorrhea and pancreatic pain, with improved quality of life and decreased impact of their disease on work and school responsibilities.
The second phase 3 trial studying volanesorsen was the COMPASS trial (A Study of Volanesorsen in Patients With Hypertriglyceridemia, NCT02211209), a multi-center, randomized, double-blind, placebo-controlled trial in patients with MCS, referred to as multifactorial severe HTG [Citation62]. The study inclusion criteria included fasting plasma TG >5.65 mmol/L (>500 mg/dL). This trial spanned 26 weeks and enrolled 224 patients, 76 of whom received volanesorsen 300 mg subcutaneously once weekly and 38 of whom received matching placebo. At 3 months, the volanesorsen group had a 71% reduction in mean plasma TG levels from baseline compared to a 0.9% reduction in the placebo group (p < 0.0001). Adverse events included injection-site reactions: 24% vs 0.2% in volanesorsen vs placebo group, respectively. One patient had thrombocytopenia (i.e. platelets <50,000/μL) and another had serum sickness, both of whom were in the volanesorsen group. Five episodes of acute pancreatitis occurred during the study treatment period, which occurred in three patients in the placebo group.
A subset of patients with lipodystrophy can develop severe HTG and chylomicronemia [Citation63]. In this regard, BROADEN (A Study of Volanesorsen [Formerly ISIS-APOCIIIRx] in Patients With Familial Partial Lipodystrophy, NCT02527343) is an ongoing randomized, double-blind, placebo-controlled trial evaluating the efficacy of volanesorsen in reducing fasting TG levels in patients with familial partial lipodystrophy.
Volanesorsen has faced some headwinds in efforts to attain approval for clinical use. The main challenge has been the high prevalence of treatment-associated thrombocytopenia, which the sponsor has attempted to mitigate by proposing a monitoring and down-titration protocol for patients with severe HTG. To date, this approach has not been accepted by North American regulators, although the drug is available through compassionate use and special access programs. Volanesorsen has been approved in Europe for patients with FCS [Citation54].
4.2. Anti-apo C-III targeted antisense oligonucleotide: olezarsen
Other pharmaceuticals targeting apo C-III are in earlier phases of development. Olezarsen (Akcea-Ionis Pharmaceuticals; formerly AKCEA-APOCIII-LRx) is an N-acetylgalactosamine-(GalNac) conjugated antisense oligonucleotide (ASO) targeting apo C-III [Citation64]. The GalNac conjugation allows the attached ASO to reach much lower concentrations than volanesorsen and to enter hepatocytes with tremendous efficiency via the asialoglycoprotein receptor. In theory, this formulation provides superior apo C-III antagonism with a much lower risk of adverse effects. A phase 1/2a, double-blind, placebo-controlled, dose–escalation study in patients with HTG showed that a single dose of olezarsen 10, 30, 60, 90, or 120 mg was associated with a median reduction in plasma TG levels by −12%, −7%, −42%, −73%, and −77% after 14 days, respectively [Citation64]. There was only one injection-site reaction of mild erythema, and no evidence of thrombocytopenia. There is currently an ongoing phase 3, multi-center, randomized, double-blind, placebo-controlled study called BALANCE (NCT04568434) evaluating its efficacy in patients with FCS.
4.3. Anti-apo C-III short interfering RNA: ARO-APOC3
Another apo C-III antagonist currently in development is ARO-APOC3 (Arrowhead Pharmaceuticals), a hepatocyte-targeted RNA interference therapeutic targeting apo C-III [Citation64,Citation65]. In preliminary results of a phase 1 trial evaluating the safety and key pharmacodynamics and lipid parameters in patients with severe HTG, ARO-APOC3 was associated with a maximal mean reduction of −80% to −99% in apo C-III levels, and a maximal mean reduction of −74% to −92% in plasma TG levels [Citation65]. In patients with chylomicronemia, ARO-APOC3 was associated with a maximal mean reduction of −98% in apo C-III levels, and a maximal mean reduction of −88% in TG levels. There is an ongoing phase 2b, randomized, double-blind, placebo-controlled study called AROAPOC3-2001 (NCT04720534) investigating the efficacy of ARO-APOC3 in patients with severe HTG. There is also an ongoing phase 3, randomized, double-blinded, placebo-controlled trial called AROAPOC3-3001 (NCT05089084), recently started in January 2022, evaluating the efficacy and safety of ARO-APOC3 in patients with FCS.
4.4. Anti-angiopoietin-like protein 3 monoclonal antibody: evinacumab
Angiopoietin-like protein 3 (ANGPTL3) is a circulating protein encoded by the ANGPTL3 gene on chromsome 1p31 that is made in the liver and regulates lipid metabolism in part by inhibiting LPL and endothelial lipase activity [Citation66–68]. There is also possibly an intracellular function of ANGPTL3 that may govern the production and secretion of apo B containing lipoproteins. Rare, naturally occurring loss-of-function DNA variants of ANGPTL3 have been associated with reduced plasma levels of both TG levels and LDL cholesterol, and with protection from ASCVD [Citation68].
Evinacumab (brand name Evkeeza, Regeneron) is a monoclonal antibody that targets circulating ANGPTL3 [Citation69]. It is currently approved by the U.S. Food and Drug Administration for patients with HoFH based on the results of the ELIPSE HoFH trial [Citation70]. This randomized study was performed in 65 participants with HoFH, a condition that is characterized by severely elevated LDL cholesterol with basically normal TG levels. The drug was administered intravenously on a monthly basis. The investigators observed a 47% reduction in LDL cholesterol in the evinacumab group compared to a 2% increase in the placebo group at 24 weeks [Citation69]. There was also a 55% decrease in TG levels (from an essentially normal baseline) in patients receiving evinacumab compared to a 4.6% reduction in the placebo group. This indicates remarkable biochemical efficacy in an orphan condition, which responds poorly to traditional therapy and requires lifelong serial apheresis. There is currently an ongoing phase 3, open-label study assessing the long-term efficacy of this drug in HoFH (NCT03409744).
To evaluate the potential of evinacumab in patients with HTG, a phase 2, randomized, double-blind, placebo-controlled trial of evinacumab in patients with severe HTG with at least one prior hospitalization for acute pancreatitis was performed [Citation71]. One of the cohorts in this study included only patients with FCS; in that cohort, evinacumab was not associated with a reduction in TG levels compared to placebo. However, there were meaningful reductions in TG levels with evinacumab in the MCS cohorts, who do retain some residual LPL activity since these patients are not completely LPL deficient. This suggests that targeting circulating ANGPTL3 extracellularly requires the presence of at least some functional LPL activity. It is also possible that much higher doses of evinacumab may be required when LPL activity is absent. In this study, there were a similar number of adverse events between the evinacumab and placebo groups, including abdominal pain, headache, and HLAP. Most of the HLAP events in the evinacumab group occurred during the off-drug period >4 weeks after the last dose of evinacumab, when TG levels had begun rising to pre-treatment levels and evinacumab levels were subtherapeutic. A study is planned to evaluate evinacumab in MCS patients (NCT03175367).
4.5. Anti-angiopoietin-like protein 3 targeted ASO: vupanorsen
Vupanorsen (IONIs-ANGPTL3-LRx, Akcea, and Pfizer) is an GalNac conjugated ASO therapy targeting ANGPTL3 mRNA in hepatocytes by efficient delivery through the cell surface asialoglycoprotein receptor [Citation72,Citation73]. There was some initial excitement about the potential value of vupanorsen in patients with moderate HTG and insulin resistance, as suggested by a phase 2, randomized, double-blind, placebo-controlled study, which showed a reduction of TG and LDL cholesterol by up to 63.1% and 32.9%, respectively [Citation73]. However, in a phase 3 dose-finding study among patients with combined hyperlipidemia, vupanorsen was observed to have only modest lipid lowering effects that were associated with elevations in liver enzymes (alanine aminotransferase and aspartate aminotransferase) and increased hepatic fat, leading to the termination of the drug development program in early 2022 [Citation74]. These adverse effects were not observed with evinacumab, which only interacts with extracellular ANGPTL3, suggesting that interfering with intracellular ANGPTL3 through RNA silencing may target an additional mechanism that promotes a net negative metabolic outcome.
4.6. Anti-ANGPTL3 short interfering RNA: ARO-ANG3
ARO-ANG3 (Arrowhead Pharmaceuticals) is an RNA interference therapeutic targeting ANGPTL3 [Citation75]. In a phase 1 trial of patients with heterozygous familial hypercholesterolemia, there was a 25–43% reduction in TG levels in the ARO-ANG3 group [Citation75]. There is currently an ongoing phase 2b, randomized, double-blind, placebo-controlled trial to evaluate the efficacy and safety of ARO-ANG3 in patients with mixed dyslipidemia (NCT04832971). There is another phase 2 study of ARO-ANG3 planned to begin in June 2022 in patients with HoFH (NCT05217667).
4.7. Other suspended therapies
In addition to vupanorsen, we mention briefly additional discontinued therapies that were initially thought to be promising for severe HTG (see ). Alipogene tiparvovec (Glybera, uniQure) was an LPL gene therapy that used adeno-associated viral vector 1 engineered to express a common gain-of-function LPL variant [Citation76–79]. It required intramuscular injection and was transiently approved for use in Europe until 2017 at which time the sponsor did not pursue marketing authorization renewal due to limited usage, high cost, and transient efficacy [Citation17,Citation18].
Table 2. Discontinued treatments for chylomicronemia
Pradigastat (Novartis) is an oral, small-molecule inhibitor of diacylglycerol O-acyltransferase 1 (DGAT1), an enzyme essential for TG synthesis found in intestine, liver, and adipose tissue. Pradigastat reduced the fasting TG levels by 70% in six FCS patients [Citation80–84], but unfortunately, it was poorly tolerated with severe gastrointestinal side effects, which seems to be a common feature among agents that target DGAT1 [Citation84,Citation85].
4.8. Other potential targets for TG reduction
Potential targets for future therapies for severe HTG include apo C-II and angiopoietin-like protein 4 (ANGPTL4). Apo C-II is a co-factor for LPL, but therapies to enhance apo-CII expression would primarily be useful for the extremely small subgroup of patients (2–3%) with FCS who have apo C-II deficiency [Citation20,Citation86]. It is theoretically possible that stimulating or supplement apo C-II can positively affect the apo C-II to apo C-III ratio and thus promote LPL activity, but effective antagonism of apo C-III would seem to be a more efficient strategy. Finally, ANGPTL4 is a protein that regulates LPL, similar to ANGPTL3; however, preclinical trials have shown an association between ANGPTL4 inhibition and the development of mesenteric adenitis, so this target has not been pursued further [Citation87]. Finally, APOA5 endcoding apo A-V has been considered as another drug target that when upregulated theoretcialy could reduce TG levels [Citation56].
5. Conclusion
Primary chylomicronemia refers to the pathological accumulation of chylomicrons in the plasma, resulting in severe HTG [Citation1]. There are two main types of primary chylomicronemia: ultra-rare FCS and the more prevalent MCS [Citation2]. The treatment of MCS relies on addressing secondary causes, and standard TG-lowering therapies have shown some efficacy in this population [Citation2]. In contrast, FCS patients lack lipolytic capacity, and have a minimal response to conventional therapies [Citation19]. Therefore, the treatment of FCS relies on a strict low-fat diet, which can be difficult to adhere to and leads to a reduced quality of life [Citation5,Citation19]. Apo C-III antagonists, specifically volanesorsen, olezarsen, and ARO-APOC3, are most promising in reducing TG levels in patients with FCS and patients with more common MCS. Anti-ANGPTL3 therapies appear to be effective in MCS but are perhaps less so in FCS. Although FCS is an ultra-rare disease, successful development of safe and effective therapies for these patients will likely be translatable for other more common and etiologically complex forms of severe HTG.
6. Expert opinion
The development of novel pharmacological agents for chylomicronemia and particularly FCS has been an area of great interest for endocrinologists, lipidologists, and pharmaceutical researchers and developers in recent years. There is a disconcerting lack of effective therapies for this indication to help reduce the significant risk of pancreatitis in this condition [Citation1,Citation2]. The most promising target being studied is apo C-III. The value of apo C-III as a target has been demonstrated by the efficacy of the apo C-III ASO antagonist, volanesorsen, which is currently approved for the treatment of FCS in Europe, but not in other jurisdictions [Citation61]. Volanesorsen has shown excellent efficacy in lowering TG levels in patients with FCS (TG reduction of approximately 70–80%) plus improvement in the rest of the lipid profile and in quality of life; however, it has also been associated with thrombocytopenia, limiting its use [Citation10–12]. In our opinion, other apo C-III antagonists, such as the ASO olezarsen and siRNA ARO-APOC3, show promise for this difficult to manage medical condition. Both agents are in earlier stages of development. Both have also shown excellent efficacy in lowering TG levels in HTG, and phase 3 trials are underway investigating their efficacy in patients with severe HTG and with FCS specifically [Citation65]. Olezarsen and ARO-APOC3 also appear to carry a lower risk of thrombocytopenia compared to volanesorsen. We suspect that if these agents ultimately show efficacy in improving severe HTG in FCS patients they might also be prove to be useful in the more heterogeneous and more common MCS subtype of chylomicronemia. There are theoretical reasons to consider that these agents will help reduce ASCVD risk in patients with MCS or with less severe forms of HTG, but such studies will need to be undertaken specifically. Alleviating the blocked processing of chylomicrons could increase levels downstream of smaller atherogenic lipoproteins; a randomized clinical trial would sort out risks versus benefits.
Another molecular target, ANGPTL3, is theoretically appealing, but both monoclonal antibody and RNA silencing antagonists have faced some challenges in recent clinical trials. Specifically, the anti-ANGPTL3 monoclonal antibody evinacumab shows good efficacy in patients with severe HTG due to MCS, but attenuated (sometimes neutral) efficacy in FCS. Furthermore, development of the anti-ANGPTL3 ASO vupanorsen was recently terminated due to only modest efficacy with respect to TG lowering combined with adverse hepatic effects, including development of fatty liver. It will be important to evaluate whether the siRNA ARO-ANG3 has similar efficacy and adverse effects to vupanorsen. If similar adverse effects are seen, it might suggest that targeting intracellular ANGPTL3 via RNA silencing may yield less net clinical benefit than targeting extracellular ANGPTL3 via a monoclonal antibody, although the latter approach may still possibly have limited efficacy in patients with FCS and perhaps better efficacy in MCS. The totality of evidence from ANGPTL3 antagonist clinical studies to date suggest that 1) some degree of LPL activity is needed when targeting extracellular circulating ANGPTL3; and 2) targeting intrahepatocellular ANGPTL3 may inhibit some intracellular function of ANGPTL3 that may affect lipoprotein assembly and secretion. The clearest and most consistent clinical benefit of evinacumab has been observed in patients with severe hypercholesterolemia rather than in patients with severe HTG.
Finally, liver-targeted genome-editing approaches using clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR associated protein 9 (Cas9) technology are being actively investigated as a potential permanent corrective treatment for homozygous FH, focusing on knocking out functional proprotein convertase subtilisin kexin type 9 [Citation88]. Since some portion of apo C-III is liver derived, an analogous approach might in theory be applicable to FCS. However, this treatment modality applied to humans is in its pre-infancy stages and while it is extremely exciting and holds great promise for the future, technical issues related to both safety and efficacy need to be clearly established before it can be considered for FCS patients.
Article highlights
Primary chylomicronemia refers to the pathological accumulation of chylomicrons in plasma resulting in severe hypertriglyceridemia (i.e. triglycerides [TG] >10 mmol/L or >875 mg/dL).
Familial chylomicronemia syndrome (FCS), an ultra-rare subtype of chylomicronemia, occurs in patients with two copies of a loss-of-function variant in one of the five canonical lipolysis genes, most commonly LPL, resulting in the absence of lipolytic capability.
Multifactorial chylomicronemia syndrome (MCS), a genetically complex disorder, is ~100-times more prevalent than FCS, but patients can have comparably elevated TG levels.
FCS patients respond minimally to standard TG-lowering therapies; treatment to prevent acute pancreatitis relies on a very low-fat diet, which compromises quality of life.
MCS patients respond somewhat better to TG-lowering therapies and management of secondary factors, but TG levels and pancreatitis risk remain high.
Emerging therapies include apo C-III antagonists, i.e. volanesorsen, olezarsen, and ARO-APOC3, to which patients respond consistently well, and ANGPTL3 antagonists, i.e. evinacumab and ARO-ANG3, to which FCS patients respond inconsistently.
Investigational treatments in FCS require further study of efficacy and safety; if confirmed, their use may encompass patients with more common MCS.
Declaration of interest
RA Hegele reports consulting fees from Acasti, Aegerion, Akcea/Ionis, Amgen, Arrowhead, HLS Therapeutics, Novartis, Pfizer, Regeneron and Sanofi. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
A reviewer on this manuscript has disclosed consulting/advisory fees: Ionis, Akcea and Novartis; and being an investigator: Akcea, Amgen, AstraZeneca, Ionis, Novartis, Regeneron and RegenXBio. Another reviewer on this manuscript has disclosed being a site clinical investigator for trials sponsored by Amgen, Biomarin, Regeneron and Akcea. Peer reviewers on this manuscript have no other relevant financial relationships or otherwise to disclose.
Additional information
Funding
References
- Brahm AJ, Hegele RA. Chylomicronaemia - current diagnosis and future therapies. Nat Rev Endocrinol. 2015;11:352–362.
- Paquette M, Bernard S, Hegele RA, et al. Chylomicronemia: differences between familial chylomicronemia syndrome and multifactorial chylomicronemia. Atherosclerosis. 2019;283:137–142.
- Chait A, Eckel RH. The chylomicronemia syndrome is most often multifactorial. Ann Intern Med. 2019;170:626–634.
- Paragh G, Á N, and Harangi M, et al. Causes, clinical findings and therapeutic options in chylomicronemia syndrome, a special form of hypertriglyceridemia. Lipids Health Dis. 2022;21:21.
- Williams L, Rhodes KS, Karmally W, et al. Familial chylomicronemia syndrome: bringing to life dietary recommendations throughout the life span. J Clin Lipidol. 2018;12:908–919.
- Hegele RA, Pollex RL. Hypertriglyceridemia: phenomics and genomics. Mol Cell Biochem. 2009;326:35–43.
- Hegele RA, Borén J, Ginsberg HN, et al. Rare dyslipidaemias, from phenotype to genotype to management: a European Atherosclerosis Society task force consensus statement. Lancet Diabetes Endocrinol. 2020;8(1):50–67.
- Hegele RA, Ginsberg HN, Chapman MJ, et al. The polygenic nature of hypertriglyceridaemia: implications for definition, diagnosis, and management. Lancet Diabetes Endocrinol. 2014;2(8):655–666.
- Nurmohamed NS, Dallinga–Thie GM, Stroes ESG. Targeting apoC-III and ANGPTL3 in the treatment of hypertriglyceridemia. Expert Rev Cardiovasc Ther. 2020;18(6):355–361.
- Reeskamp LF, Tromp TR, Stroes ESG. The next generation of triglyceride-lowering drugs: will reducing apolipoprotein C-III or angiopoietin like protein 3 reduce cardiovascular disease? Curr Opin Lipidol. 2020;31:140–146.
- Lazarte J, and Hegele RA. Volanesorsen for treatment of familial chylomicronemia syndrome. Expert Rev Cardiovasc Ther. 2021;19:685–693.
- Witztum JL, Gaudet D, and Freedman SD, et al. Volanesorsen and triglyceride levels in familial chylomicronemia syndrome. N Engl J Med. 2019;381:531–542.
- Kersten S. ANGPTL3 as therapeutic target. Curr Opin Lipidol. 2021;32:335–341.
- Dron JS, Hegele RA. Genetics of hypertriglyceridemia. Front Endocrinol. 2020;11:455.
- Hegele RA, Berberich AJ, Ban MR, et al. Clinical and biochemical features of different molecular etiologies of familial chylomicronemia. J Clin Lipidol. 2018;12:920–927.
- Berberich AJ, Hegele RA. A modern approach to dyslipidemia. Endocr Rev. 2021;2:bnab037.
- Laufs U, Parhofer KG, Ginsberg HN, et al. Clinical review on triglycerides. Eur Heart J. 2020;41:99–109.
- Goldberg RB, Chait A. A comprehensive update on the chylomicronemia syndrome. Front Endocrinol. 2020;11:593931.
- Baass A, Paquette M, Bernard S, et al., Familial chylomicronemia syndrome: an under-recognized cause of severe hypertriglyceridaemia. 2020;J Inter Med. 287:340–348.
- Brown WV, Gaudet D, Goldberg I, et al. Roundtable on etiology of familial chylomicronemia syndrome. J Clin Lipidol. 2018;12:5–11.
- Valdivielso P, Ramírez-Bueno A, Ewald N. Current knowledge of hypertriglyceridemic pancreatitis. Eur J Intern Med. 2014;25:689–694.
- Gaudet D, Signorovitch J, Swallow E, et al. Medical resource use and costs associated with chylomicronemia. J Med Econ. 2013; 16: 657–666.
- Sisman G, Erzin Y, Hatemi I, et al. Familial chylomicronemia syndrome related chronic pancreatitis: a single-center study. Hepatobiliary Pancreatic Dis Intl. 2014; 13: 209–214.
- Nordestgaard BG, Benn M, Schnohr P, et al. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. 2007;298:299–308.
- Stroes E, Moulin P, Parhofer KG, et al. Diagnostic algorithm for familial chylomicronemia syndrome. Atherosclerosis Suppl. 2017;23:1–7.
- Moulin P, Dufour R, Averna M, et al. Identification and diagnosis of patients with familial chylomicronaemia syndrome (FCS): expert panel recommendations and proposal of an “FCS score.” Atherosclerosis. 2018;275:265–272.
- Bhatt DL, Steg PG, Miller M, et al. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N Engl J Med. 2019;380(1):11–22.
- Nicholls SJ, Lincoff AM, Garcia M, et al. Effect of high-dose omega-3 fatty acids vs corn oil on major adverse cardiovascular events in patients at high cardiovascular risk: the STRENGTH randomized clinical trial. JAMA. 2020;324(22):2268–2280.
- Berberich AJ, Hegele RA. Secondary causes of chylomicronemia: defining the underside of the iceberg. J Intern Med. 2018;283:405–407.
- Rouis M, Dugi KA, Previato L, et al. Therapeutic response to medium-chain triglycerides and ω-3 fatty acids in a patient with the familial chylomicronemia syndrome. Arterioscler Thromb Vasc Biol. 1997;17:1400–1406.
- Ahmad Z, Wilson DP. Familial chylomicronemia syndrome and response to medium-chain triglyceride therapy in an infant with novel mutations in GPIHBP1. J Clin Lipidol. 2014;8:635–639.
- Yadav D, Hawes RH, Brand RE, et al. Alcohol consumption, cigarette smoking, and the risk of recurrent acute and chronic pancreatitis. Arch Intern Med. 2009;169:1035–1045.
- Jacobson TA, Maki KC, Orringer CE, et al. National Lipid Association recommendations for patient-centered management of dyslipidemia: part 2. J Clin Lipidol. 2015; 9: S1–S122.e1.
- Berberich AJ, Ziada A, Zou GY, et al. Conservative management in hypertriglyceridemia-associated pancreatitis. J Intern Med. 2019;286:644–650.
- Inayat F, Zafar F, Baig AS, et al. Hypertriglyceridemic pancreatitis treated with insulin therapy: a comparative review of 34 cases. Cureus. 2018;10(10):548.
- Chen Z, Huang X, Zhang M, et al. Rapid reduction in triglyceride levels by therapeutic plasma exchange in patients with hypertriglyceridemic pancreatitis. J Clin Apheresis. 2022;37:82–90.
- Wolska A, Dunbar RL, Freeman LA, et al. Apolipoprotein C-II: new findings related to genetics, biochemistry, and role in triglyceride metabolism. Atherosclerosis. 2017;267:49–60.
- Tremblay AJ, Lamarche B, Hogue J-C, et al. Effects of ezetimibe and simvastatin on apolipoprotein B metabolism in males with mixed hyperlipidemia. J Lipid Res. 2009;50(7):1463–1471.
- Yuan G, Al-Shali KZ, Hegele RA. Hypertriglyceridemia: its etiology, effects and treatment. Can Med Assoc J. 2007;176:1113–1120.
- Wolska A, Yang ZH, Remaley AT. Hypertriglyceridemia: new approaches in management and treatment. Curr Opin Lipidol. 2020;31:331–339.
- Nordestgaard BG, Varbo A. Triglycerides and cardiovascular disease. Lancet. 2014;384:626–635.
- Kowa Research Institute, Inc. Kowa to discontinue K-877 (Pemafibrate) “Prominent” Cardiovascular outcomes study [Internet]. Chicago IL: Cision PR Newswire; April 2022. [cited 2022 May 28]. Available from: https://www.prnewswire.com/news-releases/kowa-to-discontinue-k-877-pemafibrate-prominent-cardiovascular-outcomes-study-301520956.html
- Ganda OP. When to lower triglycerides? Curr Opin Lipidol. 2020;31:238–245.
- Goldberg A, Alagona P, Capuzzi DM, et al. Multiple-dose efficacy and safety of an extended-release form of niacin in the management of hyperlipidemia. Am J Cardiol. 2000;85:1100–1105.
- Gotto AM, Moon JE. Pharmacotherapies for lipid modification: beyond the statins. 2013;Nature Rev Cardiol. 10:560–570.
- Sherratt SCR, Lero M, Mason RP. Are dietary fish oil supplements appropriate for dyslipidemia management? A review of the evidence. Curr Opin Lipidol. 2020;31:94–100.
- Pschierer V, Richter WO, Schwandt P. Primary chylomicronemia in patients with severe familial hypertriglyceridemia responds to long-term treatment with (n-3) fatty acids. J Nutrition. 1995;125:1490–1494.
- Richter WO, Jacob BG, Ritter MM, et al. Treatment of primary chylomicronemia due to familial hypertriglyceridemia by omega-3 fatty acids. Metabolism. 1992;41:1100–1105.
- Bajaj A, Cuchel M. Homozygous familial hypercholesterolemia: what treatments are on the horizon? Curr Opin Lipidol. 2020;31:119–124.
- Sacks FM, Stanesa M, Hegele RA. Severe hypertriglyceridemia with pancreatitis: thirteen years’ treatment with lomitapide. JAMA Intern Med. 2014;174(3):443–447.
- Cuchel M, Meagher EA, du Toit Theron H, et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381:40–46.
- deGoma EM. Lomitapide for the management of homozygous familial hypercholesterolemia. Rev Cardiovasc Med. 2014;15:109–118.
- Basu D, Goldberg IJ. Regulation of lipoprotein lipase-mediated lipolysis of triglycerides. Curr Opin Lipidol. 2020;31:154–160.
- Hegele RA. Apolipoprotein C-III inhibition to lower triglycerides: one ring to rule them all? Eur Heart J. 2022;43:1413–1415.
- Gouni-Berthold I. The role of antisense oligonucleotide therapy against apolipoprotein-CIII in hypertriglyceridemia. Atherosclerosis Suppl. 2017;30:19–27.
- Hegele RA. Multidimensional regulation of lipoprotein lipase: impact on biochemical and cardiovascular phenotypes. J Lipid Res. 2016;57:1601–1607.
- Qin W, Sundaram M, Wang Y, et al. Missense mutation in APOC3 within the C-terminal lipid binding domain of human ApoC-III results in impaired assembly and secretion of triacylglycerol-rich very low density lipoproteins: evidence that ApoC-III plays a major role in the formation of lipid precursors within the microsomal lumen. J Biol Chem. 2011;286:27769–27780.
- Pollin TI, Damcott CM, Shen H, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science. 2008;322:1702–1705.
- Crosby J, Peloso GM, Auer PL, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371:22–31.
- Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG, et al. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med. 2014;371:32–41.
- Arca M, Hsieh A, and Soran H, et al. The effect of volanesorsen treatment on the burden associated with familial chylomicronemia syndrome: the results of the ReFOCUS study. Expert Rev Cardiovasc Ther. 2018;16:537–546.
- Gouni-Berthold I, Alexander VJ, and Yang Q, et al. Efficacy and safety of volanesorsen in patients with multifactorial chylomicronaemia (COMPASS): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2021;9:264–275.
- Hegele RA, Joy TR, Al-Attar SA, et al. Lipodystrophies: windows on adipose biology and metabolism. J Lipid Res. 2007;48:1433–1444.
- Alexander VJ, Xia S, Hurh E, et al. N-acetyl galactosamine-conjugated antisense drug to APOC3 mRNA, triglycerides and atherogenic lipoprotein levels. Eur Heart J. 2019;40(33):2785–2796.
- Clifton P, Sullivan D, Baker J, et al., Abstract 12594: pharmacodynamic effect of ARO-APOC3, an investigational hepatocyte-targeted RNA interference therapeutic targeting apolipoprotein C3, in patients with hypertriglyceridemia and multifactorial chylomicronemia. Circulation. 142(Suppl_3): 12594. 2020.
- Ng DS. Evolving ANGPTL-based lipid-lowering strategies and beyond. Curr Opin Lipidol. 2021;32(4):271–272.
- Musunuru K, Pirruccello JP, Do R, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. New Engl J Med. 2010;363(23):2220–2227.
- Dewey FE, Gusarova V, Dunbar RL, et al. Genetic and pharmacologic inactivation of ANGPTL3 and cardiovascular disease. New Engl J Med. 2017;377(3):211–221.
- Raal FJ, Rosenson RS, Reeskamp LF, et al. Evinacumab for homozygous familial hypercholesterolemia. New Engl J Med. 2020;383(8):711–720.
- Markham A. Evinacumab: first approval. Drugs. 2021;81(9):1101–1105.
- Rosenson RS, Gaudet D, Ballantyne CM, et al. A phase 2 trial of the efficacy and safety of evinacumab in patients with severe hypertriglyceridemia. Atherosclerosis. 2021;331:e293.
- Graham MJ, Lee RG, Brandt TA, et al. Cardiovascular and metabolic effects of ANGPTL3 antisense oligonucleotides. New Engl J Med. 2017;377(3):222–232.
- Gaudet D, Karwatowska-Prokopczuk E, Baum SJ, et al. Vupanorsen, an N-acetyl galactosamine-conjugated antisense drug to ANGPTL3 mRNA, lowers triglycerides and atherogenic lipoproteins in patients with diabetes, hepatic steatosis, and hypertriglyceridaemia. Eur Heart J. 2020;41(40):3936–3945.
- Pfizer Inc., Ionis Pharmaceuticals, Inc. Pfizer and ionis announce discontinuation of vupanorsen clinical development program [Internet]. New York NY and Carlsbad CA: Pfizer; January 2022. cited 2022 May 28]. Available from: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-and-ionis-announce-discontinuation-vupanorsen
- Watts GF, Schwabe C, Scott R, et al., Abstract 15751: pharmacodynamic effect of ARO-ANG3, an investigational RNA interference targeting hepatic angiopoietin-like protein 3, in patients with hypercholesterolemia. Circulation. 142(Suppl_3): 15751. 2020.
- Rip J, van Dijk KW, Sierts JA, et al. AAV1-LPLS447X gene therapy reduces hypertriglyceridemia in apoE2 knock in mice. Biochim Biophys Acta. 2006;1761(10):1163–1168.
- Gaudet D, Méthot J, Kastelein J. Gene therapy for lipoprotein lipase deficiency. Curr Opin Lipidol. 2012;23(4):310–320.
- Carpentier AC, Frisch F, Labbé SM, et al. Effect of alipogene tiparvovec (AAV1-LPL S447X) on postprandial chylomicron metabolism in lipoprotein lipase-deficient patients. J Clin Endocrinol Metab. 2012;97(5):1635–1644.
- Gaudet D, Méthot J, Déry S, et al. Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: an open-label trial. Gene Ther. 2013;20(4):361–369.
- Cao J, Zhou Y, Peng H, et al. Targeting acyl-CoA:diacylglycerol acyltransferase 1 (DGAT1) with small molecule inhibitors for the treatment of metabolic diseases. J Biol Chem. 2011;286(48):41838–41851.
- DeVita RJ, Pinto S. Current status of the research and development of diacylglycerol O -acyltransferase 1 (DGAT1) inhibitors. J Med Chem. 2013;56(24):9820–9825.
- Schober G, Arnold M, Birtles S, et al. Diacylglycerol acyltransferase-1 inhibition enhances intestinal fatty acid oxidation and reduces energy intake in rats. J Lipid Res. 2013;54(5):1369–1384.
- Meyers CD, Tremblay K, Amer A, et al. Effect of the DGAT1 inhibitor pradigastat on triglyceride and apoB48 levels in patients with familial chylomicronemia syndrome. Lipids Health Dis. 2015;14(1):8.
- Meyers CD, Amer A, Majumdar T, et al. Pharmacokinetics, pharmacodynamics, safety, and tolerability of pradigastat, a novel diacylglycerol acyltransferase 1 inhibitor in overweight or obese, but otherwise healthy human subjects. J Clin Pharmacol. 2015;55(9):1031–1041.
- Meyers C, Gaudet D, Tremblay K, et al. The DGAT1 inhibitor LCQ908 decreases triglyceride levels in patients with the familial chylomicronemia syndrome. J Clin Lipidol. 2012;6(3):266–267.
- Wolska A, Reimund M, Remaley AT. Apolipoprotein C-II: the re-emergence of a forgotten factor. Curr Opin Lipidol. 2020;31(3):147–153.
- Dewey FE, Gusarova V, O’Dushlaine C, et al. Inactivating variants in ANGPTL4 and risk of coronary artery disease. New Engl J Med. 2016;374(12):1123–1133.
- Whittaker MN, Musunuru K. Therapeutic application of genome editing in dyslipidemia. Curr Opin Lipidol. 2022;33(2):133–138.