
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Latently infected CD T cells represent one of the major obstacles to HIV eradication even after receiving prolonged highly active anti-retroviral therapy (HAART). Long-term use of HAART causes the emergence of drug-resistant virus which is then involved in HIV transmission. In this paper, we develop mathematical HIV models with staged disease progression by incorporating entry inhibitor and latently infected cells. We find that entry inhibitor has the same effect as protease inhibitor on the model dynamics and therefore would benefit HIV patients who developed resistance to many of current anti-HIV medications. Numerical simulations illustrate the theoretical results and show that the virus and latently infected cells reach an infected steady state in the absence of treatment and are eliminated under treatment whereas the model including homeostatic proliferation of latently infected cells maintains the virus at low level during suppressive treatment. Therefore, complete cure of HIV needs complete eradication of latent reservoirs.
1. Introduction
Human immunodeficiency virus (HIV) attacks the immune system by invading CD T cells, which are critical for energizing regulated immune responses to fight infections. As we see in Figure , the virus contains an enzyme essential for infectivity called protease. There is also a protein shell surrounding the nucleus of the virus called a capsid, which involves two enzymes necessary for HIV replication, reverse transcriptase and integrase, and two strands of RNA. The protease enzyme and the capsid are covered with the envelope, and this is covered with spikes of glycoproteins gp120 and gp41.
Figure 1. The HIV replication cycle [Citation36].
![Figure 1. The HIV replication cycle [Citation36].](/cms/asset/d9b4c8db-a96a-449a-b9eb-4b3501f6ca0f/tjbd_a_2113828_f0001_oc.jpg)
The virus uses CD T-cell components to release new viruses and initiate the next rounds of replication. This replication cycle goes through four main infection stages as shown in Figure ; viral entry into the cell, reverse transcription from viral RNA to DNA, integration of viral DNA into the host cell DNA, and transcription and translation to release infectious virus from the infected cell. During the viral entry stage, with the help of glycoprotein gp120, the virus first attaches to the primary receptor CD4, followed by the CCR5 or CXCR4 co-receptors on the surface of CD
T cells. Then the virus merges its envelope with the cell wall by the glycoprotein gp41. This process of fusion allows the HIV capsid to enter the CD
T cell. Inside the cell, the reverse transcription stage starts, and the virus uses its reverse transcriptase enzyme to reverse the order of the transcription process of generating messenger RNA from nuclear DNA to generate proviral DNA. The viral DNA in the integration stage enters the cell nucleus and binds to the cell DNA using HIV integrase enzyme. Once the viral DNA is integrated into the CD
T-cell DNA, the provirus begins using the cell machinery to generate the viral RNA. This RNA provides instructions for making new viral proteins in long chains. The new HIV proteins and RNA move to the cell surface to release immature (noninfectious) viruses. The newly formed noninfectious virus releases protease enzyme which break up the long protein chains into smaller chains that assemble to create a fully mature (infectious) virus at the cell wall. The infectious virus buds from the cell wall to initiate the next round of replication.
In HIV replication cycle, anti-HIV drug classes act at different stages of infection progression. They can slow the virus progression, reduce the transmission rate and suppress the viral load to below the detection limit. Entry inhibitors block the processes of virus attachment and entry to the target CD T cell. Reverse transcriptase inhibitors stop the action of reverse transcriptase and the creation of proviral DNA. Integrase inhibitors prevent the viral DNA from being incorporated into the host CD
T-cell DNA. Protease inhibitors block the production of infectious virus particles by preventing noninfectious virus from being fully matured. Even if the antiretroviral therapy can eliminate the virus, one of the latently infected CD
T cells could be activated and rebound the virus spread. These latently infected CD
T cells present a major barrier to HIV cure and play an important role in low viral load persistence in patients receiving prolonged antiretroviral therapy [Citation8,Citation23,Citation67]. On the other hand, latently infected CD
T cells may not be completely affected by current anti-HIV therapy and can be activated to rejoin the replication cycle and release new infectious virus particles. Studying and understanding the properties of these cells are therefore important to eradicate HIV. Many HIV models including latent infection with a single infection stage have been developed to investigate the dynamics of HIV [Citation2,Citation3,Citation28,Citation38,Citation43,Citation68,Citation71–74,Citation79,Citation81,Citation83,Citation87]. Most of the models consist of one compartment of productively infected CD
T cells. They assumed that the HIV replication cycle goes through a single infection stage to produce infectious viruses. In fact, HIV goes through multiple distinct stages to release new infectious virus particles: viral entry, reverse transcription, integration, and transcription and translation. Many mathematical models have been introduced to study the dynamics of HIV during multiple infection stages, and these models better predict the virus dynamics [Citation24,Citation31–33,Citation39,Citation45,Citation57,Citation85,Citation86,Citation88,Citation92].
Many studies including mathematical models, cell culture experiments and patients data introduced the effect of different drug classes on the dynamics of HIV decay. These studies showed that the viral load decay depends on the inhibited stage in HIV replication cycle. Wang et al. and Sedaghat et al. [Citation76,Citation77,Citation85] studied mathematical models and introduced that when drug classes are operative, the viral load goes through a single phase of decay while when drug classes are not
effective or act at later stages of the viral replication cycle, the viral load decline can have two phases. Moreover, later in HIV replication cycle an inhibitor acts, the more rapid the viral load decay under a
effective treatment [Citation21,Citation24,Citation49,Citation57,Citation76,Citation77,Citation85], whereas inhibitors acting later in the cycle may not result in a faster viral load decay than reverse transcription inhibitor if the drug efficacy is not a
[Citation21,Citation85].
In fact, viral entry stage is different from other stages in the replication cycle since during which the cell has not been HIV infected yet and to infect the cell, the proteins on HIV surface must bind to the proteins on the surface of CD T cell, triggering the fusion of the viral envelope with the cell wall. Entry inhibitors work by attaching themselves to proteins on the surface of CD
T cells or proteins on the surface of HIV to prevent viral entry into the cells. Some entry inhibitors target the proteins on HIV surface (gp120 and gp41) and some others target the receptor (CD4) and co-receptors (CCR5 and CXCR4) on the cell surface [Citation91]. Thus if entry inhibitors successfully block the first stage in HIV replication cycle, HIV would be unable to enter the cells. Furthermore, entry inhibitors help prevent resistance to other two drug classes (nucleoside and non-nucleoside reverse transcriptase inhibitors [NRTIs and NNRTIs and protease inhibitors PIs)Citation10,Citation12,Citation25,Citation26,Citation47,Citation50,Citation55,Citation65]. Patients who have tried and failed to respond to NRTIs, NNRTIs and/or PIs will likely benefit from entry inhibitor drugs because they are from different class [Citation10,Citation12,Citation25,Citation26,Citation37,Citation41,Citation47,Citation50,Citation55,Citation65]. Entry inhibitors like post-attachment inhibitor ibalizumab, CCR5 inhibitor maraviroc and fusion inhibitor enfuvirtide are FDA approved [Citation17,Citation54,Citation64,Citation78,Citation94]. Therefore, the inclusion of entry inhibitors class into the models is beneficial for HIV patients and more useful in studying the influence of drug classes.
Many mathematical models have been developed to describe the effect of the entry inhibitors on HIV progression. Mathematical models in [Citation11,Citation14] suggest that a combination of fusion inhibitors (enfuvirtide) and protease inhibitors gives a better outcome than single-drug activity in suppressing the viral load and inhibiting viral entry into the host cell. Magombedze et al. [Citation48] developed mathematical models of the immuno-pathogenesis of HIV infection by incorporating three different stages of the HIV replication cycle: viral entry, transcription from viral RNA to DNA, and production of HIV viral particles to study the effect of their inhibitors. They showed that any therapy that includes a combination of entry inhibitor and protease inhibitor is the most potent against HIV replication. Mathematical models in [Citation46,Citation80] demonstrated the importance of perfect patient compliance during HIV treatment and interruption in taking enfuvirtide can be worse than no therapy at all.
Wang et al. [Citation86] developed a multi-stage latent infection model, based on the model presented in [Citation85], with all drug classes except entry inhibitors. They incorporated latently infected cells which established in the stage of integration and a compartment of cells that have un-integrated viral DNA with two copies of the viral long-terminal repeat (2-LTR circles) to study the effect of treatment intensification with raltegravir on both viral load and 2-LTR dynamics in patients with sustained viral suppression. They found that treatment intensification induces a minor decrease in the viral load and a minor increase in 2-LRT. Adding additional raltegravir into treatment regimens did not help improve the treatment outcomes. However, the drug class of entry inhibitors has not been incorporated into the model and investigated.
This paper develops an HIV latent infection model with four drug classes: entry, reverse transcriptase, integrase and protease inhibitors. The model is an extension of the models introduced by Wang et al. [Citation85,Citation86] and Gilmore et al. [Citation24], and it includes both entry inhibitors and latently infected CD T cells. We investigate the local stability of the steady states. The basic reproduction number
of the model is derived to determine whether the infection will die out or persist. We show that chronic disease will be established if
and will be eradicated if
. Numerical simulations of the model are performed to explain the influence of drug classes on the dynamics of the model. We find that the impact of entry inhibitor on the model's dynamics is the same as protease inhibitor. We also find that integrase inhibitor is the most effective drug in reducing the viral load and latently infected cells compared with the other inhibitors and uninfected CD
T cells attain the highest level under the effect of entry inhibitor. The minimum effectiveness of each drug class to eliminate the virus is calculated. Moreover, we show that combination therapy of four drug classes doesn't give better outcomes. We also introduce the influence of the latency fraction on the dynamics of the latently infected CD
T cells and viral load decay. The model with homeostatic proliferation of latently infected cells is shown to be able to produce the low viral load persistence observed in HIV patients receiving lengthy suppressive treatment. We also evaluate the effect of proliferation rate and carrying capacity of latent infected cells on the virus level.
2. HIV latent infection model with four infection stages and four drug classes
We develop an HIV latent infection model with four infection progression stages and four drug classes based on the models in refs. [Citation24,Citation85,Citation86]. Wang et al. introduced the model without latently infected CD T cells in [Citation85] to analyse the effect of different drug classes on the dynamics of HIV decay and in [Citation86] they introduced the model without entry inhibitors including the population of cells that contain 2-LTR DNA circles to evaluate the influence of raltegravir intensification on the viral load dynamics. We incorporate both entry inhibitors and latently infected CD
T cells into the model. Considering that HIV infection inside the CD
T cells progresses in four major phases: viral entry, reverse transcription, integration and creation infectious virus, the model can be described by the following system of differential equations:
(1)
(1) In the model,
is the population of uninfected CD
T cells at time t and
, i = 1, 2, 3 represent the population of infected CD
T cells that have finished stages 1 up to i in the virus life cycle.
is the concentration of infected cells that have finished the process of integration and gone to the latency state. In the fourth phase of HIV life cycle, protease enzyme causes infected cells to produce infectious virus particles.
is the concentration of infectious virus particles (that have not been affected by the enzyme inhibitors), whereas
is the concentration of non-infectious virus particles (that have been influenced by protease inhibitors). Parameters
,
,
and
denote the efficacy of four drug classes: entry inhibitors, reverse transcriptase inhibitors, integrase inhibitors and protease inhibitors, respectively. Table gives a brief definition and the values of the parameters used in model Equation1
(1)
(1) .
Table 1. Parameter values of model Equation1(1)
(1) .
We derive the basic reproductive number using the next-generation method in [Citation19,Citation84], we consider the infection and viral production term in the model and obtain the nonnegative matrix F and the nonsingular matrix V as follows:
(2)
(2)
(3)
(3) The basic reproduction number can be calculated by the spectral radius of the next-generation matrix
:
.
(4)
(4) where
(5)
(5) Therefore,
is given as follows:
(6)
(6) Model (Equation1
(1)
(1) ) has two equilibria: disease-free equilibrium
and chronic disease equilibrium
, where
(7)
(7)
3. Local stability analysis of model (1)
In this section, we study the local stability of the disease-free equilibrium and chronic disease equilibrium.
Theorem 3.1
The disease-free equilibrium of model (Equation1
(1)
(1) ) is locally asymptotically stable if
and unstable when
.
Proof.
Using Lemma 1 and Theorem 2 of the paper by van den Driessche and Watmough [Citation84], we find that the disease-free steady state is locally asymptotically stable when and unstable when
.
Theorem 3.2
The chronic disease equilibrium of model (Equation1
(1)
(1) ) is locally asymptotically stable when it exists, i.e.
.
Proof.
We start by calculating the Jacobian matrix at the infected steady state, it follows the characteristic equation:
(8)
(8) where λ is the eigenvalue.
The characteristic equation can be rewritten as
(9)
(9) where
(10)
(10) It is obvious that 0<g<1. Suppose that the eigenvalue λ has a non-negative real part, using Cauchy–Schwartz inequality and the condition of
we obtain
(11)
(11) The modulus of the left-hand side of Equation (Equation11
(11)
(11) ) is greater than the modulus of the right-hand side. This is a contradiction and means that all eigenvalues have negative real parts. Thus the endemic steady state
is locally asymptotically stable when
.
4. Numerical results
In this section we observe numerical simulations using the ode23s solver in MATLAB. In all simulations we plot the total viral concentration V of infectious and non-infectious virus particles, that is , because non-infectious virus can still be detected by the reverse transcriptase polymerase chain reaction test (RT-PCR) and counted in the total viral load.
4.1. Numerical simulations of model (1)
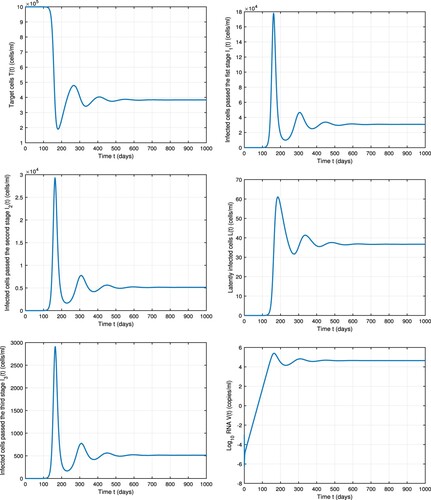
We first present numerical simulations of model (Equation1(1)
(1) ) without treatment in Figure (). We chose the initial value of the chronic disease steady state to be
. Parameter values used are all chosen from Table (). We assume that the death rate of infected cells increases as the infection stages progress
,
and
. The basic reproduction number with these parameter values is 2.6>1. Numerical simulation in Figure confirms our result obtained in Theorem 3.2 and shows that the solution approaches the chronic disease steady state
. Thus in the absence of treatment, the infection is predicted to persist.
Figure 2. Dynamics of model Equation1(1)
(1) without treatment. Parameter values are chosen from Table .
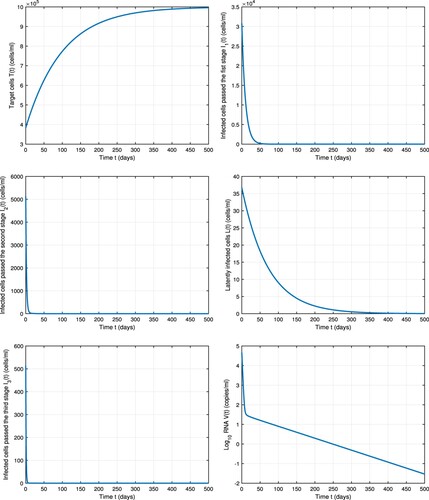
In Figure , numerical simulations of model (Equation1(1)
(1) ) are performed under treatment. The four drug classes are assumed to be
effective (i.e.
). The same parameters in Table are used and the endemic steady state
is chosen to be the initial value of the model under treatment. The basic reproduction number with these drug values is
. Numerical simulations show that the uninfected cells (T) rebound to the pre-infection level
and all other model compartments: infected cells
and
, latently infected cells (L) and viral load (V ) are predicted to decline to 0 under treatment. It agrees with the stability result in Theorem 3.1. Therefore, the chronic disease is predicted to die out after treatment. We compare the influence of each drug class administration on HIV eradication taking in consideration eliminating the other drug classes. We find that if entry/protease inhibitor is at least
effective, it can efficiently block the viral entry into the cell and therefore eradicate the virus, whereas the efficacy of reverse transcriptase inhibitor and integrase inhibitor should be greater than or equal to
and
, respectively to eliminate the virus.
Figure 3. Dynamics of model (Equation1(1)
(1) ) with treatment. The four drug classes
,
,
and
are assumed to be
effective. Parameter values are chosen from Table .
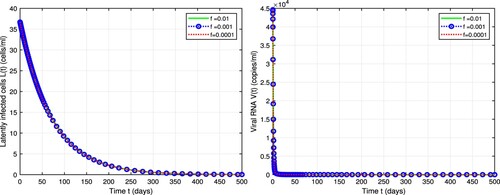
We also examine the sensitivity of latent reservoir and viral load in model (Equation1(1)
(1) ) on the fraction of latency (f). The parameter values and the initial condition are the same as Figure (), but we test the model for a variety of f. We find that the level of latently infected cells is very sensitive to f before treatment and Figure shows that, as the fraction latency increases from 0.0001 to 0.01 [Citation2], the latently infected cells level increases because a higher fraction of latency activates more latently infected cells while in Figure () there is no difference in the latent infection levels under treatment because
efficacy of integrase inhibitor reduces the inflow of new latently infected cells, that is
, by
to a small fraction
. Since the proportion of infection that went to latency can be activated and rejoin the HIV replication cycle to release new virus particles, the fraction of latency (f) doesn't affect the dynamics of
cells and therefore the viral load V as we see in Figures and .
Figure 4. Dynamics of latently infected cells and viral load in model (Equation1(1)
(1) ) before treatment with different fractions of latency, that is, f = 0.01, 0.001, and 0.0001 [Citation2]. Parameter values are chosen from Table .
![Figure 4. Dynamics of latently infected cells and viral load in model (Equation1(1) dTdt=s−dTT−(1−ηE)βVIT,dI1dt=(1−ηE)βVIT−δ1I1−(1−ηRT)β1I1,dI2dt=(1−ηRT)β1I1−δ2I2−(1−ηI)β2I2,dLdt=f(1−ηI)β2I2−δLL−αL,dI3dt=(1−f)(1−ηI)β2I2−δ3I3+αL,dVNIdt=ηPNδ3I3−cVNI,dVIdt=(1−ηP)Nδ3I3−cVI.(1) ) before treatment with different fractions of latency, that is, f = 0.01, 0.001, and 0.0001 [Citation2]. Parameter values are chosen from Table 1.](/cms/asset/a58ad56e-f006-44f8-80bc-e8d584a0f604/tjbd_a_2113828_f0004_oc.jpg)
Figure 5. Dynamics of latently infected cells and viremia in model (Equation1(1)
(1) ) with different fractions of latency f = 0.01, 0.001, and 0.0001. Drugs are assumed to be
,
,
, and
. Parameter values are chosen from Table .
In Figure , we show that the dynamics of the model depend on the inhibited stages in the viral replication cycle (see Table ). We assume the initial values are the same as those in Figure . Protease inhibitor and entry inhibitor act to block two consecutive stages in the HIV replication cycle (last and first stages). Protease inhibitor prevents infectious virus production and entry inhibitor hinders viral entry into the cell. Both works to protect uninfected CD T cells from viral entry. Therefore, there is almost no difference in the dynamics of model Equation1
(1)
(1) whether with protease inhibitor or entry inhibitor treatment (Figure ). That means entry inhibitor is beneficial for HIV patients who have become resistant to PIS or even NRTIs or NNRTIs because it is from different class of drugs. We also find that the most effective treatment in reducing the viral load and latently infected cells is integrase inhibitor followed by reverse transcriptase inhibitor and then entry inhibitor (which has the same effect of protease inhibitor). For the compartments
, i = 1, 2, 3, inhibitor acting at a stage closer to its stage in the cycle leads to a faster decay, that is, the lists of inhibitors in order of the efficacy on
,
and
are
transcriptase, reverse
, and
, respectively. The list of drug classes in order of effectiveness on target CD
T cells is as same as those on
cells and preventing earlier stages doesn't make a better effect than protease inhibitor because it blocks the viral production in the very last stage of HIV replication cycle. Numerical simulations in Figure () suggest that treatment intensification of the four drug classes (entry inhibitor, reverse transcriptase inhibitor, integrase inhibitor and protease inhibitor) doesn't have better results.
Figure 6. Dynamics of model (Equation1(1)
(1) ) with different drug classes. Parameter values are chosen from Table .
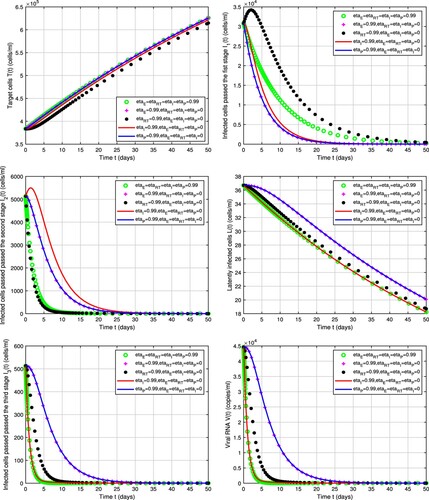
Table 2. The drugs in order of effectiveness on model (Equation1(1)
(1) ).
4.2. Numerical results of the model with homeostatic proliferation of latently infected CD T cells
T cells
In this section, we only consider integrase inhibitor in the treatment because it is shown in the previous section to be the most effective drug class on latently infected cells and viremia. Using parameter values in Table , we find that the virus would be eliminated if the efficacy of integrase inhibitor is or more. In fact, the virus in clinical observations persists at a low level between 1 and 50 RNA copies/ml during lengthy HAART. Here we extend model Equation1
(1)
(1) to describe this viral persistence by incorporating homeostatic proliferation of latently infected cells. Homeostatic proliferation is described by the logistic expression
where p is the maximum proliferation rate and
is the carrying capacity of latent cells. Equations of the basic model Equation1
(1)
(1) stay the same except the latent cells' equation becomes the following
(12)
(12) In the equation,
represents latently infected cells generation by the ongoing viral replication cycle while
is the proliferation by existing latent cells. Parameters p and
are chosen to be 0.02
and 1 cell/ml [Citation2], respectively. The other parameter values are the same as those in Table . The infected steady state
is taken to be the initial value for the model under
effective integrase inhibitor. Numerical simulations for 500 days are shown in Figure () where p = 0 represents the basic model and a proliferation rate of p = 0.02 per day is used for the new model. Figure (a and b) show that the model including homeostatic proliferation of latent cells can generate the persistence of low-level viremia during suppressive integrase inhibitor and drug intensification leads to a reduction in the level of persistent viremia without eradicating the virus. We evaluate the sensitivity of the new model with respect to the parameters used in the logistic term of homeostatic proliferation. As p or
increases, the viremia level increases (see Figure b and c). Homeostatic proliferation of latently infected cells was included in many mathematical models [Citation2,Citation60,Citation71,Citation86] and experimental study [Citation13] to describe the virus persistence at low level below the detection limit of clinical inspections.
Figure 7. Effect of parameters , p and
on the viral decay dynamics using the model with homeostatic proliferation of latently infected cells. (
) Viral load decay with different integrase inhibitor effectiveness,
, 0.99, 0.9 and 0.8. Homeostatic proliferation rate and carrying capacity of latently infected cells are fixed at 0.02 and 1, respectively. (
) Viremia dynamics with different rates of proliferation, p = 0, 0.01, 0.02, and 0.03 day
[Citation2]. The carrying capacity of latently infected cells is fixed at
cell/ml and integrase inhibitor is
effective. (
) Viral load with different carrying capacities of latently infected cells,
, 0.3, 0.5 and 1 cell/ml [Citation2]. Homeostatic proliferation rate and integrase inhibitor are fixed, p = 0.02 and
. All other parameter values are as same as those listed in Table . The HIV detection limit is 50 RNA copies/ml.
![Figure 7. Effect of parameters ηI, p and Lmax on the viral decay dynamics using the model with homeostatic proliferation of latently infected cells. (a) Viral load decay with different integrase inhibitor effectiveness, ηI=1, 0.99, 0.9 and 0.8. Homeostatic proliferation rate and carrying capacity of latently infected cells are fixed at 0.02 and 1, respectively. (b) Viremia dynamics with different rates of proliferation, p = 0, 0.01, 0.02, and 0.03 day−1 [Citation2]. The carrying capacity of latently infected cells is fixed at Lmax=1 cell/ml and integrase inhibitor is 99% effective. (c) Viral load with different carrying capacities of latently infected cells, Lmax=0.1, 0.3, 0.5 and 1 cell/ml [Citation2]. Homeostatic proliferation rate and integrase inhibitor are fixed, p = 0.02 and ηI=0.99. All other parameter values are as same as those listed in Table 1. The HIV detection limit is 50 RNA copies/ml.](/cms/asset/56a34073-6976-49cd-9e74-261713a7e4e3/tjbd_a_2113828_f0007_oc.jpg)
5. Conclusion and discussion
HAART employs a combination of NRTIs, NNRTIs and/or PIs. Durable suppression of HIV can improve the immune system and HIV-patient health, prevent HIV-associated mortality and reduce the risk of transmission [Citation4,Citation6,Citation7,Citation16,Citation27,Citation69,Citation70]. However, the virus can be suppressed by HAART rather than eradicated [Citation15,Citation23,Citation93] due to the presence of latent infection [Citation8,Citation23,Citation67]. Furthermore, long-term use of anti-HIV therapies causes the emergence of multi-drug resistant HIV strains [Citation5,Citation9,Citation64,Citation65]. The drug-resistant virus is then involved in HIV transmission and more than of newly infected individuals carrying HIV that are resistant to at least one drug of HAART [Citation29,Citation89]. Drug class of entry inhibitors can be used to address this issue [Citation10,Citation12,Citation25,Citation26,Citation47,Citation50,Citation55,Citation65]. Moreover, entry inhibitor has mechanisms of action different from those of HAART. They act outside the cell targeting the extracellular viral infection steps before the cell is infected. The first peptide-based HIV entry inhibitor, enfuvirtide, was approved by the U.S. FDA in 2003 to treat HIV patients who have become resistant to HAART. Entry inhibitors are potentially more valuable among HIV medications and the search for new therapies for patients who have failed to respond to HAART is a very active area. There are many studies on developing new therapies to counter HIV entry into the cells [Citation1,Citation10,Citation12,Citation18,Citation22,Citation25,Citation26,Citation34,Citation35,Citation37,Citation40,Citation42,Citation47,Citation50,Citation52,Citation53,Citation55,Citation58,Citation62,Citation63,Citation65,Citation82,Citation90]. In this paper, we develop an HIV model of infection progression stages and their inhibitors by including both entry inhibitor treatment and latently infected cells.
Model Equation1(1)
(1) without latent infection compartment has been introduced by Wang et al. [Citation85] to study viral load decline dynamics under the effect of different drug classes and various drug efficacy. Infected CD
T cells that have un-integrated viral DNA with two copies of the viral long-terminal repeat were incorporated into the model to explain the influence of raltegravir intensification on the viral load and 2-LTR in patients on suppressive antiretroviral therapy that doesn't include inhibitors of viral entry [Citation86]. They found that adding additional raltegravir into treatment regimens would not help to improve the treatment outcomes. We incorporate both entry inhibitors and latently infected CD
T cells into the model. Using sensitivity test, we find that viral load and latent infection are not sensitive to the latency fraction despite that the higher fraction of latency activates more latent cells in the absence of treatment. We compare the influence of the four drug classes on the dynamics of the model. We find that entry inhibitors have the same effect as protease inhibitors on the dynamics of the model (Figure ) and would benefit HIV patients who have tried and failed many of current HIV medications. We show that the dynamics of the model depend on the drug used and they in order of effectiveness are summarized in Table . The class of protease/entry inhibitors shows the best effect on uninfected CD
T cells and preventing earlier stages doesn't make better outcomes. Integrase inhibitor is shown to be the most effective drug in suppressing viral load and latently infected cells. This prediction agrees with the modelling results obtained in [Citation24,Citation76,Citation77,Citation85]. It is also consistent with clinical data [Citation49,Citation56] and experimental data in cell culture [Citation21]. Numerical simulations also show that targeting more than one stage in the HIV replication cycle by adding new drug classes wouldn't increase the chance of eradicating the virus over integrase inhibitors. This is consistent with several clinical trials [Citation20,Citation30,Citation44,Citation51] and previous modelling results [Citation2, Citation86].
The basic model analysis predicts that the infection persists in the absence of treatment but if treatment is very effective, the basic reproductive number becomes less than 1, and both latently infected cells and viremia will die out. When each drug class is taken alone in the basic model, we find that entry/protease inhibitor can efficiently prevent HIV from entering the cell and then eradicate the virus if it is at least effective while the efficacy of reverse transcriptase inhibitor and integrase inhibitor should be greater than or equal to
and
, respectively to eliminate the virus. Numerical simulations show that the model with homeostatic proliferation of latently infected cells can maintain the viral load at low level and treatment intensification can only lower the virus stability level. Moreover, increase the value of parameters used in the logistic term
leads to a higher level of viral load. This shows that eliminating latent cells in HIV patients is essential to achieve the goal of HIV eradication.
In summary, we develop mathematical models of HIV replication cycle including latency state to study the impact of different drug classes on the infection progression and explain the role of latently infected cells in HIV persistence during prolonged potent treatment. We find that entry inhibitor has the same effect as protease inhibitor and would benefit HIV patients who became drug-resistant due to the long-term use of treatment. Integrase inhibitor is shown to be the most potent drug against the replication of latent infected cells and virus particles. Using the basic model, we show that the virus and latently infected cells reach an infected steady state in the absence of treatment and are eliminated under treatment while the model including homeostatic proliferation of latent infected cells maintains the virus at a low level below 50 RNA copies/ml even under lengthy HAART. Therefore, HIV latency is a major source contributing to the viral persistence in patients receiving prolonged HAART and complete cure of HIV needs complete removal of latent reservoirs.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- K.W. Ahn and M.J. Root, Complex interplay of kinetic factors governs the synergistic properties of HIV-1 entry inhibitors, J. Biol. Chem. 292 (40) (2017), pp. 16498–16510.
- A. Alshorman, C. Samarasinghe, W. Lu, and L. Rong, An HIV model with age-structured latently infected cells, J. Biol. Dyn. 11 (sup1) (2017), pp. 192–215.
- A. Alshorman, X. Wang, M.J. Meyer, and L. Rong, Analysis of HIV models with two time delays, J. Biol. Dyn. 11 (sup1) (2017), pp. 40–64.
- Antiretroviral Therapy Cohort Collaboration, Survival of HIV-positive patients starting antiretroviral therapy between 1996 and 2013: a collaborative analysis of cohort studies, Lancet HIV. 4 (8) (2017), pp. e349–e356.
- S. Ávila-Ríos, C. García-Morales, M. Matías-Florentino, K.A. Romero-Mora, and D. Tapia-Trejo, et al. Pretreatment HIV-drug resistance in Mexico and its impact on the effectiveness of first-line antiretroviral therapy: a nationally representative 2015 WHO survey, Lancet HIV. 3 (12) (2016), pp. e579–e591.
- G. Barbaro, A. Scozzafava, A. Mastrolorenzo, and C.T. Supuran, Highly active antiretroviral therapy: current state of the art, new agents and their pharmacological interactions useful for improving therapeutic outcome, Curr. Pharm. Des. 11 (14) (2005), pp. 1805–1843.
- B.R. Bavinton, A.N. Pinto, N. Phanuphak, B. Grinsztejn, and G.P. Prestage, et al. Viral suppression and HIV transmission in serodiscordant male couples: an international, prospective, observational, cohort study, Lancet HIV. 5 (8) (2018), pp. e438–e447.
- J.N. Blankson, D. Persaud, and R.F. Siliciano, The challenge of viral reservoirs in HIV-1 infection, Annu. Rev. Med. 53 (1) (2002), pp. 557–593.
- D. Boden, A. Hurley, L. Zhang, Y. Cao, and Y. Guo, et al. HIV-1 drug resistance in newly infected individuals, JAMA 282 (12) (1999), pp. 1135–41.
- V. Briz, E. Poveda, and V. Soriano, HIV entry inhibitors: mechanisms of action and resistance pathways, J. Antimicrob. Chemother. 57 (4) (2006), pp. 619–627.
- X. Cao, F.A. Basir, X. Li, and P.K. Roy, Impact of combined therapy in HIV-1 treatment: A double impulsive approach, Int. J. Appl. Comput. Math. 6 (2020), pp. 106.
- A. Castagna, P. Biswas, A. Beretta, and A. Lazzarin, The appealing story of HIV entry inhibitors, CNS Drugs. 65 (7) (2005), pp. 879–904.
- N. Chomont, M. El-Far, P. Ancuta, L. Trautmann, and F.A. Procopio, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation, Nat. Med. 15 (8) (2009), pp. 893–900.
- S. Chowdhury, P.K. Roy, and R.J. Smith, Mathematical modelling of enfuvirtide and protease inhibitors as combination therapy for HIV, Int. J. Nonlinear Sci. Numer. Simul. 17 (6) (2016), pp. 259–275.
- T.W. Chun, L. Carruth, D. Finzi, X. Shen, and J.A. DiGiuseppe, et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection, Nature. 387 (6629) (1997), pp. 183–188.
- M.S. Cohen, Y.Q. Chen, M. McCauley, T. Gamble, and M.C. Hosseinipour, et al. Antiretroviral therapy for the prevention of HIV-1 transmission, N. Engl. J. Med. 375 (9) (2016), pp. 830–839.
- F. Curreli, Y.D. Kwon, D.S. Belov, R.R. Ramesh, and A.V. Kurkin, et al. Synthesis, antiviral potency, in vitro ADMET, and X-ray structure of potent CD4 mimics as entry inhibitors that target the Phe43 cavity of HIV-1gp120, J. Med. Chem. 60 (7) (2017), pp. 3124–3153.
- F. Curreli, D.S. Belov, Y.D. Kwon, R. Ramesh, and A.M. Furimsky, et al. Structure-based lead optimization to improve antiviral potency and ADMET properties of phenyl-1H-pyrrole-carboxamide entry inhibitors targeted to HIV-1 gp120, Eur. J. Med. Chem. 154 (2018), pp. 367–391.
- O. Diekmann, J.A.P. Heesterbeek, and J.A.J. Metz, On the definition and the computation of the basic reproduction ratio R0 in models for infectious diseases in heterogeneous populations, J. Math. Biol.28 (4) (1990), pp. 365–382.
- J.B. Dinoso, S.Y. Kim, A.M. Wiegand, S.E. Palmer, and S.J. Gange, et al. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy, PNAS 106 (23) (2009), pp. 9403–9408.
- D.A. Donahue, R.D. Sloan, B.D. Kuhl, T. Bar-Magen, and S.M. Schader, et al. Stage-dependent inhibition of HIV-1 replication by antiretroviral drugs in cell culture, Antimicrob. Agents Chemother.54 (3) (2010), pp. 1047–1054.
- X. Fengyan, P.A. Edward, L. Liyu, H. Yingchun, and Y. Juan, et al. Current status of the pharmacokinetics and pharmacodynamics of HIV-1 entry inhibitors and HIV therapy, Curr. Drug Metab. 18 (8) (2017), pp. 769–781.
- D. Finzi, M. Hermankova, T. Pierson, L.M. Carruth, and C. BUCK, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy, Sciences. 278 (1997), pp. 1295–1300.
- J.B. Gilmore, A.D. Kelleher, D.A. Cooper, and J.M. Murray, Explaining the determinants of first phase HIV decay dynamics through the effects of stage-dependent drug action, PLoS Comput. Biol. 9 (3) (2013), pp. e1002971.
- W.C. Greene, The brightening future of HIV therapeutics, Nat. Immunol. 5 (9) (2004), pp. 867–871.
- R.M. Gulick, New antiretroviral drugs, Clin. Microbiol. Infect. 9 (3) (2003), pp. 186–193.
- R.M. Gulick, A. Meibohm, D. Havlir, J.J. Eron, and A. Mosley, et al. Six-year follow-up of HIV-1-infected adults in a clinical trial of antiretroviral therapy with indinavir, zidovudine, and lamivudine, AIDS. 17 (16) (2003), pp. 2345–2349.
- T. Guol, Z. Qiu, M. Shen, and L. Rong, Dynamics of a new HIV model with the activation status of infected cells, J. Math. Biol. 82 (6) (2021), pp. 51.
- S.M. Hammer and L. Pedneault, Antiretroviral resistance testing comes of age, Antivir. Ther. 5 (1) (2000), pp. 23–26.
- H. Hatano, R. Scherzer, Y. Wu, K. Harvill, and K. Maka, et al. A randomized controlled trial assessing the effects of raltegravir intensification on endothelial function in treated HIV infection, J. Acquir. Immune Defic. Syndr. 61 (3) (2012), pp. 317–325.
- T.D. Hollingsworth, R.M Anderson, and C. Fraser, HIV-1 transmission, by stage of infection, J. Infect. Dis. 198 (5) (2008), pp. 687–693.
- J.M. Hyman, J. Li, and E.A. Stanley, The differential infectivity and staged progression models for the transmission of HIV, Math. Biosci. 155 (2) (1999), pp. 77–109.
- J.M. Hyman and J. Li, The reproductive number for an HIV model with differential infectivity and staged progression, Linear Algebra Its Appl. 398 (2005), pp. 101–116.
- M.O. Ilomuanya, A.T. Hameedat, E.N. Akang, S.O. Ekama, and B.O. Silva, et al. Development and evaluation of mucoadhesive bigel containing tenofovir and maraviroc for HIV prophylaxis, Future J. Pharm. Sci. 6 (1) (2020), pp. 81.
- M.C. Jamjian and I.R. McNicholl, Enfuvirtide: first fusion inhibitor for treatment of HIV infection, Am. J. Health Syst. Pharm. 61 (12) (2004), pp. 1242–1247.
- Jmarchn, HIV-Wikipedia [Internet]. Available at https://en.wikipedia.org/wiki/HIV.
- J.M. Kilby and J.J. Eron, Novel therapies based on mechanisms of HIV-1 cell entry, N. Engl. J. Med.348 (22) (2003), pp. 2228–2238.
- H. Kim and A.S. Perelson, Viral and latent reservoir persistence in HIV-1-Infected patients on therapy, PLoS Comput. Biol. 2 (10) (2006), pp. e135. pp. 1232–1247.
- M. v. Kleist, S. Menz, and W. Huisinga, Drug-class specific impact of antivirals on the reproductive capacity of HIV, PLoS Comput. Biol. 6 (3) (2010), pp. e1000720.
- J.P. Lalezari, K. Henry, M. O'Hearn, J.S.G. Montaner, and P.J. Piliero, et al. Enfuvirtide, an HIV-1 fusion inhibitor, for Drug-Resistant HIV infection in north and South America, N. Engl. J. Med. 348 (22) (2003), pp. 2175–2185.
- A. Lazzarin, B. Clotet, D. Cooper, J. Reynes, and K. Arastéh, et al. Efficacy of enfuvirtide in patients infected with drugresistant HIV-1 in Europe and Australia, N. Engl. J. Med. 348 (22) (2003), pp. 2186–2195.
- W. Li, L. Lu, W. Li, and S. Jiang, Small-molecule HIV-1 entry inhibitors targeting gp120 and gp41: a patent review (2010-2015), Expert. Opin. Ther. Pat. 27 (6) (2017), pp. 707–719.
- H. Liu and J. Zhang, Dynamics of two time delays differential equation model to HIV latent infection, Phys. A. Stat. Mech. Appl. 514 (2019), pp. 384–395.
- J. Llibre, M. Buzon, M. Massanella, A. Esteve, and V. Dahl, et al. Treatment intensification with raltegravir in subjects with sustained HIV-1 viraemia suppression: a randomized 48-week study, Antivir. Ther. 17 (2) (2012), pp. 355–364.
- A.L. Lloyd, The dependence of viral parameter estimates on the assumed viral life cycle: limitations of studies of viral load data, Proc. Biol. Sci. 268 (1469) (2001), pp. 847–854.
- J. Lou and R. J.Smith, Modelling the effects of adherence to the HIV fusion inhibitor enfuvirtide, J. Theor. Biol. 268 (1) (2011), pp. 1–13.
- L. Lu, P. Tong, X. Yu, C. Pan, and P. Zou, et al. HIV-1 variants with a single-point mutation in the gp41 pocket region exhibiting different susceptibility to HIV fusion inhibitors with pocket- or membrane-binding domain, Biochim. Biophys. Acta. 1818 (12) (2012), pp. 2950–2957.
- G. Magombedze, W. Garira, and E. Mwenje, Modelling the immunopathogenesis of HIV-1 infection and the effect of multidrug therapy: the role of fusion inhibitors in HAART, Math. Biosci. Eng. 5 (3) (2008), pp. 485–504.
- M.H. Markowitz, B.Y. Nguyen, E. Gotuzzo, F.A. Mendo, and W. Ratanasuwan, et al. Rapid and durable antiretroviral effect of the HIV-1 integrase inhibitor raltegravir as part of combination therapy in treatment-naive patients with HIV-1 infection, J. Acquir. Immune Defic. Syndr. 46 (2) (2007), pp. 125–133.
- K. Marks and R.M. Gulick, New antiretroviral agents for the treatment of HIV infection, Curr. Infect. Dis. Rep. 6 (4) (2004), pp. 333–339.
- D. McMahon, J. Jones, A. Wiegand, S.J. Gange, and M. Kearney, et al. Short-course raltegravir intensification does not reduce persistent low-level viremia in patients with HIV-1 suppression during receipt of combination antiretroviral therapy, Clin. Infect. Dis. 50(6) (2000), pp. 912–919.
- G.B. Melikyan, How entry inhibitors synergize to fight HIV, J. Biol. Chem. 292 (40) (2017), pp. 16511–16512.
- M.U. Mirza, A. Saadabadi, M. Vanmeert, O.M.H. Salo-Ahen, and I. Abdullah, et al. Discovery of HIV entry inhibitors via a hybrid CXCR4 and CCR5 receptor pharmacophore-based virtual screening approach, Eur. J. Pharm. Sci. 162 (2021), pp. 105827.
- T. Mizuguchi, S. Harada, T. Miura, N. Ohashi, and T. Narumi, et al. A minimally cytotoxic CD4 mimic as an HIV entry inhibitor, Bioorg. Med. Chem. Lett. 26 (2) (2016), pp. 397–400.
- J.P. Moore and R.W. Doms, The entry of entry inhibitors: a fusion of science and medicine, Proc. Natl. Acad. Sci. USA. 100 (19) (2003), pp. 10598–10602.
- J.M. Murray, S. Emery, A.D. Kelleher, M. Law, and J. Chen, et al. Antiretroviral therapy with the integrase inhibitor raltegravir alters decay kinetics of HIV, significantly reducing the second phase, AIDS. 21 (17) (2007), pp. 2315–2321.
- J.M. Murray, A.D. Kelleher, and D.A. Cooper, Timing of the components of the HIV life cycle in productively infected CD 4+ T cells in a population of HIV-Infected individuals, J. Virol. 85 (20) (2011), pp. 10798–10805.
- G.P. Pattnaik and H. Chakraborty, Entry Inhibitors: Efficient means to block viral infection, J. Membr. Biol. 253 (5) (2020), pp. 425–444.
- K.A. Pawelek, S. Liu, F. Pahlevani, and L. Rong, A model of HIV-1 infection with two time delays: Mathematical analysis and comparison with patient data, Math. Biosci. 235 (1) (2012), pp. 98–109.
- A.S. Perelson and P.W. Nelson, Mathematical analysis of HIV-1 dynamics in vivo, SIAM Rev. 41 (1) (1999), pp. 3–44.
- A.S. Perelson, D.E. Kirschner, and R.D. Boer, Dynamics of HIV infection of CD 4+ T cells, Math. Biosci.114 (1) (1993), pp. 81–125.
- H.H. Pharm and P.R. Skolnik, Enfuvirtide, a new fusion inhibitor for therapy of human immunodeficiency virus infection, Am. Coll. Clin. Pharm. 24(2) (2012), pp. 198–211.
- J. Pu, Q. Wang, W. Xu, L. Lu, and S. Jiang, Development of protein-and peptide-based HIV entry inhibitors targeting gp120 or gp41, Viruses 11 (8) (2019), pp. 705.
- K. Qian, S.L. Morris-Natschke, and K. Lee, HIV entry inhibitors and their potential in HIV therapy, Med. Res. Rev. 29 (2) (2009), pp. 369–393.
- T.M. Rad, L. Saghaie, and A. Fassihi, HIV-1 entry inhibitors: A review of experimental and computational studies, Chem. Biodivers. 15 (10) (2018), pp. e1800159.
- B. Ramratnam, S. Bonhoeffer, J. Binley, A. Hurley, and L. Zhang, et al. Rapid production and clearance of HIV-1 andhepatitis C virus assessed by large volume plasma apheresis, Lancet. 354 (9192) (1999), pp. 1782–1785.
- B. Ramratnam, J.E. Mittler, L. Zhang, D. Boden, and A. Hurley, et al. The decay of the latent reservoir of replication-competent HIV-1 is inversely correlated with the extent of residual viral replication during prolonged anti-retroviral therapy, Nat. Med. 6 (1) (2000), pp. 82–85.
- W.C. Roda, S. Liu, C. Power, and M.Y. Li, Modeling the effects of latency reversing drugs during HIV-1 and SIV brain infection with implications for the shock and kill strategy, Bull. Math. Biol. 83 (4) (2021), pp. 39.
- A.J. Rodger, V. Cambiano, T. Bruun, P. Vernazza, and S. Collins, et al. Sexual activity without condoms and risk of HIV transmission in serodifferent couples when the HIV-Positive partner is using suppressive antiretroviral therapy, JAMA. 316 (2) (2016), pp. 171–181.
- A.J. Rodger, V. Cambiano, T. Bruun, P. Vernazza, and S. Collins, et al. Risk of HIV transmission through condomless sex in serodifferent gay couples with the HIV-positive partner taking suppressive antiretroviral therapy (PARTNER): final results of a multicentre, prospective, observational study, Lancet. 393 (10189) (2019), pp. 2428–2438.
- L. Rong and A.S. Perelson, Modeling latently infected cell activation: Viral and latent reservoir persistence, and viral blips in HIV-infected patients on potent therapy, PLoS Comput. Biol. 5 (10) (2009a), pp. e1000533. pp. 1–18.
- L. Rong and A.S. Perelson, Modeling HIV persistence, the latent reservoir, and viral blips, J. Theor. Biol. 260 (2) (2009b), pp. 308–331.
- L. Rong and A.S. Perelson, Modeling latently infected cell activation: Viral and latent reservoir persistence, and viral blips in HIV-infected patients on potent therapy, PLoS Comput. Biol. 5 (10) (2009c), pp. e1000533.
- L. Rong and A.S. Perelson, Asymmetric division of activated latently infected cells may explain the decay kinetics of the HIV-1 latent reservoir and intermittent viral blips, Math. Biosci. 217 (1) (2009d), pp. 77–87.
- L. Rong, Z. Feng, and A.S. Perelson, Emergence of HIV-1 drug resistance during antiretroviral treatment, Bull. Math. Biol. 69 (6) (2007), pp. 2027–2060.
- A.R. Sedaghat, J.B. Dinoso, L. Shen, C.O. Wilke, and R.F. Siliciano, Decay dynamics of HIV-1 depend on the inhibited stages of the viral life cycle, Proc. Natl. Acad. Sci. USA. 105 (12) (2008), pp. 4832–4837.
- A.R. Sedaghat, R.F. Siliciano, and C.O. Wilke, Constraints on the dominant mechanism for HIV viral dynamics in patients on raltegravir, Antivir. Ther. 14 (2) (2009), pp. 263–271.
- S. Sepehri, S. Soleymani, R. Zabihollahi, M.R. Aghasadeghi, and M. Sadat, et al. Synthesis, biological evaluation and molecular docking studies of novel 4-[4-Arylpyridin-1(4H)-yl]benzoic acid derivatives as anti-HIV-1 agents, Chem. Biodivers. 14 (12) (2017), pp. e1700295.
- R.J. Smith and B.D. Aggarwala, Can the viral reservoir of latently infected CD4+ T cells be eradicated with antiretroviral HIV drugs?, J. Math. Biol. 59 (5) (2009), pp. 697–715.
- B. Song, J. Lou, and Q. Wen, Modelling two different therapy strategies for drug T-20 on HIV-1 patients, Appl. Math. Mech. Engl. Ed. 32 (4) (2011), pp. 419–436.
- N.E. Tarfulea, A mathematical model for CTL effect on a latently infected cell inclusive HIV dynamics and treatment, AIP. Conf. Proc. 1895(1) (2017), pp. 070005.
- J.C. Tilton and R.W. Doms, Entry inhibitors in the treatment of HIV-1 infection, Antiviral Res. 85 (1) (2010), pp. 91–100.
- N.K. Vaidya and L. Rong, Modeling pharmacodynamics on HIV latent infection: choice of drugs is key to successful cure via early therapy, SIAM J. Appl. Math. 77 (5) (2017), pp. 1781–1804.
- P. van den Driessche and J. Watmough, Reproduction numbers and sub-threshold endemic equilibria for compartmental models of disease transmission, Math. Biosci. 180 (1-2) (2002), pp. 29–48.
- X. Wang, X. Song, S. Tang, and L. Rong, Dynamics of an HIV model with multiple infection stages and treatment with different drug classes, Bull. Math. Biol. 78 (2016), pp. 323–349.
- X. Wang, G. Mink, D. Lin, X. Song, and L. Rong, Influence of raltegravir intensification on viral load and 2-LTR dynamics in HIV patients on suppressive antiretroviral therapy, J. Theor. Biol. 416 (2017), pp. 16–27.
- X. Wang, S. Tang, X. Song, and L. Rong, Mathematical analysis of an HIV latent infection model including both virus-to-cell infection and cell-to-cell transmission, J. Biol. Dyn. 11 (sup2) (2017), pp. 455–483.
- M.J. Wawer, R.H. Gray, N.K. Sewankambo, D. Serwadda, and X. Li, et al. Rates of HIV-1 transmission per coital act, by stage of HIV-1 infection, in Rakai, Uganda, J. Infect Dis. 191 (9) (2005), pp. 1403–1409.
- S.A. Wegner, S.K. Brodine, J.R. Mascola, S.A. Tasker, and R.A. Shaffer, et al. Prevalence of genotypic and phenotypic resistance to anti-retroviral drugs in a cohort of therapy-naïve HIV-1 infected US military personnel, AIDS. 14 (8) (2000), pp. 1009–1015.
- B.D. Welch, J.N. Francis, J.S. Redman, S. Paul, and M.T. Weinstock, et al. Design of a potent D-peptide HIV-1 entry inhibitor with a strong barrier to resistance, J. Virol. 84 (21) (2010), pp. 11235–11244.
- C.B. Wilen, J.C. Tilton, and R.W. Doms, HIV: cell binding and entry, Cold Spring Harb. Perspect. Med. 2(8) (2012), pp. a006866.
- D. Wodarz and M.A. Nowak, Mathematical models of HIV pathogenesis and treatment, BioEssays.24 (12) (2002), pp. 1178–1187.
- J.K. Wong, M. Hezareh, H.F. Günthard, D.V. Havlir, and C.C. Ignacio, et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia, Sciences. 278(5341) (1997), pp. 1291–5.
- H. Zhang, D. Kang, B. Huang, N. Liu, and F. Zhao, et al. Discovery of non-peptide small molecular CXCR4 antagonists as anti-HIV-1 agents: recent advances and future opportunities, Eur. J. Med. Chem. 114 (2016), pp. 65–78.