
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Introduction
Developing and evaluating novel compounds for treatment or prophylaxis of emerging infectious diseases is costly and time-consuming. Repurposing of already available marketed compounds is an appealing option as they already have an established safety profile. This approach could substantially reduce cost and time required to make effective treatments available to fight the COVID-19 pandemic. However, this approach is challenging since many drug candidates show efficacy in in vitro experiments, but fail to deliver effect when evaluated in clinical trials. Better approaches to evaluate in vitro data are needed, in order to prioritize drugs for repurposing.
Areas covered
This article evaluates potential drugs that might be of interest for repurposing in the treatment of patients with COVID-19 disease. A pharmacometric simulation-based approach was developed to evaluate in vitro activity data in combination with expected clinical drug exposure, in order to evaluate the likelihood of achieving effective concentrations in patients.
Expert opinion
The presented pharmacometric approach bridges in vitro activity data to clinically expected drug exposures, and could therefore be a useful compliment to other methods in order to prioritize repurposed drugs for evaluation in prospective randomized controlled clinical trials.
1. Introduction
During the last 2 years, the Coronavirus disease 2019 (COVID-19) has spread rapidly worldwide. The World Health Organization reported ~445 million confirmed cases and 5.9 million deaths worldwide (as of 8 March 2022). Development and evaluation of novel therapeutic agents in the treatment and prevention of COVID-19 is time-consuming and costly. Repurposing of existing marketed drugs has been proposed to quickly and cheaply get effective medicine to clinical use [Citation1]. Drug repurposing comprises of several steps; identification of the new therapeutic target, evaluation of pre-clinical efficacy (in vitro and in vivo animal models), evaluation of effectiveness in prospective randomized controlled clinical trials, regulatory registration, and post-marketing monitoring [Citation2]. The evaluation and prioritization of repurposed drugs can be facilitated by applying in silico approaches, e.g. pharmacometric modeling and simulation, network-based models, structure-based models, and AI-based approaches. Some of these approaches can be used to identify new potential biological targets of already approved drugs, based on their chemical structure. Additionally, these approaches might predict the efficacy and undesirable effects [Citation2,Citation3]. In vitro activity evaluations toward the new biological target is a crucial experimental step to identify repurposed drugs with suitable activity against SARS-CoV-2, before studying the drug in animal models and humans to determine in vivo efficacy and safety. However, clinical trials are costly and time-consuming, and might expose patients to ineffective drugs. Thus, translation of in vitro anti-SARS-CoV-2 activity to in vivo efficacy is needed to prioritize potential drug candidates and deselect drugs that are unlikely to achieve effective clinical drug exposure.
This review focuses on the translational step by evaluating the pharmacokinetic properties of potential drugs using a pharmacometric modeling and simulation framework. The aim of this work was to review several marketed drugs with proposed in vitro activity against SARS-CoV-2, and the likelihood of them reaching clinically therapeutic concentrations in the treatment of COVID-19.
2. Framework of drug repurposing for COVID-19
In this review, population-based pharmacometric simulations were performed for several different marketed drugs to determine their suitability to be repurposed for the treatment of COVID-19. The drugs included in the current review were part of a drug screening process to identify drugs with in vitro activity against SARS-CoV-2, performed by the Drugs for Neglected Diseases Initiative (DNDi) to identify suitable clinical arms for ANTICOV. This consortium is an open-label, randomized, comparative, ‘adaptive platform trial’ that is testing the safety and efficacy of treatments in mild-to-moderate COVID-19 patients. ANTICOV aims to identify early treatments that can prevent progression of COVID-19 to severe disease and potentially limit transmission. This initiative is conducted in collaboration with the Access to COVID-19 Tools Accelerator (ACT-A) Therapeutics Partnership and co-convened by Unitaid and Wellcome. Drugs included were chloroquine, mefloquine, amodiaquine, daclatasvir, sofosbuvir, favipiravir, nitazoxanide, ivermectin, atazanavir/ritonavir, and colchicine. Details of the developed framework are described below, and also illustrated in .
Figure 1. Population-based simulation framework for drug repurposing in the treatment of COVID-19.
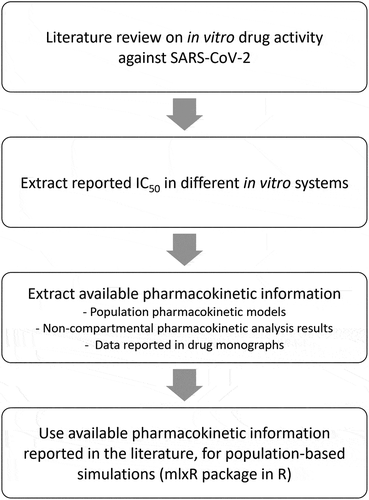
2.1. In vitro activity data against SARS-CoV-2
The drug concentration responsible for 50% of maximum in vitro effect (IC50, in vitro) of the inhibition of SARS-CoV2 growth were derived using different cell types (). In vitro IC50 values reported in molar units were converted to appropriate concentration units (e.g. ng/mL) using their molecular weights. It was assumed that negligible amounts of plasma binding protein were present in the in vitro experiments, and IC50, in vitro values should therefore be interpreted as free drug concentrations. Thus, the in vitro IC50 values were corrected for plasma protein binding by using the reported fraction of unbound drug in plasma (fu) in humans (Equation 1). The corrected IC50 values (IC50) values represent the total plasma concentration needed to achieve 50% of maximum effect, identified in the in vitro experiment.
Table 1. Basic pharmacokinetic and pharmacodynamic parameters of each drug in different cell types.
2.2. Population pharmacokinetic simulations
A literature search was performed to identify available pharmacokinetic information of the investigated drugs. If a population pharmacokinetic model was published, the reported structural pharmacokinetic parameters were used for simulations; including absorption parameters (e.g. absorption rate, lag-time, and mean-transit absorption time), elimination clearance, inter-compartmental clearance(s), and volume of distribution(s). Reported inter-individual variability and inter-occasion variability on each structural parameter in the population pharmacokinetic model was also used in the simulation. Additional covariates reported for the population were considered and included in the simulations. When more than one population pharmacokinetic model was available, the model based on a dense sampling schedule and/or a greater number of participants was selected. If a population pharmacokinetic model was not available, results from a non-compartmental pharmacokinetic analysis was used to derive key pharmacokinetic parameters used for simulations; absorption rate constant (approximated by using the time to peak concentration), elimination clearance and total volume of distribution. A one-compartment disposition model with first order-absorption was assumed since compartment-specific parameters are not derived in a non-compartmental analysis. Additionally, an inter-individual variability of 30% was assumed for all pharmacokinetic parameters to reflect an expected population variability.
The models describing the population pharmacokinetic properties of each drug were translated to Mlxtran models using Mlxtran coding language, which allowed flexibility in implementing ordinary differential equations (ODEs) based mixed-effects models. The structural model, inter-individual variability, inter-occasion variability, residual variability, and covariate effects were all described in the Mlxtran model. An example of a Mlxtran model used is presented in the supplementary material (Text S1). The Mlxtran models were then used for simulations (i.e. SimulX function in the mlxR package) [Citation8]. The SimulX function predicts and generates the concentration–time profiles from the used Mlxtran models. The mean plasma concentration and 90% prediction interval from the simulations were plotted and compared with the in vitro IC50 of each compound. The pharmacokinetic parameters used for the simulations were summarized in the supplementary (Table S1). A total of 1000 hypothetical individuals were simulated for each dosing scenario. Simulated pharmacokinetic profiles were based on standard daily dosing and expanded for a total treatment duration of 10 days. Only favipiravir was simulated by using a loading dose (i.e. 1600 mg BID for the first day followed by 600 mg BID for 9 days).
3. Drugs of interest in drug repurposing for COVID-19
3.1. Antimalarial agents
3.1.1. Chloroquine
Chloroquine/hydroxychloroquine was one of the first antimalarials proposed for treatment and prevention of COVID-19. There has been an extensive evaluation of chloroquine efficacy and effectiveness against COVID-19, and it has been demonstrated that this drug has no benefit in the treatment of hospitalized patients with COVID-19 [Citation9]. It is still unclear if this drug has a role in the prevention of COVID-19, but a large prospective clinical trial is underway (NCT04303507). Chloroquine is cardiotoxic, resulting in a delay in ventricular repolarization, but data from chloroquine self-poisoning studies demonstrated that standard clinical dose of chloroquine used in the treatment of COVID-19 is unlikely to cause serious cardiovascular toxicity [Citation10,Citation11]. The standard chloroquine dose for malaria treatment (10 mg/kg daily over 3 days) has been shown to be safe and well tolerated in all groups of patients, including children and pregnant women. Chloroquine can also be prescribed as a long-term treatment for rheumatoid arthritis and system lupus erythematosus (400 mg daily for the first month, 200 mg daily for the second month, and 100 mg daily thereafter). Chloroquine’s bioavailability is almost complete (86 ± 16% for oral tablets), and its elimination profile is multi-phasic [Citation12]. Chloroquine is ~60% bound to plasma protein and it is more concentrated in the erythrocytes, i.e. blood–plasma ratio of 3:1 [Citation13]. Pharmacokinetic properties of chloroquine and its major metabolite, desethyl-chloroquine, have been well characterized in most populations. Both drug and metabolite are slowly eliminated, resulting in long terminal elimination half-lives of 20–60 days. Published studies describe the population pharmacokinetic profile of chloroquine by using multi-compartmental models, having distinct distribution and elimination phases [Citation14–16]. Linear drug-metabolite models are commonly used to describe the profiles of chloroquine and desethyl-chloroquine when modeled simultaneously [Citation16].
Published in vitro studies have presented IC50 values of chloroquine in SARS-CoV-2 Vero cell lines of ~1.1–1.3 μM [Citation17–19]. However, a drug screening study using the available FDA approved drug reported a higher IC50 of 7.28 μM against SARS-CoV-2 in a Vero cell line [Citation20]. In an in vivo hamster study, hydroxychloroquine did not show anti-viral activity against COVID-19 when given alone at 50 mg/kg daily (equivalent to 39 mg/kg base) or in combination with azithromycin at 10 mg/kg daily [Citation21]. To date, no data are published on the inhibitory effect of desethyl-chloroquine against SARS-CoV-2. Two randomized clinical studies in hospitalized COVID-19 patients (RECOVERY and SOLIDARITY) [Citation9,Citation22] evaluated hydroxychloroquine at an adult maintenance dose of 400 mg/day for 10 days after an initial loading dose of 800 mg given twice. A standard dosing of chloroquine (i.e. 600 mg once daily) was evaluated, for a total duration of 10 days to investigate the potential clinical benefit in the treatment of COVID-19. Modeling and simulation presented here suggest that it is unlikely that standard dosing results in sufficient blood concentrations, when corrected for 60% protein binding, to inhibit the SARS-CoV-2 viruses ( and S1), the above clinical trials demonstrated that chloroquine/hydroxychloroquine is not beneficial in the treatment of COVID-19 patients.
Figure 2. Comparing the simulated plasma concentration–time profile of antimalarial drugs (n = 1000) with their in vitro IC50 against SARS-CoV-2. (A) Simulated plasma concentrations of chloroquine were based on a published population pharmacokinetic model [Citation23] and compared with its IC50 in different cell lines [Citation17–19]. Chloroquine was assumed to be 60% bound to plasma proteins. (B) Simulated plasma concentrations of mefloquine were based on a population pharmacokinetic model and compared with its in vitro IC50 in different cell lines [Citation20,Citation29]. Mefloquine was assumed to be 98% bound to plasma proteins. Simulated plasma concentrations of amodiaquine (C) and its metabolite desethylamodiaquine (D) were based on a published population pharmacokinetic drug-metabolite model [Citation33] and compared with their in vitro IC50 in difference cell lines [Citation20,Citation29]. Both amodiaquine and desethylamodiaquine were assumed to be 90% bound to plasma proteins. Solid black lines represent the mean population plasma concentration–time profiles, the shaded area represents the 90% prediction interval of the simulated concentrations, blue lines represent uncorrected in vitro IC50 values, and red lines represent in vitro IC50 values corrected for plasma protein binding.
![Figure 2. Comparing the simulated plasma concentration–time profile of antimalarial drugs (n = 1000) with their in vitro IC50 against SARS-CoV-2. (A) Simulated plasma concentrations of chloroquine were based on a published population pharmacokinetic model [Citation23] and compared with its IC50 in different cell lines [Citation17–19]. Chloroquine was assumed to be 60% bound to plasma proteins. (B) Simulated plasma concentrations of mefloquine were based on a population pharmacokinetic model and compared with its in vitro IC50 in different cell lines [Citation20,Citation29]. Mefloquine was assumed to be 98% bound to plasma proteins. Simulated plasma concentrations of amodiaquine (C) and its metabolite desethylamodiaquine (D) were based on a published population pharmacokinetic drug-metabolite model [Citation33] and compared with their in vitro IC50 in difference cell lines [Citation20,Citation29]. Both amodiaquine and desethylamodiaquine were assumed to be 90% bound to plasma proteins. Solid black lines represent the mean population plasma concentration–time profiles, the shaded area represents the 90% prediction interval of the simulated concentrations, blue lines represent uncorrected in vitro IC50 values, and red lines represent in vitro IC50 values corrected for plasma protein binding.](/cms/asset/cc6b85c9-3cbb-4b7d-98e7-1b146a9af08b/ierj_a_2113388_f0002_oc.jpg)
3.1.2. Mefloquine
Mefloquine in combination with artesunate is a recommended first-line treatment for acute uncomplicated malaria, administered at 200/440 mg daily for 3 days. A higher dose of 1500 mg of mefloquine was recommended when mefloquine was used as single-dose monotherapy. Mefloquine is also suitable for malaria prevention, and have been used as a weekly administered malaria chemoprophylactic agent in nonimmune travelers (i.e. 250 mg daily for 3 days followed by 250 mg weekly). It is safe and well tolerated in various populations, including children and pregnant women. Neuropsychiatric side effects were raised as a concern after mefloquine administration, but there was inconclusive evidences of this side effect [Citation24]. Mefloquine is readily absorbed, but its absolute oral bioavailability has not been determined as it cannot be administered intravenously. Drug administration with food increases its relative bioavailability by ~40%. Mefloquine is highly protein bound (~98%) and has a large volume of distribution (~200 L). It is slowly eliminated with a terminal elimination half-life of 2–4 weeks. Hepatic metabolism is the major route of elimination, resulting in the formation of carboxy-mefloquine, which has 3-5-fold larger exposure compared to mefloquine due to a longer terminal elimination half-life. Approximately 10% of the dose is excreted unchanged via urine.
A two-compartment distribution model has commonly been used to describe the pharmacokinetic properties of mefloquine [Citation25–28]. Mefloquine absorption kinetics have been explained by a delayed transit absorption compartment model. The pharmacokinetic properties of mefloquine are relatively constant across different populations, including pregnant women and children. The exposure to mefloquine in pregnant women tends to be higher but not significantly different compared to non-pregnant women.
An in vitro study reported mefloquine IC50 and IC90 values against SARS-CoV-2 in a Vero cell line at 1.8 ± 0.1 and 8.1 ± 3.7 µM, respectively [Citation29]. Another study reported somewhat higher IC50 values at 4.33 μM [Citation20]. An in vitro combination of artesunate-mefloquine also showed promising antiviral activity, resulting in 72.1 ± 18.3% inhibition of the virus replication at expected clinical peak concentrations of mefloquine (i.e. 2.5–4.1 μM) [Citation30]. No study has evaluated the effect of carboxy-mefloquine on SARS-CoV-2. A standard dosing of mefloquine (i.e. 440 mg once daily) was evaluated, for a total duration of 10 days to investigate the potential clinical benefit in the treatment of COVID-19. Modeling and simulation presented here suggest that it is unlikely that standard dosing of mefloquine results in sufficient blood concentrations, when corrected for 98% protein binding, to inhibit the SARS-CoV-2 virus ( and S1). However, the mefloquine concentration in lung tissue is predicted to be ten times higher than that in blood [Citation31], which has been suggested to be sufficient to inhibit the SARS-CoV-2 virus clinically.
3.1.3. Amodiaquine
Amodiaquine in combination with artesunate is a recommended first-line treatment of acute uncomplicated malaria. It is effective against both P. falciparum and P. vivax infections. It can also be used in combination with sulfadoxine-pyrimethamine as malaria chemoprevention in risk groups, such as young children and pregnant women [Citation32]. Amodiaquine is safe and well tolerated in different populations, including children and pregnant women. For both treatment and prevention of malaria, amodiaquine is administered as a daily dose (540 mg) for 3 days. Absolute bioavailability of amodiaquine cannot be determined since no parenteral administered formulation is available. It has a rapid absorption and its major route of elimination is through hepatic metabolism via CYP2C8 to form desethyl-amodiaquine. Both parent and metabolite are extensively bound to plasma proteins, ~90%. The parent drug is more rapidly eliminated compared to its metabolite, resulting in a terminal elimination half-life of 10–20 hours and 10–20 days, respectively [Citation33]. Due to the long half-life, the metabolite accumulates with multiple dosing while this not seen for the parent drug. Amodiaquine and its metabolite follow a multi-phasic elimination profile.
Two-compartment and three-compartment disposition models have been used commonly to described the pharmacokinetic profile of amodiaquine and desethyl-amodiaquine, respectively [Citation33,Citation34]. A transit absorption model has been used to explain its absorption profiles. Amodiaquine’s clearance is depended on weight and age (i.e. body-size and enzymatic maturation in infants), and it therefore requires dose adjustment in children. Additionally, bioavailability is relatively higher at the convalescence phase of malaria indicating a disease effect.
An in vitro study reported an amodiaquine IC50 value of ~5.15 μM [Citation20], and IC50 and IC90 values of desethyl-amodiaquine of 0.52 ± 0.2 and 1.93 ± 1.0 µM, respectively, against SARS-CoV-2 in Vero E6 cell lines [Citation29]. The combination of artesunate-amodiaquine showed a promising in vitro antiviral activity resulting in 32.3 ± 9.9% inhibition of SARS-CoV-2 replication in Vero E6 cell lines at the expected peak concentrations of desethyl-amodiaquine [Citation30]. A standard dosing of amodiaquine (i.e. 810 mg once daily) was evaluated, for a total duration of 10 days to investigate the potential clinical benefit in the treatment of COVID-19. Modeling and simulation presented here suggest that it is unlikely that standard dosing of amodiaquine results in sufficient blood concentrations, when corrected for 90% protein binding, to inhibit the SARS-CoV-2 virus ( and S1). In addition, only a small fraction of the total amodiaquine dose (0.07%) were recovered in rat lung tissue [Citation35], indicating that it is not likely to result in sufficient concentrations to inhibit the SARS-CoV-2 viruses clinically.
3.2. Anti-hepatitis C agents
3.2.1. Daclatasvir
Daclatasvir is an antiviral drug used in the treatment of hepatitis C, and interferes and prevents conformational changes of the virus protein 5A (NS5A). In hepatitis C patients, the typical dose of daclatasvir is 60 mg per day for several weeks of treatment in combination with 400 mg sofosbuvir. The drug is rapidly absorbed, reaching maximal concentrations within 1–2 hours after dose. Daclatasvir has an oral bioavailability of about 67% and a high plasma protein binding of ~99% [Citation36]. Daclatasvir is excreted primarily unchanged via feces (53%), and ~7% of the dose is excreted via renal elimination. CYP3A4 is a key enzyme in the metabolism of daclatasvir. As a result, when daclatasvir is used with a strong CYP3A4 inhibitor (e.g. rifampin), the dosage of daclatasvir should be adjusted. One study showed that daclatasvir steady state exposure (AUCss) increased by 30% in female subjects when co-administered with rifampin [Citation37]. No other patient’s demographic characteristics show clinically relevant impact on daclatasvir exposure [Citation36–39].
The population pharmacokinetic properties of daclatasvir has commonly been described by a one-compartment disposition model with the first-order absorption [Citation38,Citation39]. However, a pooled pharmacokinetic study from phase II and III clinical trials (n = 2135) concluded a two-compartment disposition kinetics for daclatasvir [Citation37].
Daclatasvir has been suggested to interfere with the viral RNA SARS-CoV-2 replication and viral assembly. It was reported to inhibit the SARS-CoV-2 replication in Vero, HuH-7, Calu-3 cells resulting in an IC50 of 0.8, 0.6, and 1.1 μM, respectively [Citation18]. Sofosbuvir/daclatasvir combinations have demonstrated a higher potency against SARS-CoV-2, suggesting synergistic effects between the two drugs. A standard dosing of daclatasvir (i.e. 60 mg once daily) was evaluated, for a total duration of 10 days to investigate the potential clinical benefit in the treatment of COVID-19. Modeling and simulation presented here suggest that it is unlikely that standard dosing results in sufficient blood concentrations, when corrected for 99% protein binding, to inhibit the SARS-CoV-2 virus ( and S2).
Figure 3. Comparing the simulated plasma concentration–time profiles of anti-hepatitis B drugs (n = 1000) with their in vitro IC50 against SARS-CoV-2. (A) Simulated plasma concentrations of daclatasvir were based on a published population pharmacokinetic model of daclatasvir [Citation37] and compared with its IC50 in different cell lines [Citation18]. Daclatasvir was assumed to be 99% bound to plasma proteins. Simulated plasma concentrations of sofosbuvir (B) and its major metabolite, GS-331007 (C) were based on the population pharmacokinetic drug-metabolite model [Citation41] and compared with their in vitro IC50 in different cell lines [Citation18]. Sofosbuvir and GS-331007 was assumed to be 65% and 0% bound to plasma proteins, respectively. Solid black lines represent the mean population plasma concentration–time profiles, the shaded area represents the 90% prediction interval of the simulated concentrations, blue lines represent uncorrected in vitro IC50 values, and red lines represent in vitro IC50 values corrected for plasma protein binding.
![Figure 3. Comparing the simulated plasma concentration–time profiles of anti-hepatitis B drugs (n = 1000) with their in vitro IC50 against SARS-CoV-2. (A) Simulated plasma concentrations of daclatasvir were based on a published population pharmacokinetic model of daclatasvir [Citation37] and compared with its IC50 in different cell lines [Citation18]. Daclatasvir was assumed to be 99% bound to plasma proteins. Simulated plasma concentrations of sofosbuvir (B) and its major metabolite, GS-331007 (C) were based on the population pharmacokinetic drug-metabolite model [Citation41] and compared with their in vitro IC50 in different cell lines [Citation18]. Sofosbuvir and GS-331007 was assumed to be 65% and 0% bound to plasma proteins, respectively. Solid black lines represent the mean population plasma concentration–time profiles, the shaded area represents the 90% prediction interval of the simulated concentrations, blue lines represent uncorrected in vitro IC50 values, and red lines represent in vitro IC50 values corrected for plasma protein binding.](/cms/asset/ece69970-8a54-4c4d-9620-fa56b4425049/ierj_a_2113388_f0003_oc.jpg)
3.2.2. Sofosbuvir
Sofosbuvir is another antiviral agent used in the treatment of hepatitis C. Sofosbuvir is rapidly absorbed and it reaches maximum concentrations within 0.5–2 hours after dose. Sofosbuvir is metabolized intra-cellularly to a hepatitis C-active uridine tri-phosphate compound (i.e. GS-461203) in the hepatic cells. This metabolite is confined to the hepatic cells and therefore undetectable in human plasma. GS-331007 is another major metabolite that can be found in human plasma. Sofosbuvir has a short terminal elimination half-life of 30 minutes, while the GS-331007 metabolite has a longer terminal elimination half-life of ~24 hours [Citation40]. Also, the peak drug concentrations and total exposure of the metabolite are higher than the parent compound. Sofosbuvir binds to plasma proteins (61–65%), whereas GS-331007 is minimally bound to plasma proteins.
The population pharmacokinetic properties of sofosbuvir and its metabolites have previously been described by a mechanistic drug-metabolite model [Citation41]. The parent drug, sofosbuvir, was described by a one-compartment disposition model, while the metabolite, GS-331007, followed two-compartment disposition kinetics.
Sofosbuvir has been suggested to terminate the SARS-CoV-2 RNA polymerase activity. Sofosbuvir IC50 was 5.1 and 7.3 μM for the HuH-7 and Calu-3 cells, respectively. Sofosbuvir alone is less potent than daclatasvir, but shown to have synergistic effects when given in combination with daclatasvir, i.e. IC50 = 0.5–0.7 μM in the Calu-3 cell line [Citation18]. This should be interpreted as a 10- to 35-fold increase in potency of the combination drugs. The sofosbuvir nucleoside metabolite (GS-331007) was less potent compared to the parent drug, i.e. IC50 = 9.3 ± 0.2 μM in the Calu-3 cell line. A standard dosing of sofosbuvir (i.e. 400 mg once daily) was evaluated, for a total duration of 10 days to investigate the potential clinical benefit in the treatment of COVID-19. Modeling and simulation presented here suggest that it is unlikely that standard dosing of sofosbuvir alone results in sufficient blood concentrations, when corrected for 65% plasma protein binding, to inhibit the SARS-CoV-2 virus ( and S2). The treatment of sofosbuvir plus daclatasvir in combination with ribavirin, or sofosbuvir plus ledipasvir in hospitalized COVID-19 patients was not significantly superior compared to standard of care [Citation42,Citation43]. However, these randomized controlled trials were relatively small and larger clinical trials are needed to fully understand if these drugs could be used in the treatment of COVID-19. A systemic review and meta-analysis showed that patients receiving sofosbuvir in combination with daclatasvir had a lower risk of mortality (RR: 0.31, 95% CI: 0.12–0.78) and reduced need for ICU or invasive mechanical ventilator (IMV) (RR: 0.33, 95% CI: 0.18–0.69) compared to standard of care [Citation44].
3.3. Anti-influenza agents
3.3.1. Favipiravir
Favipiravir is a purine analogue and was developed as an anti-influenza drug. It is a prodrug and it undergoes intracellular ribosylation and phosphorylation to form the active compound, favipiravir-ribofuranosyl-5’-triphosphate (FAVI-RTP). The triphosphate metabolite can bind and inhibit the RNA-dependent RNA polymerase (RdRp) and block the viral RNA genome transcription and replication. This mechanism of action is novel compared to the current anti-influenza agents (i.e. amantadine and rimantadine are M2 channel ion inhibitors, and oseltamivir, zanamivir, peramivir, and laninamivir are neuraminidase inhibitors). Several studies showed that favipiravir possesses a broad-spectrum of antiviral activity, including influenza, Rift Valley fever, hepatitis C, and Chikungunya. Drug administration vary depending on the disease; favipiravir treatment in influenza consist of 1600 mg twice daily on the first day followed by 600 mg twice daily for 4 days. Favipiravir is extensively and rapidly absorbed (bioavailability of ~97% and Tmax of 1 hour). Its terminal elimination half-life is relatively short, i.e. 4.8–5.6 hours. Approximately, 54% of favipiravir binds to plasma proteins. Its elimination pathway occurs primarily through a liver oxidation and conjugation metabolism. An inactive hydroxide metabolite, M1, is primarily generated by the hepatic metabolism pathway. Its active intracellular triphosphate metabolite (FAVI-RTP) cannot be measured in the systematic circulation. A mechanistic population pharmacokinetic model has been developed and explains favipiravir and its active intracellular metabolites, FAVI-RTP, plasma concentration–time profiles [Citation45]. The FAVI-RTP metabolite is concentrated intracellularly and has a longer half-life compared to the parent compound.
A Phase 2a clinical trial of favipiravir for the treatment of severe influenza investigated its population pharmacokinetic properties extensively to suggest an appropriate dose [Citation46]. The population pharmacokinetic properties of favipiravir and its M1 metabolite were described by a drug-metabolite model. Both favipiravir and M1 metabolite kinetics were best described by a single disposition compartment. Favipiravir clearance was time-dependent (i.e. 6.12% increase in clearance per day of treatment), and the clearance of the M1 metabolite was affected by the creatinine clearance.
Favipiravir has been suggested to inhibit RdRp of RNA viruses and therefore block viral replication of SARS-CoV-2. In vivo studies in hamster demonstrated that a low dose of favipiravir was associated with a minor reduction of viral titers, while a high dose significantly reduced viral titers in the lung and improved lung histopathology [Citation21]. In patients with mild-to-moderate COVID-19, favipiravir reduced the time of viral shedding, time to clinical cure, and time to hospital discharge when compared to patients receiving standard of care [Citation47], but these finding lacked statistical significance. A loading dose (i.e. 1600 mg twice daily for the first day followed by 600 mg BID for 9 days) was evaluated, to investigate the potential clinical benefit in the treatment of COVID-19. Modeling and simulation presented here suggest that it is likely that standard dosing of favipiravir results in sufficient blood concentrations, even when corrected for 54% protein binding, to inhibit the SARS-CoV-2 virus ( and S3). Large prospective clinical trials are needed to evaluate its potential clinical benefit in the treatment of COVID-19.
Figure 4. Comparing the simulated plasma concentration–time profile of favipiravir (n = 1000) with its in vitro IC50 against SARS-CoV-2. Simulated plasma concentrations of favipiravir were based on a published population pharmacokinetic model of favipiravir [Citation46] and compared with its IC50 in Vero E6 cell line [Citation17]. Favipiravir was assumed to be 54% bound to plasma proteins. Solid black line represents the mean population plasma concentration–time profile, the shaded area represents the 90% prediction interval of the simulated concentrations, the blue line represents the uncorrected in vitro IC50 value, and the red line represents the in vitro IC50 value corrected for plasma protein binding.
![Figure 4. Comparing the simulated plasma concentration–time profile of favipiravir (n = 1000) with its in vitro IC50 against SARS-CoV-2. Simulated plasma concentrations of favipiravir were based on a published population pharmacokinetic model of favipiravir [Citation46] and compared with its IC50 in Vero E6 cell line [Citation17]. Favipiravir was assumed to be 54% bound to plasma proteins. Solid black line represents the mean population plasma concentration–time profile, the shaded area represents the 90% prediction interval of the simulated concentrations, the blue line represents the uncorrected in vitro IC50 value, and the red line represents the in vitro IC50 value corrected for plasma protein binding.](/cms/asset/cd50f2fc-cda4-4d44-b9b9-970678945f07/ierj_a_2113388_f0004_oc.jpg)
3.4. Antiparasitic agents
3.4.1. Nitazoxanide
Nitazoxanide is a broad spectrum parasiticidal agent, indicated for the treatment of diarrhea caused by Giardia lamblia or Cryptosporidium parvum. Additionally, nitazoxanide has been described in the literature to have an antiviral effect in adults with acute uncomplicated influenza and is also expected to have an anti-inflammatory effect, as it has shown to inhibit the production of several pro-inflammatory cytokines [Citation48]. The standard recommended dose of oral nitazoxanide in adults is 500 mg twice daily. Following oral administration, nitazoxanide is not detected in the plasma as it is rapidly deacetylated to its active metabolite, tizoxanide. More than 99% of tizoxanide is bound to plasma proteins. Tizoxanide is metabolized further via glucuronidation, and excreted in the urine, bile and feces [Citation49]. A half-life of 6 minutes for nitazoxanide [Citation50] and ~1.3 hours for tizoxanide has been reported [Citation51]. At the time of this review, no population pharmacokinetic models of nitazoxanide and/or tizoxanide were available. The pharmacokinetic parameters used for population-based simulations (elimination clearance and volume of distribution) were extracted from a published physiologically based pharmacokinetic (PBPK) model, developed to describe pharmacokinetic data of tizoxanide plasma concentrations in healthy participants receiving single doses of 500–4000 mg of nitazoxanide with or without food [Citation52]. An absorption rate constant of 0.45 h−1 was approximated to receive a Tmax of 2 hours, similar to what has been reported in healthy volunteers [Citation53,Citation54]. A one-compartment disposition model with first-order absorption was used to simulate and predict tizoxanide exposure in human. The model assumed 30% inter-individual variability in all pharmacokinetic parameters.
Nitazoxanide has been reported to have antiviral activity against a broad range of viruses as well as human and animal coronaviruses. The IC50 value of nitazoxanide against SARS-CoV-2 was reported to be 2.12 μM (0.562 mg/L) in Vero E6 cells [Citation17]. A standard dosing of nitazoxanide (i.e. 500 mg twice daily) was evaluated, for a total duration of 10 days to investigate the potential clinical benefit in the treatment of COVID-19. Modeling and simulation presented here suggest that it is unlikely that standard dosing of nitazoxanide results in sufficient blood concentrations, when corrected for 99% protein binding, to inhibit the SARS-CoV-2 virus ( and S4).
Figure 5. Comparing the simulated plasma concentration–time profiles of antiparasitic drugs (n = 1000) with their in vitro IC50 against SARS-CoV-2. (A) Simulated plasma concentrations of tizoxanide were based on a published PBPK model of nitazoxanide [Citation52] and compared with its IC50 value reported in Vero E6 cells [Citation17]. Tizoxanide was assumed to be 99% bound to plasma proteins. (B) Simulated plasma concentrations of ivermectin were based on a published population pharmacokinetic model of ivermectin in healthy volunteers [Citation60] and compared with its IC50 values reported in Vero-hSLAM cells [Citation62]. Ivermectin was assumed to be 93% bound to plasma proteins. (C) Simulated atazanavir plasma concentrations were based on a published population pharmacokinetic model of atazanavir in HIV infected patients [Citation68] and compared with its IC50 in different cell lines [Citation64]. Atazanavir was assumed to be 86% bound to plasma proteins. (D) Simulated colchicine plasma concentrations were based on subject-specific nonlinear regression model [Citation75] and compared with plasma concentrations inhibiting neutrophil chemotaxis [Citation79]. Solid black lines represent the mean population concentration–time profiles, the shaded area represents the 90% prediction interval of the simulated concentrations, blue lines represent uncorrected in vitro IC50 values, and red lines represent in vitro IC50 values corrected for plasma protein binding.
![Figure 5. Comparing the simulated plasma concentration–time profiles of antiparasitic drugs (n = 1000) with their in vitro IC50 against SARS-CoV-2. (A) Simulated plasma concentrations of tizoxanide were based on a published PBPK model of nitazoxanide [Citation52] and compared with its IC50 value reported in Vero E6 cells [Citation17]. Tizoxanide was assumed to be 99% bound to plasma proteins. (B) Simulated plasma concentrations of ivermectin were based on a published population pharmacokinetic model of ivermectin in healthy volunteers [Citation60] and compared with its IC50 values reported in Vero-hSLAM cells [Citation62]. Ivermectin was assumed to be 93% bound to plasma proteins. (C) Simulated atazanavir plasma concentrations were based on a published population pharmacokinetic model of atazanavir in HIV infected patients [Citation68] and compared with its IC50 in different cell lines [Citation64]. Atazanavir was assumed to be 86% bound to plasma proteins. (D) Simulated colchicine plasma concentrations were based on subject-specific nonlinear regression model [Citation75] and compared with plasma concentrations inhibiting neutrophil chemotaxis [Citation79]. Solid black lines represent the mean population concentration–time profiles, the shaded area represents the 90% prediction interval of the simulated concentrations, blue lines represent uncorrected in vitro IC50 values, and red lines represent in vitro IC50 values corrected for plasma protein binding.](/cms/asset/3b4f5ff2-eb17-4df9-aa92-d001531afd1e/ierj_a_2113388_f0005_oc.jpg)
3.4.2. Ivermectin
Ivermectin is an antiparasitic agent approved for the treatment of intestinal strongyloidiasis and onchocerciasis [Citation55]. The recommended dose for the treatment of onchocerciasis and strongyloidiasis is a single oral dose of 150–200 µg/kg [Citation55]. Following oral administration, ivermectin is moderately well absorbed, which is improved when administered with a high fat meal [Citation56]. Ivermectin is widely distributed in the body, attributable to the high lipophilicity of the drug, and a high plasma protein binding of 93% has been reported [Citation57]. Ivermectin is primarily metabolized by CYP3A4 in the liver, forming three major metabolites; one demethylated and one hydroxylated metabolite, as well as the combination of demethylation and hydroxylation [Citation58]. Ivermectin and its metabolites are excreted mainly in the feces, and only 1% of the drug is excreted in the urine [Citation56]. The terminal elimination half-life of ivermectin is ~18 hours following oral administration. Several population pharmacokinetic models of ivermectin have been published [Citation59–61]. The pharmacokinetic properties of ivermectin have been described by a two-compartment disposition model with first-order elimination in all reported studies. We used the published population pharmacokinetic model of ivermectin in healthy volunteers for these simulations, consisting of a two-compartment disposition model with six transit absorption compartments [Citation60]. Body weight was implemented as a covariate on clearance and volume of distribution using allometric scaling.
Ivermectin has been reported to have broad antiviral activity. The in vitro IC50 value of ivermectin against SARS-CoV-2 has been reported to be 2.8 μM (2.45 mg/L) in Vero-hSLAM cells [Citation62]. A standard dosing of ivermectin (i.e. 0.2 mg/kg once daily, for a 70 kg bodyweight) was evaluated over a total treatment duration of 10 days, to investigate potential clinical benefits in the treatment of COVID-19. Modeling and simulation presented here suggest that it is unlikely that standard dosing of ivermectin results in sufficient blood concentrations, when corrected for 93% protein binding, to inhibit the SARS-CoV-2 virus ( and S4).
3.5. Antiretroviral and other therapeutic agents
3.5.1. Atazanavir
Atazanavir is a protease inhibitor, used as part of a combination therapy in the treatment of HIV. The recommended dose of atazanavir is 300 mg taken once daily with ritonavir 100 mg [Citation63]. For patients who cannot tolerate ritonavir, atazanavir 400 mg is recommended to be taken once daily with food. Following oral administration, atazanavir is rapidly absorbed with a Tmax of ~2.5 hours. There is no data on the absolute bioavailability of atazanavir. Based on the area under the concentration–time curve, the relative bioavailability of atazanavir capsule has been reported to be ~60% compared with an oral solution [Citation56,Citation66]. Administration of atazanavir with a light or high fat meal increased drug exposure compared to administration in the fasting state [Citation63]. Plasma protein binding of atazanavir is 86%, and the drug binds to both alpha-1-acid glycoprotein (AAG) and albumin. Atazanavir is metabolized primarily by CYP3A4 and metabolites excreted mainly in the feces [Citation63]. The terminal elimination half-life of atazanavir in healthy volunteers and HIV patients were reported to be 7 hours following 400 mg of atazanavir administration. Several population pharmacokinetic studies of atazanavir have been published [Citation67–72]. The pharmacokinetic properties of atazanavir have been described by a one-compartment disposition model with first-order absorption in most of these studies [Citation67–70,Citation72]. The pharmacokinetic parameter estimates were similar among studies, and body weight, sex, ethnicity, and enzyme polymorphisms have all been reported as significant covariates. The population pharmacokinetic model we used for the simulations was a one-compartment disposition model with first-order absorption and lag-time [Citation68]. Atazanavir concentrations, when administered with ritonavir, were used for the pharmacokinetic simulations. This model included body weight as a covariate on volume of distribution and sex as a covariate on atazanavir clearance.
Activity against SARS-CoV2 has been reported when atazanavir was given alone and in combination with ritonavir. The IC50 values reported in Vero cells were 2.00–9.36 μM (1.41–6.60 mg/L)[Citation64,Citation65] when atazanavir was given alone and 0.5 μM (0.352 mg/L) when atazanavir was given with ritonavir[Citation64] . The IC50 reported in human pulmonary epithelial cells was 0.22 μM (0.155 mg/L) when atazanavir was given alone and 0.6 μM (0.423 mg/L) when atazanavir was given with ritonavir [Citation64]. A standard dosing of 300 mg of atazanavir with 100 mg ritonavir once daily was evaluated over a total treatment duration of 10 days, to investigate potential clinical benefits in the treatment of COVID-19. Modeling and simulation presented here suggest that it is unlikely that standard dosing of atazanavir results in sufficient blood concentrations, when corrected for 86% protein binding, to inhibit the SARS-CoV-2 virus in the majority of patients ( and S4).
3.5.2. Colchicine
Colchicine is an anti-inflammatory drug that is normally used to reduce pain associated with gout. It is also indicated for familial Mediterranean fever treatment [Citation56]. The recommended dose for gout flares prophylaxis is 0.6 mg once or twice daily with a maximum dose of 1.2 mg/day [Citation73]. For the treatment of acute gout flares, 1.2 mg of colchicine is recommended at the first sign of gout flares followed by 0.6 mg one hour later. The maximum recommended dose for acute gout flares is 1.8 mg over one-hour period. The dose should be reduced to a prophylactic dose after 12 hours [Citation73]. Following oral administration, colchicine is rapidly absorbed from the gastrointestinal tract, with a Tmax of 1–2 hours reported in healthy volunteers [Citation74,Citation75]. Approximately 39% of colchicine binds to plasma proteins, mainly albumin. Colchicine is metabolized in the liver mainly by CYP3A4, and colchicine is also a substrate of the efflux transporter P-glycoprotein [Citation56]. A study in healthy volunteers showed that 40–65% of colchicine was recovered unchanged in the urine after oral administration. Enterohepatic recirculation and biliary excretion are hypothesized to be involved in colchicine elimination as well [Citation56]. The terminal elimination half-life of colchicine has been reported to be 16–58 hours in healthy volunteers [Citation74–76]. At the time of this review, no population pharmacokinetic model of colchicine was available. Therefore, we used the pharmacokinetic parameters reported in healthy volunteers from a subject-specific nonlinear regression model, using weighted least-squares to fit individual subject data [Citation75]. The pharmacokinetics of colchicine was described by a three-compartment model after intravenous administration. A lag time of 14 minutes was reported after oral absorption with a Tmax of ~1 hour. The model assumed 30% inter-individual variability in all pharmacokinetic parameters.
The anti-inflammatory properties of colchicine suggest a potential role in the treatment of COVID-19, especially in pneumonia patients with a cytokine storm [Citation77]. The effective drug concentration for an anti-inflammatory effect in COVID-19 is unknown, but several randomized control trials are currently evaluating the safety and efficacy of colchicine in patients with COVID-19 [Citation77]. Peak plasma concentrations ranging from 2 to 17 ng/ml has been reported in healthy volunteers after oral administration of 0.6 to 1 mg colchicine [Citation74,Citation75,Citation78]. Colchicine at a concentration of 10 ng/mL [Citation79] has been reported to inhibit neutrophil chemotaxis, which is the main mechanism of action of colchicine for reducing inflammation [Citation79,Citation80].
A standard colchicine dosing to prevent gout flares (i.e. 0.6 mg twice daily) was evaluated over a total treatment duration of 10 days, to investigate potential clinical benefits in the treatment of COVID-19. Modeling and simulation presented here showed resulting plasma concentrations below 10 ng/mL, the concentrations needed to inhibit neutrophil chemotaxis ( and S4). As the effective drug concentration for an anti-inflammatory effect in COVID-19 is unknown, results from ongoing clinical trials are needed to evaluate if colchicine shows potential clinical benefit in the treatment of COVID-19.
4. Conclusions
This work presents a pharmacometric modeling and simulation approach to evaluate drugs suggested to be repurposed for the treatment of COVID-19. By using published pharmacokinetic models and applying population-based simulations of standard clinical doses, we assessed the likelihood of reaching plasma concentrations associated with SARS-CoV-2 activity in in vitro systems. Based on these simulations, only favipiravir, resulted in plasma drug concentrations associated with in vitro activity in the majority of patients. Using a population-based pharmacometric modeling and simulation approach is an efficient, cheap, ethical and fast approach to assess different molecules before commencing expensive clinical trials. This can de-risk repurposing efforts, but prospective randomized controlled clinical trials are still crucial to evaluate drug efficacy and safe in the treatment of patients with COVID-19.
The study has several limitations, such as translation of in vitro efficacy to in vivo efficacy, which is challenging as there are several factors to considers, including magnitude of protein binding, drug concentration at the target site, and differences between the in vitro cell lines and the human target site (i.e. lung epithelial cells). Another limitation is that population pharmacokinetic models were not available for all drugs. The structural distribution model and the magnitude of inter-individual variability was assumed for drugs which had only non-compartmental pharmacokinetic parameters reported. The assumption of 30% variability for all pharmacokinetic parameters might resulted in an unexpectedly low or high variability in drug concentrations compared to a real-life clinical treatment setting.
In conclusion, simulated population pharmacokinetic profiles were successfully compared to adjusted in vitro IC50 values to evaluate a range of different repurposed drugs that might be of interest to investigate further in large-scale prospective randomized controlled clinical trials in the treatment of COVID-19. We present here a framework that could be used to prioritize and de-risk the development of repurposed compounds in the treatment of COVID-19.
5. Expert opinion
Repurposing of drugs for emerging infectious diseases can shorten the drug discovery and development process substantially, which is especially important in the very rapidly evolving landscape of an ongoing pandemic. Repurposing of already available and marketed drugs is quick, safe, cheap, and ethical compared to traditional drug development, and could de-risk the development of drugs for novel diseases. It is relatively quick and easy to develop screening tools for in vitro efficacy toward emerging diseases, and a crucial first step to select drugs for repurposing. A sensitive assay to evaluate already available drugs for in vitro efficacy can be applied to screen a broad range of available compounds and drug classes to quickly generate a library of potential drugs for repurposing. However, the translation between in vitro efficacy and clinical efficacy is far from straightforward.
In this review, we present a simple framework for a pharmacometric evaluation of potential drug candidates, to evaluate the likelihood of reaching efficacious drug levels with standard clinical dosing. Several marketed drugs were selected for potential use in the treatment of COVID-19, based on in vitro efficacy in different cell lines. These reported target values should be interpreted as free drug concentrations (i.e. unbound drug concentrations) associated with efficacy, and the IC50 values were therefore corrected for plasma protein binding reported in humans, to generate a target total concentration in plasma/blood. The pharmacokinetic properties of the different drugs were reviewed and a suitable pharmacometric model used for population-based simulations. Standard daily dosing was extended for an assumed total duration of 10 days of treatment, and population-based pharmacokinetic simulations were compared to the corrected target values reported in the in vitro system. Only favipiravir showed simulated total plasma concentrations associated with effective inhibition of SARS-CoV-2 in in vitro systems. This provides a relatively unbiased framework for prioritizing and de-risking the selection process of drugs to evaluate further in prospective randomized controlled clinical trials. It should be emphasized that large randomized controlled clinical trials are generally lacking in the repurposing of drugs for the treatment of COVID-19. Many of the reported trials are not controlled and under-powered to demonstrate clinical efficacy and therefore not suitable to be used for clinical guidance in the treatment of COVID-19. There is also a drastic increase of reports in the pre-print literature on the subject of small molecules to be used in the treatment of COVID-19, which makes it extremely difficult to interpret the many conflicting reports on different drug molecules. The current pandemic has also been highly politicized and different drug molecules have been promoted based on very little scientific evidence.
The presented framework could help to guide the selection process, but there are still many limitations and further improvements that can be implemented. A limitation is the simplification of simulating total plasma/blood concentrations and comparing this to in vitro efficacy data in an isolated cell line. The difficulties with translation of in vitro to in vivo include the inherent differences of the cell culture (certain cell lines) and the target human tissue, as well as the unknown drug concentration at the target site. In addition, several anti-viral drugs have complex intra-cellular metabolism pathways to generate active metabolites (e.g. sofosbuvir and favipiravir). Simple pharmacokinetic models derived from plasma/blood samples will only describe circulating metabolites present in plasma/blood and might therefore not correspond to pharmacokinetic properties of the active intracellular metabolites. Furthermore, in vitro systems can only identify drugs that have an impact on SARS-CoV-2 in the experimental cell culture. Therefore, drugs that could promote host immune modulation could be missed (e.g. dexamethasone have shown to decrease the 28-day mortality in patients with severe COVID-19 by reducing the inflammatory cascade). Thus, we propose to evaluate the in vitro activity in several different types of experimental cell lines (if possible) and particularly focus on experimental cell lines that represent the tissue type at the site of action. If models are available, it would also be a clear advantage to use physiologically-based pharmacokinetic models to simulate drug concentrations at the site of action, instead of relying on the assumption that plasma/blood concentrations are similar to that at the site of action. Results from animal models could also be incorporated into the development framework to assess the therapeutic potential of selected drugs when applied in an animal model experiencing the disease of interest. Lastly, if several small, and possibly non-randomized, studies are conducted to generate results in patients, we advocate strongly for open data sharing and pooled pharmacometric analyses to maximize the amount of information that can be extracted from these trials. Pooled model-based analyses could also generate the evidence needed to produce clinical guidance on the use of small molecules in patients with COVID-19.
Article highlights
Several marketed drugs were evaluated, using in vitro activity data and population-based pharmacokinetic simulations to determine if they could achieve effective drug concentrations in the treatment of COVID-19.
Only favipiravir showed promising clinical drug exposures, as compared to in vitro activity data, and could be a promising candidate for further evaluation in prospective randomized clinical trials in the treatment of COVID-19.
Several drugs showed unsuitable pharmacokinetic exposure profiles, and they are unlikely to generate sufficient clinical drug concentrations for the effective treatment of patients with COVID-19.
Repurposing marketed therapeutic agents in the treatment of COVID-19 is a challenging task and population pharmacometrics could be a useful tool to identify drugs of interest.
Declaration of interest
I Scandale is an employee of the Drugs for Neglected Diseases initiative. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Supplemental Material
Download PDF (999.5 KB)Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
References
- Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov. 2004 Aug;3(8):673–683.
- Dotolo S, Marabotti A, Facchiano A, et al. A review on drug repurposing applicable to COVID-19. Brief Bioinform. 2021 Mar 22;22(2):726–741.
- Wu Z, Li W, Liu G, et al. Network-based methods for prediction of drug-target interactions. Front Pharmacol. 2018;9:1134.
- Na-Bangchang K, Limpaibul L, Thanavibul A, et al. The pharmacokinetics of chloroquine in healthy Thai subjects and patients with Plasmodium vivax malaria. Br J Clin Pharmacol. 1994 Sep;38(3):278–281.
- Boudreau EF, Fleckenstein L, Pang LW, et al. Mefloquine kinetics in cured and recrudescent patients with acute falciparum malaria and in healthy volunteers. Clin Pharmacol Ther. 1990 Oct;48(4):399–409.
- Rijken MJ, McGready R, Jullien V, et al. Pharmacokinetics of amodiaquine and desethylamodiaquine in pregnant and postpartum women with Plasmodium vivax malaria. Antimicrob Agents Chemother. 2011 Sep;55(9):4338–4342.
- Custodio JM, Chuck SK, Chu H, et al. Lack of clinically important PK interaction between coformulated ledipasvir/sofosbuvir and rilpivirine/emtricitabine/tenofovir alafenamide. Pharmacol Res Perspect. 2017 Oct;5(5):e00353.
- Lavielle M, Ilinca E, Kuate R. mlxR: simulation of longitudinal data. R package version 4.2.0. 2021. [cited 2022 Mar 8]. Available from: https://cran.r-project.org/web/packages/mlxR/index.html
- RECOVERY Collaborative Group, Horby P, Mafham M, Linsell L, et al. Effect of hydroxychloroquine in hospitalized patients with covid-19. N Engl J Med. 2020 Nov 19;383(21):2030–2040.
- Watson JA, Tarning J, Hoglund RM, et al. Concentration-dependent mortality of chloroquine in overdose. Elife. 2020 Jul 8;9. DOI: 10.7554/eLife.58631
- Chotsiri P, Tarning J, Hoglund RM, et al. Pharmacometric and electrocardiographic evaluation of chloroquine and azithromycin in healthy volunteers. Clin Pharmacol Ther. 2022 May 22. DOI: 10.1002/cpt.2665
- Gustafsson LL, Rombo L, Alvan G, et al. On the question of dose-dependent chloroquine elimination of a single oral dose. Clin Pharmacol Ther. 1983 Sep;34(3):383–385.
- Gustafsson LL, Walker O, Alvan G, et al. Disposition of chloroquine in man after single intravenous and oral doses. Br J Clin Pharmacol. 1983 Apr;15(4):471–479.
- Hoglund R, Moussavi Y, Ruengweerayut R, et al. Population pharmacokinetics of a three-day chloroquine treatment in patients with Plasmodium vivax infection on the Thai-Myanmar border. Malar J. 2016 Feb 29;15:129.
- Obua C, Hellgren U, Ntale M, et al. Population pharmacokinetics of chloroquine and sulfadoxine and treatment response in children with malaria: suggestions for an improved dose regimen. Br J Clin Pharmacol. 2008 Apr;65(4):493–501.
- Karunajeewa HA, Salman S, Mueller I, et al. Pharmacokinetics of chloroquine and monodesethylchloroquine in pregnancy. Antimicrob Agents Chemother. 2010 Mar;54(3):1186–1192.
- Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020 Mar;30(3):269–271.
- Sacramento CQ, Fintelman-Rodrigues N, Temerozo JR, et al. In vitro antiviral activity of the anti-HCV drugs daclatasvir and sofosbuvir against SARS-CoV-2, the aetiological agent of COVID-19. J Antimicrob Chemother. 2021 Jun 18;76(7):1874–1885.
- Pizzorno A, Padey B, Dubois J, et al. In vitro evaluation of antiviral activity of single and combined repurposable drugs against SARS-CoV-2. Antiviral Res. 2020 Sep;181:104878.
- Jeon S, Ko M, Lee J, et al. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrob Agents Chemother. 2020 Jun 23;64(7). DOI: 10.1128/AAC.00819-20.
- Kaptein SJF, Jacobs S, Langendries L, et al. Favipiravir at high doses has potent antiviral activity in SARS-CoV-2-infected hamsters, whereas hydroxychloroquine lacks activity. Proc Natl Acad Sci U S A. 2020 Oct 27 117(43):26955–26965.
- WHO Solidarity Trial Consortium, Pan H, Peto R, Karim QA, et al. Repurposed antiviral drugs for covid-19 - interim WHO solidarity trial results. N Engl J Med. 2021 Feb 11;384(6):497–511.
- White NJ, Watson JA, Hoglund RM, et al. COVID-19 prevention and treatment: a critical analysis of chloroquine and hydroxychloroquine clinical pharmacology. Plos Med. 2020 Sep;17(9):e1003252.
- Weinke T, Trautmann M, Held T, et al. Neuropsychiatric side effects after the use of mefloquine. Am J Trop Med Hyg. 1991 Jul;45(1):86–91.
- Guidi M, Mercier T, Aouri M, et al. Population pharmacokinetics and pharmacodynamics of the artesunate-mefloquine fixed dose combination for the treatment of uncomplicated falciparum malaria in African children. Malar J. 2019 Apr 18 18(1):139.
- Hoglund RM, Ruengweerayut R, Na-Bangchang K. Population pharmacokinetics of mefloquine given as a 3-day artesunate-mefloquine in patients with acute uncomplicated Plasmodium falciparum malaria in a multidrug-resistant area along the Thai-Myanmar border. Malar J. 2018 Sep 3; 17(1):322.
- Reuter SE, Upton RN, Evans AM, et al. Population pharmacokinetics of orally administered mefloquine in healthy volunteers and patients with uncomplicated Plasmodium falciparum malaria. J Antimicrob Chemother. 2015 Mar;70(3):868–876.
- Valea I, Tinto H, Traore-Coulibaly M, et al. Pharmacokinetics of co-formulated mefloquine and artesunate in pregnant and non-pregnant women with uncomplicated Plasmodium falciparum infection in Burkina Faso. J Antimicrob Chemother. 2014 Sep;69(9):2499–2507.
- Gendrot M, Andreani J, Boxberger M, et al. Antimalarial drugs inhibit the replication of SARS-CoV-2: an in vitro evaluation. Travel Med Infect Dis. 2020 Sep - Oct;37:101873.
- Gendrot M, Duflot I, Boxberger M, et al. Antimalarial artemisinin-based combination therapies (ACT) and COVID-19 in Africa: in vitro inhibition of SARS-CoV-2 replication by mefloquine-artesunate. Int J Infect Dis. 2020 Oct;99:437–440.
- Jones R, Kunsman G, Levine B, et al. Mefloquine distribution in postmortem cases. Forensic Sci Int. 1994 Sep 6;68(1):29–32.
- WHO Policy Recommendation. Seasonal Malaria Chemoprevention (SMC) for Plasmodium falciparum malaria control in highly seasonal transmission areas of the Sahel sub-region in Africa: world health organization. 2012. [cited 2022 Mar 8]. Available from: https://apps.who.int/iris/bitstream/handle/10665/337978/WHO-HTM-GMP-2012.02-eng.pdf?sequence=1&isAllowed=y
- Tarning J, Chotsiri P, Jullien V, et al. Population pharmacokinetic and pharmacodynamic modeling of amodiaquine and desethylamodiaquine in women with Plasmodium vivax malaria during and after pregnancy. Antimicrob Agents Chemother. 2012 Nov;56(11):5764–5773.
- Ali AM, Penny MA, Smith TA, et al. Population pharmacokinetics of the antimalarial amodiaquine: a pooled analysis to optimize dosing. Antimicrob Agents Chemother. 2018 Oct;62(10). 10.1128/AAC.02193-17.
- Winstanley PA, Edwards G, Curtis CG, et al. Tissue distribution and excretion of amodiaquine in the rat. J Pharm Pharmacol. 1988 May;40(5):343–349.
- Gandhi Y, Eley T, Fura A, et al. Daclatasvir: a review of preclinical and clinical pharmacokinetics. Clin Pharmacokinet. 2018 Aug;57(8):911–928.
- Chan P, Li H, Zhu L, et al. Population pharmacokinetic analysis of Daclatasvir in subjects with chronic hepatitis C virus infection. Clin Pharmacokinet. 2017 Oct;56(10):1173–1183.
- Osawa M, Ueno T, Ishikawa H, et al. Population pharmacokinetic analysis for Daclatasvir and asunaprevir in Japanese subjects with chronic hepatitis C virus infection. J Clin Pharmacol. 2018 Nov;58(11):1468–1478.
- Osawa M, Ueno T, Shiozaki T, et al. Population pharmacokinetic analysis of Daclatasvir, asunaprevir, and beclabuvir combination in HCV-Infected subjects. Clin Pharmacol Drug Dev. 2019 Aug;8(6):802–817.
- Shen Z, Zhu X, Zhang H, et al. Pharmacokinetic profile of a generic formulation of sofosbuvir and its metabolite GS-331007 in healthy Chinese subjects. Clin Pharmacol Drug Dev. 2019 Nov;8(8):1073–1080.
- Jin F, Kirby B, Gao Y, et al. Population pharmacokinetic modeling of sofosbuvir, an NS5B polymerase inhibitor, and its metabolites in patients with hepatitis C virus infection. In: Population Approach Group in Europe (PAGE-meeting). Crete Greece:Hersonissos; 2015
- Abbaspour Kasgari H, Moradi S, Shabani AM, et al. Evaluation of the efficacy of sofosbuvir plus daclatasvir in combination with ribavirin for hospitalized COVID-19 patients with moderate disease compared with standard care: a single-centre, randomized controlled trial. J Antimicrob Chemother. 2020 Nov 1;75(11):3373–3378.
- Khalili H, Nourian A, Ahmadinejad Z, et al. Efficacy and safety of sofosbuvir/ ledipasvir in treatment of patients with COVID-19; A randomized clinical trial. Acta Biomed. 2020 Nov 10;91(4):e2020102.
- Zein A, Sulistiyana CS, Raffaello WM, et al. Sofosbuvir with daclatasvir and the outcomes of patients with COVID-19: a systematic review and meta-analysis with GRADE assessment. Postgrad Med J. 2021;98(1161):509–514.
- Pertinez H, Rajoli RKR, Khoo SH, et al. Pharmacokinetic modelling to estimate intracellular favipiravir ribofuranosyl-5’-triphosphate exposure to support posology for SARS-CoV-2. J Antimicrob Chemother. 2021 Jul 15;76(8):2121–2128.
- Wang Y, Zhong W, Salam A, et al. Phase 2a, open-label, dose-escalating, multi-center pharmacokinetic study of favipiravir (T-705) in combination with oseltamivir in patients with severe influenza. EBio Med. 2020 Dec;62:103125.
- Udwadia ZF, Singh P, Barkate H, et al. Efficacy and safety of favipiravir, an oral RNA-dependent RNA polymerase inhibitor, in mild-to-moderate COVID-19: a randomized, comparative, open-label, multicenter, phase 3 clinical trial. Int J Infect Dis. 2021 Feb;103:62–71.
- Kelleni MT. NSAIDs/nitazoxanide/azithromycin repurposed for COVID-19: potential mitigation of the cytokine storm interleukin-6 amplifier via immunomodulatory effects. Expert Rev Anti Infect Ther. 2022 Jan;20(1):17–21.
- ALINIA [product monograph] Tampa FL. Romark L.C. 2005. [cited 2022 Mar 8]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2005/021818lbl.pdf
- Broekhuysen J, Stockis A, Lins RL, et al. Nitazoxanide: pharmacokinetics and metabolism in man. Int J Clin Pharmacol Ther. 2000 Aug;38(8):387–394.
- Rossignol JF. Nitazoxanide: a first-in-class broad-spectrum antiviral agent. Antiviral Res. 2014 Oct;110:94–103.
- Rajoli RK, Pertinez H, Arshad U, et al. Dose prediction for repurposing nitazoxanide in SARS-CoV-2 treatment or chemoprophylaxis. medRxiv. 2020 May 6
- Marcelin-Jimenez G, Contreras-Zavala L, Maggi-Castellanos M, et al. Development of a method by UPLC-MS/MS for the quantification of tizoxanide in human plasma and its pharmacokinetic application. Bioanalysis. 2012 May;4(8):909–917.
- Balderas-Acata JI, Ríos-Rogríguez Bueno EP, Pérez-Becerril F, et al. Bioavailability of two oral-suspension formulations of a single dose of nitazoxanide 500 mg: an open-label, randomized-sequence, two-period crossover, comparison in healthy fasted Mexican adult volunteers [research article]. J Bioequivalence Bioavailability. 2011;3(3):043–047.
- STROMECTOL [product monograph] Kirkland QC. Canada: Merck Canada Inc.; 2018. [cited 2022 Mar 8]. Available from: https://www.merck.ca/static/pdf/STROMECTOL-PM_E.pdf
- Wishart DS, Knox C, Guo AC, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006 Jan 1;34( Database issue):D668–72.
- Gonzalez Canga A, Sahagun Prieto AM, Diez Liebana MJ, et al. The pharmacokinetics and interactions of ivermectin in humans–a mini-review. AAPS J. 2008;10(1):42–46.
- Tipthara P, Kobylinski KC, Godejohann M, et al. Identification of the metabolites of ivermectin in humans. Pharmacol Res Perspect. 2021 Feb;9(1):e00712.
- El-Tahtawy A, Glue P, Andrews EN, et al. The effect of azithromycin on ivermectin pharmacokinetics–a population pharmacokinetic model analysis. PLoS Negl Trop Dis. 2008 May 14;2(5):e236.
- Duthaler U, Suenderhauf C, Karlsson MO, et al. Population pharmacokinetics of oral ivermectin in venous plasma and dried blood spots in healthy volunteers. Br J Clin Pharmacol. 2019 Mar;85(3):626–633.
- Gwee A, Duffull S, Zhu X, et al. Population pharmacokinetics of ivermectin for the treatment of scabies in Indigenous Australian children. PLoS Negl Trop Dis. 2020 Dec;14(12):e0008886.
- Caly L, Druce JD, Catton MG, et al. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antiviral Res. 2020 Jun;178:104787.
- REYATAZ [product monograph]. Montreal Canada: Bristol-Myers Squibb Canada Co.; 2020. [cited 2022 Mar 8]. Available from: https://www.bms.com/assets/bms/ca/documents/productmonograph/REYATAZ_EN_PM.pdf
- Fintelman-Rodrigues N, Sacramento CQ, Lima CR,et al . (2020). Atazanavir, Alone or in Combination with Ritonavir, Inhibits SARS-CoV-2 Replication and Proinflammatory Cytokine Production. Antimicrob Agents Chemother, 64(10), 10.1128/AAC.00825-20
- Yamamoto N, Matsuyama S, Hoshino T,et al. Nelfinavir inhibits replication of severe acute respiratory syndrome coronavirus 2 in vitro. bioRxiv: 2020.04.06.026476.
- Le Tiec C, Barrail A, Goujard C, et al. Clinical pharmacokinetics and summary of efficacy and tolerability of atazanavir. Clin Pharmacokinet. 2005;44(10):1035–1050.
- Dickinson L, Boffito M, Back D, et al. Population pharmacokinetics of ritonavir-boosted atazanavir in HIV-infected patients and healthy volunteers. J Antimicrob Chemother. 2009 Jun;63(6):1233–1243.
- Punyawudho B, Thammajaruk N, Ruxrungtham K, et al. Population pharmacokinetics and dose optimisation of ritonavir-boosted atazanavir in Thai HIV-infected patients. Int J Antimicrob Agents. 2017 Mar;49(3):327–332.
- Schipani A, Dickinson L, Boffito M, et al. Simultaneous population pharmacokinetic modelling of atazanavir and ritonavir in HIV-infected adults and assessment of different dose reduction strategies. J Acquir Immune Defic Syndr. 2013 Jan 1 62(1):60–66.
- Solas C, Gagnieu MC, Ravaux I, et al. Population pharmacokinetics of atazanavir in human immunodeficiency virus-infected patients. Ther Drug Monit. 2008 Dec;30(6):670–673.
- Kile DA, MaWhinney S, Aquilante CL, et al. A population pharmacokinetic-pharmacogenetic analysis of atazanavir. AIDS Res Hum Retroviruses. 2012 Oct;28(10):1227–1234.
- Foissac F, Blanche S, Dollfus C, et al. Population pharmacokinetics of atazanavir/ritonavir in HIV-1-infected children and adolescents. Br J Clin Pharmacol. 2011 Dec;72(6):940–947.
- COLCHICINE [product monograph] Pointe-Claire, Québec: Odan laboratories Ltd. 2016. [cited 2022 Mar 8]. Available from: https://pdf.hres.ca/dpd_pm/00034804.PDF
- Ferron GM, Rochdi M, Jusko WJ, et al. Oral absorption characteristics and pharmacokinetics of colchicine in healthy volunteers after single and multiple doses. J Clin Pharmacol. 1996 Oct;36(10):874–883.
- Rochdi M, Sabouraud A, Girre C, et al. Pharmacokinetics and absolute bioavailability of colchicine after i.v. and oral administration in healthy human volunteers and elderly subjects. Eur J Clin Pharmacol. 1994;46(4):351–354.
- Girre C, Thomas G, Scherrmann JM, et al. Model-independent pharmacokinetics of colchicine after oral administration to healthy volunteers. Fundam Clin Pharmacol. 1989;3(5):537–543.
- Kaddoura M, AlIbrahim M, Hijazi G, et al. COVID-19 therapeutic options under investigation. Front Pharmacol. 2020;11:1196.
- Terkeltaub RA, Furst DE, Bennett K, et al. High versus low dosing of oral colchicine for early acute gout flare: twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum. 2010 Apr;62(4):1060–1068.
- Paschke S, Weidner AF, Paust T, et al. Technical advance: inhibition of neutrophil chemotaxis by colchicine is modulated through viscoelastic properties of subcellular compartments. J Leukoc Biol. 2013 Nov;94(5):1091–1096.
- Schlesinger N, Firestein BL, Brunetti L. Colchicine in COVID-19: an old drug, new use. Curr Pharmacol Rep. 2020 Jul;18:1–9.