Abstract
This review highlights progress made since 2000 in the development of solvent-free organic reactions for student use. It is directed at university instructors to illustrate the broad scope of solventless reactivity possible in undergraduate laboratories. Eliminating a reaction medium directly addresses the Twelve Principles of Green Chemistry and is a focal point of contemporary industrial research. Experimental conditions are straightforward to implement and include reactant grinding, room temperature stirring, microwave irradiation, and conventional heating. A wide range of functional group transformations is easily achievable. Solvent-free reactions are often complete in a matter of minutes and routinely require simplistic work-up and purification protocols. Procedures are compiled from educational resources (primarily pedagogical journals and laboratory manuals) and corollary green experimental elements emphasized where possible.
Introduction
When teaching organic reactivity principles to new chemistry undergraduates, instructors often discuss and rationalize types of solvent employed for different transformations. Examples of this include common use of dry ether for Grignard reactions Citation1 and acetonitrile for processes proceeding via an Sn2 mechanism Citation2. Students quickly appreciate that solvents are an important component of organic reactions and eventually ponder “which solvent should I select?” when faced with designing conditions for their own chemistry. Sheldon has affirmed “the best solvent is no solvent” Citation3 with the last two decades seeing remarkable strides made in the field of solvent-free synthesis where reactions are conducted in the absence of auxiliary substances. Pioneering work by Tanaka and Toda has been reviewed Citation4 Citation5 where solid reactants are often ground together to effect reactivity. This approach has spawned the technique of “grindstone chemistry” Citation6, which is adapted for large-scale production of pharmaceuticals. Much solventless reactivity is currently performed under microwave irradiation Citation7 Citation8 either in a domestic microwave oven or a commercially available dedicated reactor. Microwave heating of reagent–reactant complexes is more consistent and expeditious to that achievable using conventional hot plates, oil/sand baths, and mantles. Since 2000, chemical educators have designed a wide range of procedures facilitating application of solvent-free reactions in the undergraduate laboratory Citation9. This article compiles these experiments and is directed at college and university instructors with the aim of showcasing and promoting the principle of organic reactivity sans solvent.
Solventless reactivity has a number of advantages from the perspective of both teachers and students. Two of the Twelve Principles of Green Chemistry Citation10 are to “use safer solvents and reaction conditions” and to “prevent waste.” These axioms are both directly met by eliminating a reaction medium. A third principle is “increase energy efficiency” which is often addressed. Solvent-free processes regularly exhibit impressive rate accelerations due to increased reactant concentrations with some occurring under ambient conditions in the presence or absence of grinding. Microwave irradiation regularly affords a dramatic reduction in reaction time (often from hours to minutes) so that many procedures can be undertaken in a three or four-hour laboratory period. In addition, product work-up, isolation, and purification are straightforward for most reactions.
Reactions summarized here date from 2000 onward and are arranged according to the major experimental techniques employed in solventless reactivity. These are room temperature mechanical mixing (including reactant grinding and ambient stirring); microwave/visible light irradiation and conventional heating. Each technique is sub-divided into reaction types where applicable: C–C bond-forming reactions, C–N bond-forming reactions, and transformations involving C–H and/or C–O bond formations. Experimental conditions, reaction times, and documented student yields (where known) are emphasized along with other green chemistry aspects and reactions of interest.
Room temperature mechanical mixing
C–C bond formations
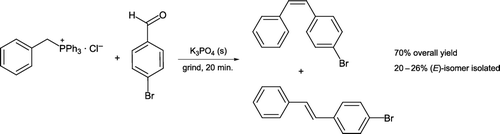
Many solvent-free pedagogical procedures feature C–C and C–N bond formation with the key mechanistic step being nucleophilic addition at an electron-deficient carbon atom. Leung and Angel Citation11 described preparation of (Z) and (E)-(4-bromophenyl)styrene by a solventless Wittig reaction. The reaction involves grinding 4-bromobenzaldehyde, benzyltriphenylphosphonium chloride, and tribasic potassium phosphate with a pestle and mortar for 20 minutes (). Washing the crude mixture with water and recrystallization from ethanol affords the pure (E)-isomer as the isolated product, engendering a green work-up procedure.
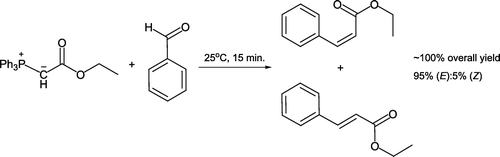
Nguyen and Weizman Citation12 recently developed a quantitative Wittig synthesis of diastereomeric ethyl cinnamate esters. This approach dispenses with dichloromethane as the usual reaction solvent as reactants are simply stirred at room temperature (). (E) and (Z)-isomers are both formed and isolated as oils after dilution with hexane and removal of insoluble triphenylphosphine oxide with a filtration pipette. Proton nuclear magnetic resonance (NMR) analysis reveals a typical 95% (E)-isomer:5% (Z)-isomer product ratio.
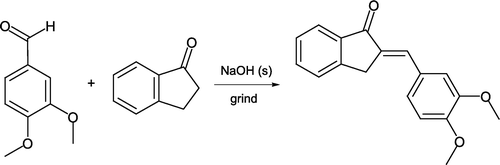
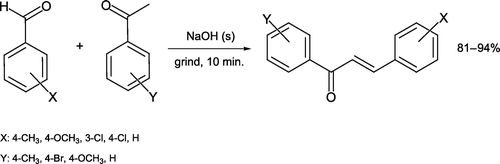
The aldol condensation between an aldehyde and ketone lends itself extremely well to grinding in the absence of solvent, and is a process with high atom economy Citation13. Such a reaction between 3,4-dimethoxybenzaldehyde and 1-indanone in the presence of catalytic sodium hydroxide has been reported [Citation14, ]. Dehydration of the initial aldol product leads to a conjugated enone that is recrystallized from 9:1 ethanol:water. Although both carbonyl reactants are solids, their direct solventless mixing leads to melting so that condensation occurs in the liquid state. Presence of a liquid or melt phase has been discovered as essential for such aldol reactions (and related “solid–solid” or “solid-state” processes) to proceed Citation15. In keeping with these results, and employing both solid and liquid reactants, Palleros Citation16 demonstrated a range of chalcone products can be synthesized by a similar procedure in excellent yields (). These reactions are extremely rapid due to high reactant concentrations in a liquid yet solvent-free medium.
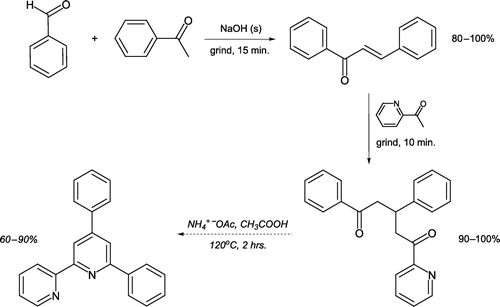
A sequential crossed-aldol condensation/Michael addition involving reactant grinding has been developed leading to a 1,5-diketone in 70–100% overall yield [Citation17, ]. The chalcone intermediate can be isolated and characterized if desired, or directly reacted further with 2-acetylpyridine to form the Michael adduct. The authors additionally describe subsequent reaction of the 1,5-diketone with ammonium acetate to generate a Kröhnke pyridine Citation18 Citation19 utilizing acetic acid as a renewable solvent.
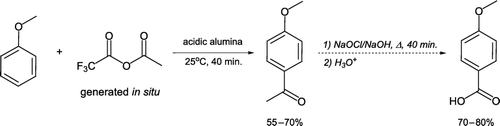
Electrophilic aromatic substitution of anisole can be achieved by solventless acylation with a mixed acid anhydride generated in situ [Citation20, ]. Reaction of trifluoroacetic anhydride with acetic acid on acidic alumina generates ethanoic trifluoroethanoic anhydride, which reacts with anisole at room temperature to form 4-methoxyacetophenone. This procedure removes the requirement of aluminium trichloride as a Lewis acid catalyst and related problematic work-up/product isolation procedures. The crude oil may be oxidized with aqueous sodium hypochlorite (commercial bleach) under basic conditions to synthesize 4-methoxybenzoic acid.
C–N bond formations
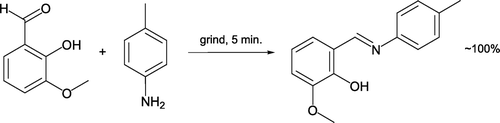
Touchette Citation21 outlined quantitative preparation of a bright orange imine on solventless reaction of ortho-vanillin (melting point, mp 40–42°C) and 4-toluidine (mp 41–46°C) (). This reaction can be easily undertaken in a beaker within one hour and includes an appealing color change. The process is considerably exothermic, aiding evaporation of the water by-product. As with many effective aldol condensations, formation of a melt phase facilitates conversion. This provides a memorable example of an impurity causing a lowered melting point of a solid compound. Inexpensive para-vanillin may be used by large classes as an alternative reactant.
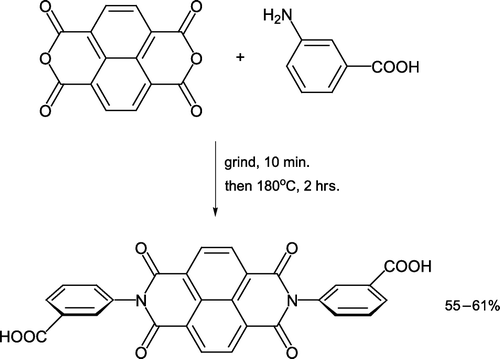
The contemporary practical technique of cocrystal controlled solid-state synthesis [abbreviated to C3S3, Citation22] is amenable to implementation in the undergraduate laboratory. Cocrystals are composed of two or more solids crystallized together generating a novel lattice arrangement, which is different from those existing in the pure substances. Within the lattice are unique interactions (e.g. hydrogen bonds) that orient functional groups close enough for reaction to occur. The C3S3 approach is in contrast to routine solid-state synthesis where compounds maintain their lattice structure and react in an amorphous environment. 1,4,5,8-Naphthalenetetracarboxylic dianhydride is ground together with 3-aminobenzoic acid [Citation23, ] without any solvents to initially form a pale yellow cocrystal. On prolonged heating, the cocrystal generates a dark yellow diimide product which is isolated by washing with methanol and characterized by (IR), NMR, X-ray diffraction (XRD), and differential scanning calorimetry (DSC). A variety of other anhydrides (e.g. phthalic anhydride) and amines (e.g. adamantylamine) can be reacted in the C3S3 approach. The product of combining these two solids together [N-(1-adamantyl)phthalimide] is a potential anti-cancer pharmaceutical ingredient Citation24.
C–H and/or C–O bond-forming reactions
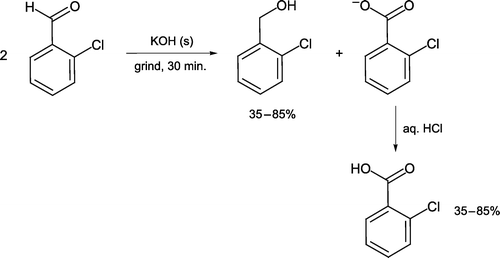
The Cannizzaro disproportionation of 2-chlorobenzaldehyde can be performed under grinding conditions with solid potassium hydroxide acting as base [Citation25, ]. The reactants are ground for 30 minutes before dilution with water. Solid 2-chlorobenzyl alcohol is isolated from the aqueous solution and 2-chlorobenzoic acid precipitated by acidification, so that organic solvents are not required during work-up. Both products are collected and analyzed as an unusual yet didactic feature of an undergraduate synthesis experiment.
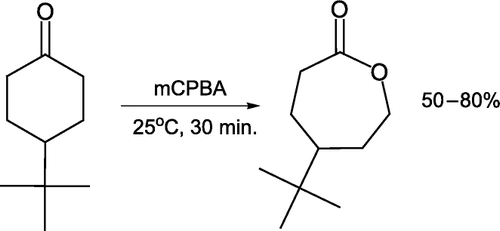
Esteb et al. Citation26 described the Baeyer–Villiger oxidation of 4-t-butylcyclohexanone with meta-chloropesoxybenzoic acid (mCPBA) under ambient conditions by stirring both reactants together for 30 minutes. An exothermic reaction leads to the caprolactone product, which is extracted into ether and isolated as a low melting point solid in good yield ().
Microwave heating and irradiation with visible light
C–C bond formations
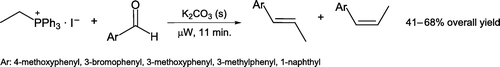
Martin and Kellen-Yuen Citation27 outlined solvent-free Wittig preparation of five β-methylstyrene derivatives by heating an aromatic aldehyde with ethyltriphenylphosphonium iodide in a domestic microwave oven (), rather than a laboratory-grade microwave reactor. Potassium carbonate is used as a weak, environmentally benign base. (Z) and (E)-alkenes are generally synthesized in almost equimolar amounts and may be separated from unreacted starting material by flash chromatography. Relative product ratios are readily determined by proton NMR and GC-MS techniques.
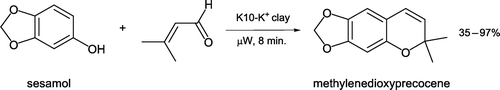
The facile solventless synthesis of methylenedioxyprecocene, a naturally occurring insecticide Citation28 Citation29 in a domestic microwave oven has been described (Citation30, ). The transformation occurs in three steps: initial electrophilic addition of 3-methyl-2-butenal to sesamol followed by loss of water and finally an intramolecular hetero-Diels–Alder reaction to form the isolated product. The process features inexpensive, readily available Montmorillonite K10 clay in a basic form as a solid catalyst and a heating time of just eight minutes. Methylenedioxyprecocene is separated from basic Montmorillonite K10 by ethyl acetate extraction and both clay and solvent recycled, incorporating other green aspects to the experiment.
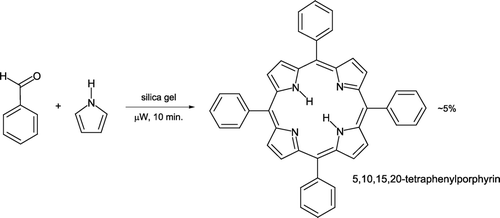
Microwave chemistry has been applied to synthesis of porphyrin derivatives Citation31 which are essential compounds for life to exist. In humans, porphyrins containing iron (hemes) ions are necessary for hemoglobin to act as an oxygen carrier within blood whereas in plants, structurally similar compounds form the basis of chlorophyll and facilitate photosynthetic pathways. The original report has been adapted for implementation in the undergraduate organic laboratory Citation32. Benzaldehyde and pyrrole are mixed neat, added to silica gel as a solid support, and irradiated in a domestic microwave oven for 10 minutes (). Extraction with ethyl acetate and column chromatography leads to 5,10,15,20-tetraphenylporphyrin of about 85% purity that is quantifiable by UV/visible spectroscopy. Typical product yields are around 5% after purification.
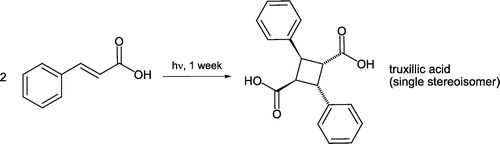
Photochemical alkene pericyclic reactions are capable of occurring in the solid-state in the absence of solvents Citation33 Citation34. When (E)-cinnamic acid is irradiated with visible light for one week a photochemical [2 + 2] cycloaddition occurs to regioselectively and stereoselectively form only one truxillic acid stereoisomer [Citation35, ]. Truxillic acid is known as a “head-to-tail” dimer in this reaction. This notable transformation proceeds at the exclusion of forming any of 10 other potential products, and with an intrinsic atom economy of 100%. Both truxillic acid and truxinic acid (the “head-to-head” dimer), are found within plant cell walls and considered contributors to the polymeric nature of these structures Citation36.
C–N bond formations
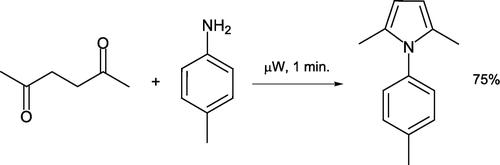
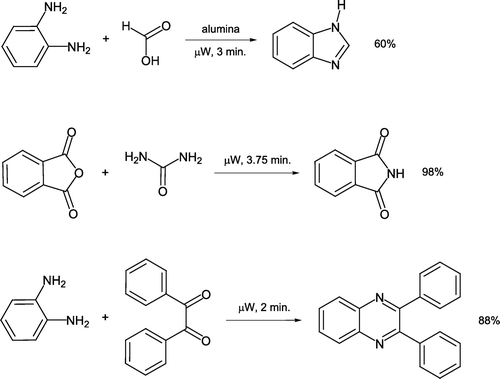
Solvent-free synthesis of a wide variety of heterocyclic compounds under microwave heating has recently been the subject of a comprehensive review Citation37. Katritzky et al. reported preparation of an N-substituted 2,5-dimethylpyrrole derivative in good yield by a classical Paal–Knorr synthesis [Citation38, ]. This remarkably fast conversion requires one minute of heating reactants sans solvent in a commercially available single-mode BenchMate microwave system Citation39 Citation40. As a comparison, 2,5-hexanedione was reacted with 4-toluidine/Montmorillonite KSF clay in dichloromethane at ambient temperature for 10–25 hours to achieve comparable product yields. Solventless synthesis of heterocyclic benzimidazole, phthalimide, and 2,3-diphenylquinoxaline has been described using a similar microwave system Citation41. Generation of benzimidazole involves reactant adsorption on alumina which acts as a carrier surface (). Products are generally recrystallized from water or ethanol after short heating times.
C–H and/or C–O bond-forming reactions
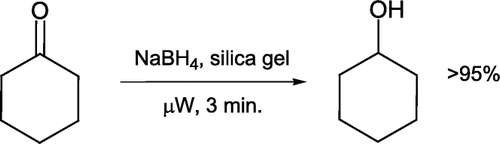
Reduction of an aldehyde or ketone with sodium borohydride is a reaction routinely discussed in introductory organic lectures and performed in many laboratory curricula. White and Kittredge Citation42 devised a cyclohexanone reduction protocol where NaBH4 is initially ground with and supported on a silica gel matrix (). Microwave irradiation in a commercial reactor for three minutes leads to cyclohexanol formation in excellent yield after work-up. Although borohydride reductions are commonly undertaken in biodegradable solvents such as ethanol Citation43, this procedure serves to minimize solvent media and drastically reduce reaction times.
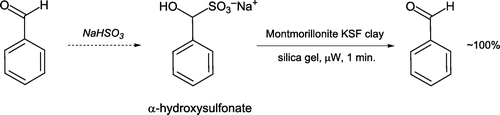
Benzaldehyde can be purified by reaction with sodium bisulfite to form an α-hydroxysulfonate and subsequent solvent-free microwave irradiation [Citation44, ]. A recyclable clay (Montmorillonite KSF) is employed on a silica gel support, in place of corrosive acidic or basic conditions. An ethyl acetate extraction facilitates isolation of regenerated pure benzaldehyde. This approach provides an alternative to other aldehyde purification techniques such as column chromatography and distillation.
Conventional heating
C–C bond formations
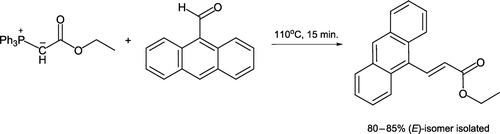
A Wittig reaction has recently been implemented in student laboratories involving conjugated ester synthesis in the absence of solvent Citation12. Conventional (hot plate/sand bath) heating of 9-anthraldehyde with a commercially available stabilized Wittig reagent [(carbethoxymethylene)triphenylphosphorane] negates the requirement of basic conditions (). At 110°C, the aldehyde has melted (mp 103–105°C) but the Wittig reagent is still a solid (mp 128–130°C). The crude product contains 5% of the minor (Z)-isomer, which is easily removed on recrystallization with methanol.
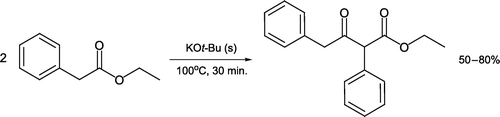
Solventless synthesis of ethyl 3-oxo-2,4-diphenylbutanoate as the product of a self-Claisen condensation has been devised [Citation45, ]. Heating ethyl (2-phenyl)ethanoate, an inexpensive and non-toxic reactant, with potassium t-butoxide for 30 minutes generates the solid β-ketoester in good yield. Performing a Claisen condensation under these conditions means the common concern of alkoxy group exchange between solvent, ester reactant, and base becomes moot.
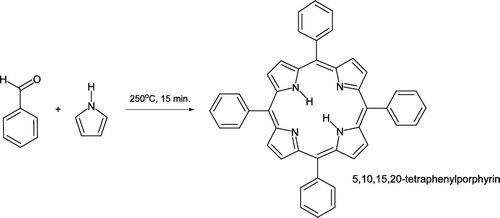
Preparation of 5,10,15,20-tetraphenylporphyrin can be achieved not only under microwave irradiation [Citation32, ], but also via more conventional heating methods. Adaptation of a novel synthetic approach Citation46 facilitates porphyrin synthesis in the gas phase Citation47. Heating benzaldehyde in a sand bath (170°C) followed by addition of pyrrole at 180°C and subsequent heating at 250°C for 15 minutes leads to product formation and isolation after column chromatography ().
C–N bond formations
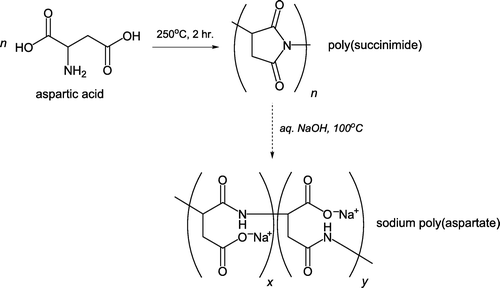
Bennett Citation48 has developed a protocol for green polymerization of an amino acid to form poly(succinimide), a precursor for sodium poly(aspartate), without use of organic solvents (). This approach represents modification of a Presidential Green Chemistry Challenge Award winning project from 1996 Citation49. L-Aspartic acid is simply heated in beaker at 250°C for two hours followed by several aqueous basic and acidic washes. After oven-drying the water-insoluble poly(succinimide) generated is hydrolyzed with 0.1M aqueous NaOH to form sodium poly(aspartate). Both L-aspartic acid and sodium poly(aspartate) are examples of biodegradable materials. The latter therefore finds use as an environmentally friendly alternative to sodium poly(acrylate) as an anti-scaling compound Citation50 and chelating agent.
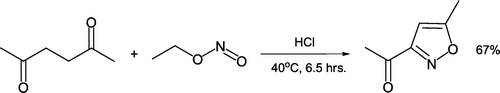
A “dry-laboratory” approach to teaching green chemistry principles of process optimization with concomitant waste reduction has been proposed Citation51. Students analyze (but do not actually perform) solvent-free synthesis of 3-acetyl-5-methylisoxazole from 2,5-hexadione and ethyl nitrite under acid-catalyzed conditions (). They consider how improvements can be made to the reaction environmental profile (e.g. elimination or diminution of work-up solvents, usage of a recyclable solid phase acid catalyst) to gain appreciation of green chemical metrics.
C–H and/or C–O bond-forming reactions
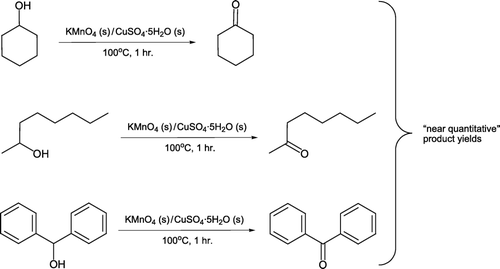
Significant efforts have recently been made to improve the “greenness” of alcohol oxidation reactions Citation52 Citation53. One strategy from a pedagogical perspective is to oxidize secondary alcohols with aqueous hydrogen peroxide in the absence of organic solvents Citation54. Esteb et al. Citation55 have employed an equimolar ground mixture of potassium permanganate and copper (II) sulfate pentahydrate as the oxidant for a range of secondary alcohols (). Each ketone is formed in “near quantitative yield” on heating for one hour by a noticeably exothermic reaction under solvent-free conditions. This method can be slightly adapted for preparation of pentanoic acid from 1-pentanol, albeit in lower isolated yields [Citation56, ].
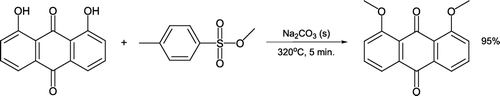
Dimethylation (dietherification) of 1,8-dihydroxy-9,10-anthracenedione has been reported at high temperatures using methyl tosylate as the alkylating agent and sodium carbonate as an environmentally benign base [Citation57, ]. The reaction takes five minutes and forms 1,8-dimethoxy-9,10-anthracenedione in excellent yield that is purified by sublimation after washing with water. Interestingly, reflux of the same reactants in tetraglyme solvent leads to exclusive monomethylation of 1,8-dihydroxy-9,10-anthracenedione, facilitating discussion of chemoselectivity principles.
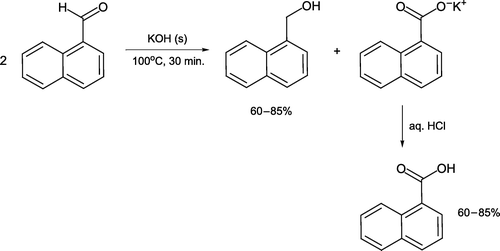
1-Naphthaldehyde can be subjected to a Cannizzaro reaction under basic solventless conditions utilizing KOH [Citation58, ]. The aldehyde and base are combined and heated for 30 minutes before cooling and aqueous dilution. 1-Naphthalenemethanol is isolated as a solid via ether extraction and 1-naphthoic acid precipitated by acidification.
Conclusions
It is clear that the last decade has produced a diverse range of solvent-free transformations for the undergraduate organic teaching laboratory. Procedures can be completed using minimal and unsophisticated apparatus within short timeframes. The current versatility of reactions without formal solvents is exemplified by the Wittig alkene synthesis. Traditionally performed in organic media (ether or tetrahydrofuran THF) and in the presence of a strong base (e.g. n-butyllithium) Citation59, students may now undertake solventless Wittig reactions by one of four approaches. These include grinding solid reactants together, microwave heating, conventional heating, or simple stirring at room temperature Citation11 Citation12 Citation27. Practical exposure to these approaches within the framework of a high-profile reaction could provide immeasurable pedagogical benefit. Nonetheless, performing a reaction without solvent does not necessarily mean the process adheres to every green chemistry principle Citation10. A cursory analysis of the aforementioned Wittig reactions reveals each takes place with very poor intrinsic atom economy. Cann and Dickneider Citation60 calculated the atom economy for Wittig synthesis of methylenecyclohexane from cyclohexanone to be only 18%, largely due to formation of triphenylphosphine oxide (Mr=278.3) as waste. The majority of reviewed reactions still require an organic solvent during work-up or for chromatographic purposes, although a few products are recrystallized from alcohol Citation11 Citation12 Citation41 or alcohol:water mixtures Citation14. Some reactions require elevated temperatures for long periods of time Citation48 Citation55 Citation56.
Debate has taken place over what actually constitutes a solvent-free reaction. Welton has asserted “a dry solid phase reaction is solvent-free, also a reaction where there is a liquid present, but it is not acting as a solvent (i.e. nothing is dissolved in it) is also solvent-free” Citation61. These two situations were presented as the only possible examples of solventless reactivity. It has been noted that many reactions declared as solvent-free in fact have a surplus of one liquid reagent present – this substance therefore behaves as both reactant and solvent Citation62. Montes et al. outlined microwave synthesis of aspirin under catalytic and non-catalytic conditions by acetylation of salicylic acid (5 mmol) with acetic anhydride (15 mmol) [Citation63 Citation64, ].

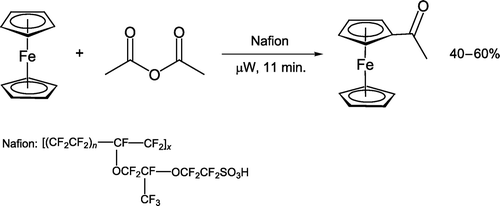
This protocol represents an efficient method of aspirin preparation but excess acetic anhydride means solvent is in fact present during all reactions. A similar position exists during a recent microwave ferrocene acetylation experiment [Citation65, ], where the authors acknowledge that acetic anhydride acts as reagent and solvent.
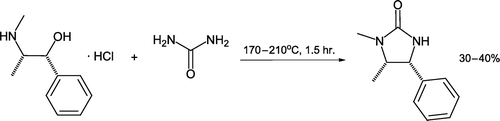
Pereira and Afonso Citation66 utilized urea (75 mmol) as an excess reagent during preparation of a chiral auxiliary from (–)-ephedrine hydrochloride (25 mmol) (). On heating to 200°C, the urea melts and additionally acts a liquid solvent. Although promoted as a solventless reaction, it is more correctly stated as one (amongst others) where no additional solvent is required beyond the reactants. Instructors and students should exercise care and consider reaction conditions/stoichiometries when interpreting processes as being “solvent-free.”
Eliminating a reaction medium furnishes occasion to discuss with undergraduates why solvents have historically been employed in organic reactivity. Beyond principles surrounding solubility and reaction homogeneity lies the concept of solvents acting as heat sinks. Many solventless reactions (including some reviewed here) are vigorously exothermic Citation21 Citation26 Citation55 Citation56 and research into calorimetric aspects of this chemistry is ongoing Citation67 . Exothermic reactions provide significant challenges when subject to scale-up in an industrial venue. Thermal “runaways” (when the rate of heat removal from a process does not correlate with the rate of heat production) can lead to catastrophic results. Improvements in solvent-free reactor design are being made at the chemical/chemical engineering interface, particularly in fields of microwave reactivity Citation68 Citation69 and photochemistry Citation70. Microreactors (where reactants combine in narrow pathways with diameters of 10 µm) are additionally now employed to deal with rapid temperature increase from solventless reactions Citation71. In conclusion, advantages to implementing solvent-free reactions into introductory and advanced organic courses are multiple and intimately linked with green chemistry principles. Drawbacks do, however, still exist and their reflection with students represents an important teaching opportunity.
References
- McMurry , J. 2008 . Organic Chemistry , 7th ed , 613 – 615 . Belmont, CA : Thomson Higher Education .
- McMurry , J. 2008 . Organic Chemistry , 7th ed , 370 – 371 . Belmont, CA : Thomson Higher Education .
- Sheldon , R.A. 2005 . Green Chem. , 7 : 267 – 278 .
- Tanaka , K. 2003 . Solvent-free Organic Synthesis , Weinheim, , Germany : Wiley-VCH .
- Tanaka , K. ; Toda , F. Chem. Rev . 2000 , 100 , 1025 1074 .
- Bose , A.K. , Pednekar , S. , Ganguly , S.N. , Chakraborty , G. and Manhas , M.S. 2004 . Tet. Lett. , 45 : 8351 – 8353 .
- Hayes , B.L. 2002 . Microwave Synthesis: Chemistry at the Speed of Light , Matthews, NC : CEM .
- Polshettiwar , V. and Varma , R.S. 2008 . Acc. Chem. Res. , 41 : 629 – 639 .
- Greener Education Materials for Chemists (GEMs) Web site . http://greenchem.uoregon.edu/gems.html ( accessed Jun 28, 2009 ).
- Anastas , P.T. and Warner , J.C. 1998 . Green Chemistry: Theory and Practice , 29 – 56 . New York : Oxford University Press .
- Leung , S.H. and Angel , S.A. 2004 . J. Chem. Educ. , 81 : 1492 – 1493 .
- Nguyen , K.C. and Weizman , H. 2007 . J. Chem. Educ. , 84 : 119 – 121 .
- Raston , C.L. and Scott , J.L. 2000 . Green Chem. , 2 : 49 – 52 .
- Doxsee , K.M. and Hutchison , J.E. 2004 . Green Organic Chemistry – Strategies, Tools and Laboratory Experiments , 115 – 119 . Brooks-Cole : Pacific Grove, CA .
- Rothenberg , G. , Downie , A.P. , Raston , C.L. and Scott , J.L. 2001 . J. Am. Chem. Soc. , 123 : 8701 – 8708 .
- Palleros , D.R. 2004 . J. Chem. Educ. , 81 : 1345 – 1347 .
- Cave , G.W.V. and Raston , C.L. 2005 . J. Chem. Educ. , 82 : 468 – 469 .
- Cave , G.W.V. ; Raston , C.L. ; Scott , J.L. Chem. Commun . 2001 21 2159 2169 .
- Kröhnke , F. Synthesis 1976 1 , 1 24 .
- French , L.G. , Stradling , S.S. , Dudley , M. , DeBottis , D. and Parisian , K. 2001 . J. Chem. Educator , 6 : 25 – 27 .
- Touchette , K.M. 2006 . J. Chem. Educator , 83 : 929 – 930 .
- (a) Cheney , M.L. ; McManus , G.J. ; Perman , J.A. ; Wang , Z. ; Zaworotko , M.J. Cryst. Growth Des . 2007 , 7 , 616 617 ; (b) Trask , A.V Jones , W. Top. Curr. Chem , 2005 254 41 70
- Cheney , M.L. , Zaworotko , M.J. , Beaton , S. and Singer , R.D. 2008 . J. Chem. Educator , 85 : 1649 – 1651 .
- Abdel-Aziz , A.A-M. 2007 . Eur. J. Med. Chem. , 42 : 614 – 626 .
- Phonchaiya , S. , Panijpan , B. , Rajviroongit , S. , Wright , T. and Blanchfield , J.T. 2009 . J. Chem. Educator , 86 : 85 – 86 .
- Esteb , J.J. , Hohman , J.N. , Schlamadinger , D.E. and Wilson , A.M. 2005 . J. Chem. Educator , 82 : 1837 – 1838 .
- Martin , E. ; Kellen-Yuen , C. J. Chem. Educator 2007 , 84 , 2004 2006 .
- Brooks , G.T. ; Pratt , G.E. ; Jennings , R.C. Nature 1979 , 281 , 570 572 .
- Dintzner , M.R. ; Lyons , T.W. ; Akroush , M.H. ; Wucka , P. ; Rzepka , A.T. Synlett 2005 , 5 , 785 788 .
- Dintzner , M.R. ; Wucka , P.R. ; Lyons , T.W. J. Chem. Educator . 2006 , 83 , 270 272 .
- Petit , A. , Loupy , A. , Maillard , P. and Momenteau , M. 1992 . Synth. Commun. , 22 : 1137 – 1142 .
- (a) Warner , M.G. ; Succaw , G.L. ; Hutchison , J.E. Green Chem . 2001 , 3 , 267 270 ; (b) Doxsee , K.M. Hutchison , J.E. Green Organic Chemistry – Strategies, Tools and Laboratory Experiments Brooks-Cole Pacific Grove CA 2004 159 162 (c) Warner M. G Succaw G. L. Doxsee K. M Hutchinson J. E. In Greener Approaches to Undergraduate Chemistry Experiments Kirchhoff M. Ryan M.A. American Chemical Society Washington DC 2002 27 31
- Cohen , M.D. ; Schmidt , G.M.J. ; Sonntag , F.I. J. Chem. Soc. 1964 , 2000 2013 .
- Morrison , I.M. , Robertson , G.W. , Stewart , D. and Wightman , F. 1991 . Phytochemistry , 30 : 2007 – 2011 .
- Doxsee , K.M. and Hutchison , J.E. 2004 . Green Organic Chemistry – Strategies, Tools and Laboratory Experiments , 206 – 210 . Pacific Grove, CA : Brooks-Cole .
- Hartley , R.D. ; Morrison III , W.H. ; Balza , F. ; Towers , G.H.N. Phytochemistry 1990 , 29 , 3699 3703 .
- Polshettiwar , V. and Varma , R.S. 2008 . Pure Appl. Chem. , 80 : 777 – 790 .
- Katritzky , A.R. , Cai , C. , Collins , M.D. , Scriven , E.F.V. , Singh , S.K. and Barnhardt , E.K. 2006 . J. Chem. Educator , 83 : 634 – 636 .
- CEM Corporation Discover BenchMate Web site . http://www.cem.com/synthesis/discoverBM.asp (accessed Jun 28, 2009) .
- Leadbeater , N.E. ; McGowan , C.B. Clean, Fast Organic Chemistry-Microwave Assisted Laboratory Experiments ; CEM : Matthews, NC , 2006 (A laboratory manual is available with purchase of a CEM Corporation microwave system) .
- Musiol , R. , Tyman-Szram , B. and Polanski , J. 2006 . J. Chem. Educator , 83 : 632 – 633 .
- White , L.L. and Kittredge , K.W. 2005 . J. Chem. Educator , 82 : 1055 – 1056 .
- McMurry , J. 2008 . Organic Chemistry , 7th edn , 609 – 610 . Belmont, CA : Thomson Higher Education .
- Cioffi , E. In Greener Approaches to Undergraduate Chemistry Experiments ; Kirchhoff , M. , Ryan , M.A. Washington, DC : American Chemical Society , 2002 18 20 .
- Esteb , J.J. ; Stockton , M.B. J. Chem. Educator 2003 , 80 , 1446 1447 .
- Drain , C.M. ; Gong , X. Chem. Commun . 1997 1 2117 2118 .
- (a) Warner , M.G. ; Succaw , G.L. ; Hutchison , J.E. Green Chem 2001 , 3 267 270 (b) Doxsee K.M Hutchison J.E. Green Organic Chemistry – Strategies, Tools and Laboratory Experiments Brooks-Cole Pacific Grove, CA 2004 152 158
- Bennett , G.D. 2005 . J. Chem. Educator , 82 : 1380 – 1381 .
- US Environmental Protection Agency Presidential Green Chemistry Small Business Challenge Award 1996 Web site. http://www.epa.gov/greenchemistry/pubs/pgcc/winners/sba96.html (accessed Jun 28, 2009) .
- Low , K.C. , Wheeler , A.P. and Koskan , L.P. 1996 . Adv. Chem. Ser. , 248 : 99 – 111 .
- Van Arnum , S.D. 2005 . J. Chem. Educator , 82 : 1689 – 1692 .
- Lenoir , D. 2006 . Angew. Chem. Int. Ed. , 45 : 3206 – 3210 .
- Sheldon , R.A. , Arends , I.W.C.E. , ten Brink , G-J. and Dijksman , A. 2002 . Acc. Chem. Res , 35 : 774 – 781 .
- Hulce , M. and Marks , D.W. 2001 . J. Chem. Educator , 78 : 66 – 67 .
- Esteb , J.J. , Schelle , M.W. and Wilson , A.M. 2003 . J. Chem. Educator , 80 : 907 – 908 .
- Barrett , J.A. , Esteb , J.J. , Hoops , G.C. and Richter , J.M. 2004 . Chem. Educator , 9 : 30 – 31 .
- Sereda , G.A. 2005 . J. Chem. Educator , 82 : 1839 – 1840 .
- Esteb , J.J. , Gligorich , K.M. , O'Reilly , S.A. and Richter , J.M. 2004 . J. Chem. Educator , 81 : 1794 – 1795 .
- Maercker , A. 1965 . Org. React. , 14 : 270 – 490 .
- Cann , M.C. and Dickneider , T.A. 2004 . J. Chem. Educator , 81 : 977 – 980 .
- Welton , T. Green Chem . 2006 , 8 , 13 .
- Blackmond , D.G. , Armstrong , A. , Coombe , V. and Wells , A. 2007 . Angew. Chem. Int. Ed , 46 : 3798 – 3800 .
- Montes , I. , Sanabria , D. , García , M. , Castro , J. and Fajardo , J. 2006 . J. Chem. Educator , 83 : 628 – 631 .
- Mirafzal , G.A. and Summer , J.M. 2000 . J. Chem. Educator , 77 : 356 – 357 .
- Birdwhistell , K.R. , Nguyen , A. , Ramos , E.J. and Kobelja , R. 2008 . J. Chem. Educator , 85 : 261 – 262 .
- Pereira , J. and Afonso , C.A.M. 2006 . J. Chem. Educator , 83 : 1333 – 1335 .
- Correa , W.H. , Edwards , J.K. , McCluskey , A. , McKinnon , I. and Scott , J.L. 2003 . Green Chem. , 5 : 30 – 33 .
- Clophax , J. , Liagre , M. , Loupy , A. and Petit , A. 2000 . Org. Proc. Res. Dev. , 4 : 498 – 504 .
- Esveld , E. , Chemat , F. and van Haveren , J. 2000 . Chem. Eng. Technol. , 23 : 279 – 283 .
- Dunk , B. and Jachuck , R. 2000 . Green Chem. , 2 : G13 – G14 .
- Anastas , P.T. and Beach , E.S. 2007 . Green Chem. Lett. Rev. , 1 : 9 – 24 .