Abstract
Alternative solvents such as supercritical carbon dioxide, water, and ionic liquids are receiving an increase of interest as better replacements for conventional solvents in chemical reactions. They have been called sustainable green solvents because they are highly promising reaction mediums for organic synthesis. This review presents an overview of some selected chemical reactions that have been developed in these green solvents with a particular emphasis on metal-catalyzed reactions.
Introduction
Green Chemistry or alternative synthetic pathways for pollution prevention is the utilization of a set of principles that reduces or eliminates the use or generation of hazardous substances in the design, manufacture, and application of chemical products. Paul T. Anastas and John C. Warner Citation1
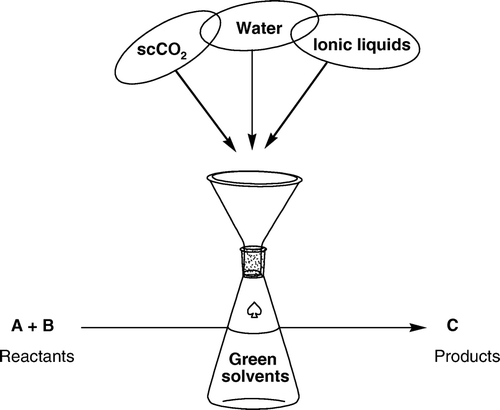
The development of a number of chemical transformations using green alternative media such as supercritical carbon dioxide (scCO2), Citation4–6 waterFootnote1 Citation7–13, and ionic liquids (ILs) Citation14–17 gave way to tremendous interest in using sustainable and environmentally friendly media (). Due to the remarkable increased number of publications in this subject, it is impossible to include all of them in this review, my apology for discarding any relevant contributions to the field. This review presents an overview of some selected chemical reactions that have been developed in scCO2, water, and ILs as sustainable solvents with particular emphasis on metal-catalyzed reactions. Selective examples using Pd, Rh, Ru, Ir, Pt, Ni, Co, Cu, Ag, and Au will be reviewed. Other green solvents such as ethanol, γ-valerolactone, ethyl lactate, poly(ethylene glycol), expanded liquids, and fluorous phase reactions will not be discussed in this review.
Figure 1. Chemical transformations in green solvents.
Selected transition metal-catalyzed reactions in supercritical carbon dioxide (scCO2)
What is supercritical carbon dioxide (scCO2)?
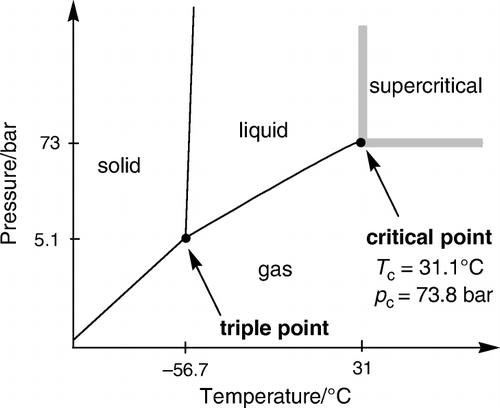
It is known that carbon dioxide (CO2) has a triple and critical point, and becomes supercritical when compressed to a pressure higher than the critical pressure (P c=73 atm) and above the critical temperature (T c=31°C; ). scCO2 has specific chemical and physical properties, which is strongly different from conventional organic solvents (by changing the pressure and temperature in particular, the density and the viscosity change drastically at conditions close to the critical point). scCO2 is non-flammable, non-toxic, non-polluting and does not contribute to global warming. In addition it has an efficient mass-transfer, is completely miscible with gaseous reactants, and is very easy to separate from the product. Taking advantage from these differences, researchers started exploring the chemical reactivity in various applications using scCO2 as an environmentally friendly new medium.
Figure 2. Schematic phase diagram of CO2.
The first experiments (polymerization of ethylene) using catalysts in supercritical fluid were reported in 1913 by Ipat'ev and Rutala Citation18. Despite the long history of catalysis in supercritical fluids, catalysis in scCO2 has been extensively studied only during the last two decades and reviewed more recently by several groups (e.g. Noyori Citation19, Jessop Citation20, Leitner Citation21, Poliakoff Citation22 Citation23, and others Citation24–28). In industry, there has been an implementation of large-scale processes, such as the extraction of caffeine Citation29. On the other hand, the use of scCO2 is still limited for specific cases in industry because of the costs associated to the high-pressure equipment and the energy requirements. Since the triarylphosphine complexes that are usually employed in liquid-phase reactions have very low solubility in scCO2 Citation30, Leitner and co-workers reported the use of phosphine ligands with fluorinated chains in transition metal reactions, which increased their solubility in scCO2 Citation31. Later, others started designing solid support substrates, polymer-based arylphosphine, and fluorinated phosphine ligands for carbon[sbnd]carbon bond formations as well as for enantioselective reactions.
Palladium
Coupling of aryl iodides with solid supported substrates
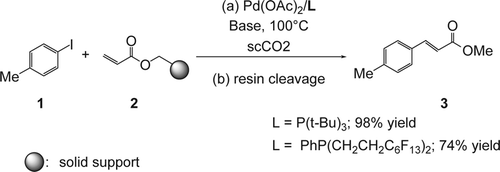
In addition to using scCO2 as an environmentally begin media, conducting reactions on a solid support provides an attractive and practical method for clean and efficient synthetic preparations, allowing convenient separation of products from the reaction mixture and forming the basis of combinatorial chemistry. Holmes and co-workers Citation32 reported that the Pd(OAc)2/P(t-Bu)3 catalyst system was highly active for the carbon[sbnd]carbon bond formation (Suzuki coupling) in scCO2 (98% yield) compared to the fluorinated palladium (Pd(OAc)2/PhP(CH2CH2C6F13)2; 74% yield) (). This system works efficiently with aryl iodides (1) while the less reactive bromide gave lower yields. Various bases such as NEt3, DIPEA were investigated and have shown to be effective in scCO2.
Coupling reaction using silicone polymer-based supported catalyst
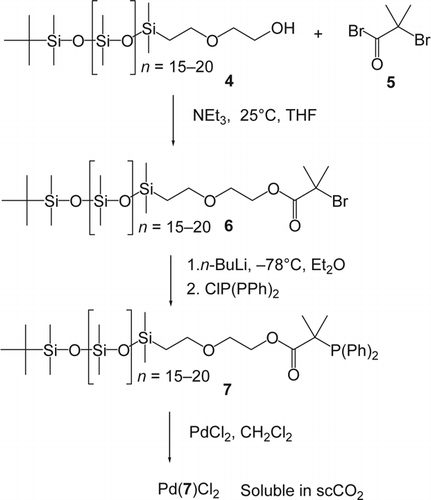
Most homogeneous catalysts are insoluble or partially soluble in scCO2. To tackle this problem, Rayner and co-workersFootnote2 Citation33 Citation34 synthesized and characterized a silicone polymer-based alkyldiarylphosphine in order to enhance the solubility of the catalyst in scCO2 (). The compound 6 was prepared in good yield from a commercially available monocarbinol-terminated polydimethylsiloxane (PDMS) and 2-bromo-2-methylpropionyl bromide in the presence of triethylamine. Lithiation of the previously prepared bromide (6) was achieved in diethyl ether at −78°C. Subsequent addition of chlorodiphenylphosphine gave the ligand (7) which was mixed with PdCl2 (1:1 ratio) to form the desired catalyst heterogeneous Pd(7)Cl2. As an application of the supported catalyst (Pd(7)Cl2), the Heck reaction was performed in scCO2 and gave in average 55% yield of the coupling products. These results were similar to those previously reported Citation35–37. Besides coupling reactions Citation38–41, there has been extensive research on other Pd-catalyzed chemical transformations such as cyclization Citation42 Citation43, carbonylation Citation44–46, etherification Citation47, oxidation Citation48 Citation49, and polymerization Citation50–52 in scCO2.
Rhodium
Intramolecular hydroaminomethylation of an unprotected secondary allylic amine
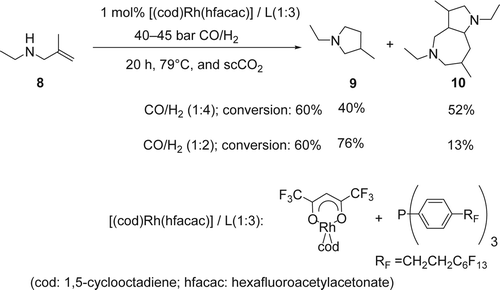
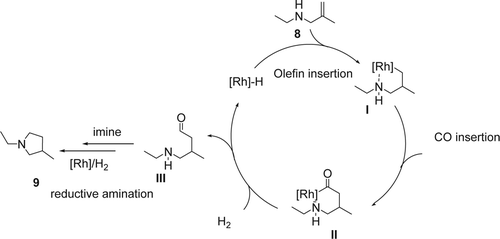
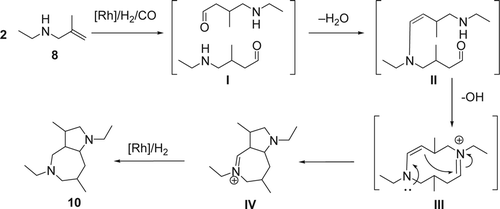
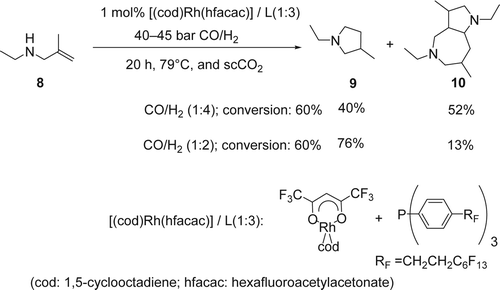
Leitner and co-workers reported Citation53 an efficient intramolecular hydroaminomethylation of an unprotected secondary allylic amine using 1 mol% of Rh catalyst and fluorinated phosphine ligand in scCO2. In this case the scCO2 was used as a temporary protecting group as well as sustainable solvent. The hydroamination of N-ethyl-2-methylprop-2-en-1-amine (8), using 45 bars of carbon monoxide (CO)/H2 with 1:4 ratio at 79°C, gave a mixture of pyrrolidine (9) and a bicyclic bisamino structure (10) with 40 and 52% yields, respectively (). On the other hand, by decreasing the amount of H2 in the system to a 1:2 ratio of CO/H2, the hydroaminomethylation of ethyl methallylic amine gave the pyrrolidine (8) as the major product (76% yield) and only 13% yield of the bicyclic bisamino. A saturated starting amine and cyclic amide were also isolated with 7 and 4% yields, respectively. Mechanistically, the [Rh]-H undergo an olefin insertion to form the intermediate I (), followed by CO insertion gave the Rh-intermediate II. The later under the presence of H2 gave the aldehyde III and regenerated the active catalyst [Rh]-H. Then the aldehyde III undergoes a subsequent intermolecular condensation reaction with the amine, followed by hydrogenation of the resulting imine or enamine, which is also catalyzed by the [Rh]-H complex giving the desired pyrolidine (9). The possible mechanism of the unusual bicyclic bisamino (10) () is the following: (1) olefin insertion of the [Rh]-H followed by CO insertion and hydrogenation gave the amino aldehydes I; (2) intermolecular condensation of the amine and the aldehyde group, and elimination of water molecule gave the enamine II. This initial dimerization by intermolecular condensation may be facilitated by solute/solute clustering of the amino aldehyde through hydrogen bonding in the non-polar scCO2 environment at higher concentrations; (3) a subsequent Mannich-type aldol addition of the resulting dimer leads to the formation of the transannular C[sbnd]C bond, creating the bicyclic skeleton IV. Finally, Rhodium-catalyzed hydrogenation of the imine IV gave the desired heterobicyclic (10). Both mechanisms were supported by high-pressure multinuclear NMR studies of the substrate/CO2 interaction.
Hydroformylation of hex-1-ene using triethylphosphine ligands
The hydroformylation reaction originally discovered by Roelen Citation54 in 1938 is an industrial process of strategic importance for the manufacture of aldehydes from olefins in the presence of CO and H2. Over the last two decades, researchers have been exploring extensively the hydroformylation of alkenes in scCO2 using Rh Citation55–60, Ru Citation61, and Co Citation62–64 as catalysts. Various phosphine ligands such as trialkylphosphines Citation65 and trialkylphosphites Citation66 were known to be sufficiently CO2-philic or at least sufficiently non-polar to be soluble or usable in scCO2. Bach and Hamilton reported Citation67 successfully the hydroformylation of hex-1-ene (11) in scCO2 using rhodium-based catalysts containing cheap, readily available triethylphosphine ligands. Using a catalyst prepared in situ from (Rh2(OAc)4) and PEt3, complete conversion to C7 aldehydes (12), with a trace of C7 alcohols (13) (total straight to branched (n:i) ratio = 2.4–2.5) is obtained within 2 hours at 100°C (). In 2002, the same group reported a comparative study on the hydroformylation of various alkenes using various ligands in scCO2 and other solvents Citation68. Two years later Fiddy and co-workers Citation69 also reported Rh/PEt3-catalyzed efficient hydroformylation of oct-1-ene in scCO2.
Table 1. Optimization of the hydroformylation conditions of hex-1-ene in scCO2.
Hydroformylation of 1-octene using fluorinated phosphine ligands
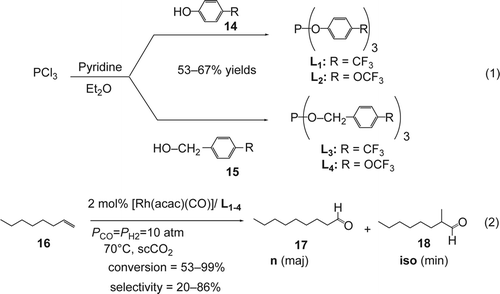
Most strategies for molecular design of ligands for catalytic hydroformylation in scCO2 must simultaneously take into account solubility, steric, and electronic considerations. Recently, Masdeu-Bultó and co-workers reported Citation70 the design, synthesis, and characterization of fluorinated phosphine ligands. The air and moisture-sensitive L 1–4 were prepared in good yields (53–67%) from the commercially available corresponding alcohols (14, 15) by reaction with phosphorus trichloride in diethyl ether in the presence of pyridine (Equation 1, ).
These ligands were used for the hydroformylation of 1-octene using catalytic precursors system [Rh(acac)(CO)2]/L 1–4 , in scCO2 (Equation 2, ). For comparison, the same hydroformylation under similar conditions were studied in toluene. Most ligands (L 1–4 ) used in scCO2 gave higher total conversion (53–99%) with modest to good selectivity (20–86%) in aldehyde. The n/iso ratio for all the systems was high (2.5–5.2), as was similarly observed for other rhodium[sbnd]phosphite systems Citation71. In general, the selectivities of aldehydes for scCO2 systems were lower than the ones obtained in toluene. The same research group showed that the presence of peracetylated β-cyclodextrin in the reaction medium increased the solubility of rhodium species modified by alkyl P-donor ligands in the catalytic hydroformylation of 1-octene in scCO2 Citation72. On the other hand, the hydroformylation was also extensively explored in water. For example, Monflier and co-workers Citation73 reported Rh-catalyzed hydroformylation of dec-1-ene in water using partially methylated β-cyclodextrins with a conversion of up to 100% and a regioselectivity of up to 95%. Shimizu and co-workers Citation74 Citation75 designed a water-soluble phosphacalix [4] arene[sbnd]rhodium complex that has acceptable levels of activity, stability, and reusability for the hydroformylation of water insoluble olefins.
Hydroboration of styrene using fluorinated phosphine ligands
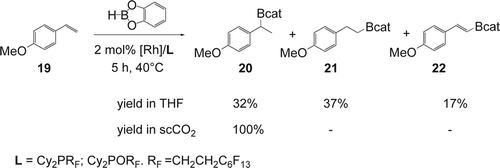
Phosphines PR2RF and phosphinites PR2(ORF) [RF=CH2CH2C6F13] are easily prepared and offer the ability not only to enhance catalyst solubility, but also to control the stereoelectronic environment of catalysts in scCO2 Citation35 Citation76–79. Boronate esters are important products in synthetic organic chemistry, medicinal chemistry, materials science, and molecular recognition. Tumas and co-workers reported Citation80 the first rhodium-catalyzed hydroboration of styrene derivatives with catecholatoborane (HBcat) in scCO2 at 40°C for five hours (). In scCO2 alkylboronate ester (20) was formed exclusively with cyclohexyl-substituted phosphine PR2(ORF) (L/Rh ratio = 2:1), demonstrating the ability to control regioselectivity and chemoselectivity by tuning the solubility and stereoelectronic properties of ligands and metal catalysts in scCO2. On the other hand using THF instead of scCO2 under the same conditions gave a mixture of alkylboronate ester isomers (21, 22, and 23) with 32%, 37%, and 17% yields, respectively ().
Asymmetric hydrogenation of dimethyl itaconate and methyl 2-acetamidoacrylate
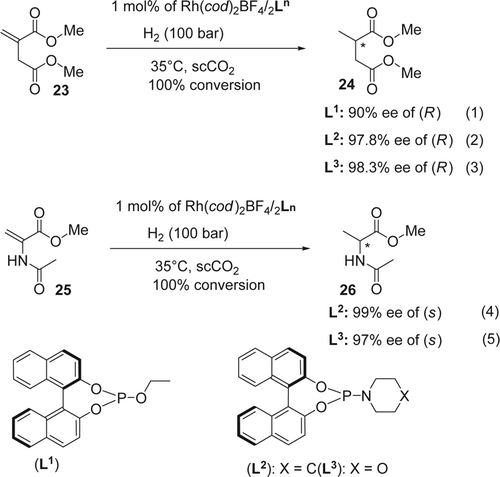
In 2007, Lyubimov and co-workers reported Citation81 the preparation of chiral organophosphorus derivatives of carboranes for the asymmetric hydrogenation of dimethyl itaconate. These monodentate carboranylphosphite ligands resulted in excellent enantioselectivities in the Rh-catalyzed asymmetric hydrogenation of dimethyl itaconate (up to 93% ee in scCO2). Recently, the same research group Citation82 Citation83 extended their synthesis to other phosphine ligands for the enantioselective hydrogenation of dimethyl itaconate (23) and methyl 2-acetamidoacrylate (25; ). The hydrogenation of dimethyl itaconate (23) using only 1 mol% of the catalysts Rh/L2 (L2=PipPhos) and Rh/L3 (L3=Morf-Phos) in scCO2 gave very good results. The saturated diester (24) was obtained with high enantioselectivity (up to 98.3%, Equations 1–3, ). The phosphine ligand L3 gave a slightly higher ee than ligands L1 and L2. On the other hand, the reduction of methyl 2-acetamidoacrylate (25) also proceeded smoothly and quantitatively to provide S-alanine derivative (26) with high enantioselectivity (up to 99%, Equations 4–5, ). In all cases the 100% conversion was achieved within 35–50 min. The high reaction rates may be attributed to the high diffusivity of gaseous hydrogen in the supercritical medium Citation84 . In addition the high concentration of H2 (100 atm) in scCO2 increased the enantioselectivity as well as the conversion in the asymmetric hydrogenation. Recently, Leitner and co-workers reported the asymmetric hydrogenation of dimethyl itaconate in chiral IL Citation85. They used a combination of a racemic ligand (BINAP) and a chiral IL ([MeProl] [NTf2]: [methyl ester of (S)-proline] [bistrifluoromethane[sbnd]sulfonimidate]) as an additive or reaction medium. In this case the ee value was up to 67% while, in the presence of a chiral BINAP ligand, the ee value increased slightly to 76%.
Ruthenium
Design of new chiral-fluorinated ligands for enantioselective hydrogenation
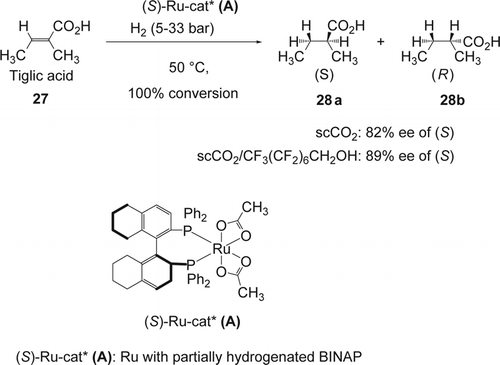
Hydrogenation reactions in environmentally friendly solvents such as scCO2 were among the first reactions that researchers studied using Rh Citation86–89 and Ru Citation90–96 as efficient catalysts. On the other hand, asymmetric homogeneous hydrogenation in scCO2 represented a tremendous challenge for chemists because of the solubility issues of most aromatic substrates, reagents, ligands, and catalysts in this reaction medium. The addition of alcohol is known to increase the solubility of aromatic compounds in scCO2 Citation97 Citation98. Noyori and co-workers reported Citation99 a clean chiral (S)-H8-BINAP-Ru(II)-catalyzed enantioselective hydrogenation of tiglic acid (27) in scCO2. In the presence of small amount of alcohol as co-solvent and using only 5 atm of H2, the desired (S)-2-methylbutanoic acid (28a) (with cis stereochemistry) was obtained with 99% yield and up to 89% ee (). In scCO2 alone, the reaction required 33 atm of H2 in order to obtain 99% yield and up to 81% ee of the desired (S)-2-methylbutanoic acid. This selectivity is lower than in polar solvent (MeOH: 82% ee) while it is higher than in non-polar solvent (hexane: 73% ee). The same enantioselective reaction was reported by Jessop and co-workers Citation100 in [emim][NTf2] as IL. They have shown an optimized ee of 95% compared with only 88% in methanol. Later other groups were exploring the same type of asymmetric hydrogenation using Ru-BINAP bearing OCF3 Citation101 (Equation 1, ), perfluoroalkylated BINAP Citation102 (Equations 2 and 3, ), as well as new perfluroalkylated-monodentate phosphorus as ligands Citation103 (Equation 4–7, ). The results for the asymmetric hydrogenation using the monodentate phosphines (Equations 4–7, ) were much less impressive in scCO2 (with the highest 65% ee and 28% conversion), compared to bidentate phosphines (Equations 1–3, ), (up to 74% ee and 100% conversion).
Iridium
Asymmetric hydrogenation of N-(1-phenylethylidene) aniline
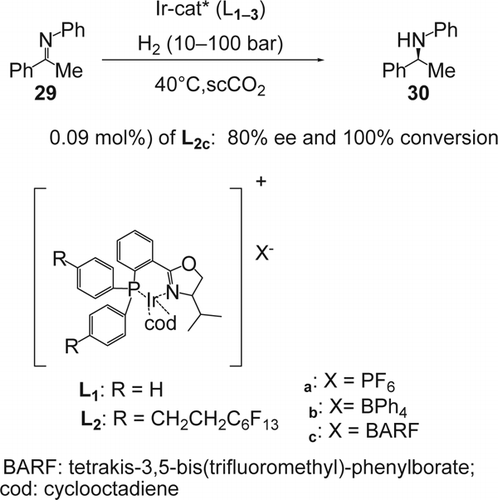
The enantioselective reduction of the C = N double bond is an important synthetic strategy for the preparation of optical-active amines and has received much attention over the past decade, in both academic and industrial research Citation104–106. Leitner and co-workers Citation107 reported a scCO2-soluble chiral iridium-catalyzed enantioselective hydrogenation of prochiral imines (29) (N-(1-phenylethylidene) aniline) at 40°C. The desired amine (30), (R)-N-phenyl-1-phenylethylamine, was formed quantitatively within 1 hour with enantiomeric excesses of up to 80% ().
Selected transition metal-catalyzed reactions in water
Water as potential solvent
An interesting question is “what is the best solvent to use in chemical reactions?” Most researchers think that the best solvent is no solvent, but in cases where a solvent is needed then a less toxic, less harmful, and more recyclable solvent may be used. Water is the cheapest solvent, is non-toxic, and is non-hazardous. Research using water as a solvent is targeted at the development of highly productive, environmentally safe, recyclable techniques, which can be promoted to large-scale applications. In addition water is useful in biphasic processes in conjunction with other solvents. However, the cleanup of aqueous waste is still difficult and the purification requires extensive energy. Since water has unique physical and chemical properties that so clearly distinguish it from other solvents, it may offer new reactivity in organic synthesis. For example, it should in principle be possible to use hydrophobic interactions as a non-bonding element of control for synthetic reactions in water. In 1980s the pioneering studies on Diels–Alder reactions in water was discovered by the research groups of Breslow Citation108 using the hydrophobic interactions effect of water. Later the research groups of Greico Citation109, Grubbs Citation110–112, Sharpless Citation113 , Kobayashi Citation114 , Li Citation115–117, and othersFootnote3 Citation118 Citation119 were exploring new chemical reactivity in aqueous media. The formation of carbon[sbnd]carbon bond formations in aqueous media becomes the central interest for many researchers Citation120–125.
Design of new water-soluble ligands
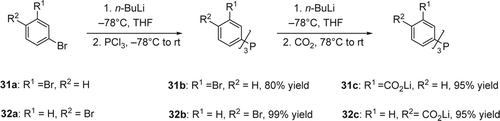
High activity and selectivity, easier product separation, and possible catalyst recycling are among the benefits gained by using homogeneous catalysts in aqueous media. Water solubility is usually reached through incorporation of strongly hydrophilic ligands bearing charges such as sulfonate (SO3Na) and carboxylate (CO2Li; ). One of the advantages of water-soluble phosphines lies in the recyclability of the catalyst that is therefore preserved in water Citation126 Citation127. In 2002, Genêt et al. Citation128 synthesized a new anionic water-soluble ligands based on triphenyl phosphine (). The water-soluble phosphine compounds 31c and 32c were obtained in two steps with high yields. Mono lithiation of the commercially available 1,3-dibromobenzene (31a) and 1,4-dibromobenzene (32a) followed by reacting with PCl3 at low temperature gave compounds 31b and 32b in 80 and 99% yields, respectively. Another lithiation of the compounds 31b and 32b at low temperature and quench by dry ice afforded both the lithiated trianions compounds 31c (tris(m-carboxyphenyl) phosphine trilithium salt (m-TPPTC)) and 32c (p-TPPTC) in 95% yield. These water-soluble phosphine compounds 31c and 32c were efficient in organo-aqueous palladium-catalyzed Heck reactions. The easily prepared ligand m-TPPTC gave excellent results, presumably because of the steric and electronic effects of the carboxylic group in meta-position.
Figure 3. Examples of water soluble ligands.
Palladium
Rapid synthesis of optically active clavicipitic acid via Heck reaction
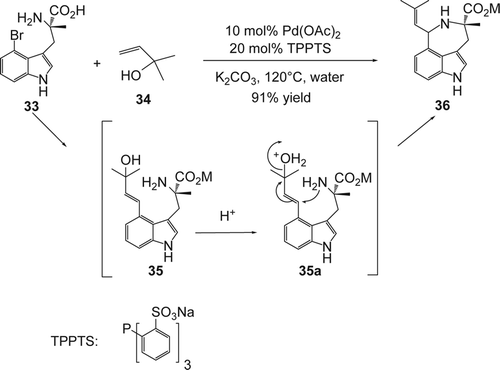
Palladium-catalyzed reactions in aqueous media have attracted much attention Citation129–136 recently because water-based synthetic processes are inherently safer as well as inexpensive. Therefore, the uses of water-soluble catalysts and water-soluble phosphine ligands, e.g. sulfonated phosphines have been explored successfully. Yokoyama and co-workers Citation137 developed a highly efficient Pd-catalyzed Heck reaction of (S)-4 bromo-tryptophan (33) with 1,1-dimethylallyl alcohol (34) in water in the presence of catalytic amount of Pd(OAc)2, tris(m-sulfonatophenyl)phosphine trisodium salt (TPPTS), as a water-soluble ligand and K2CO3 as optimal base (). The same reaction in organic solvent such as dioxane or DMF gave complex products. The compound (35) was treated without any purification with acid, which allowed the intramolecular cyclization/dehydration and the formation of the optically active clavicipitic acid (36) in 91% yield (based on the compound 1; ).
Coupling of aryl iodides with terminal alkynes
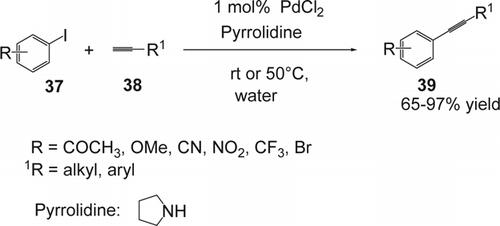
The Sonogashira cross-coupling reaction Citation138 Citation139 has found many applications in the synthesis of scaffolds leading to molecular-scale electronic devices Citation140, dendrimers Citation141, estradiol Citation142, endiyne antibiotics Citation143, and carbohydrate sensors Citation144. Classically, mediated by a dual catalytic system of a palladium[sbnd]phosphine complex and copper (I) iodide, it has been shown to be a mild and high-yielding reaction, tolerant to a wide variety of functional groups. However, a long-reaction time and contamination of the product with the catalyst are the main drawbacks of the reaction. Therefore, there has recently been a lot of interest in improving the conditions for the Sonogashira reaction by designing a suitable catalyst systems and exploring the reaction in aqueous media. In 2005, Yang and co-workers Citation145 reported a simple carbon[sbnd]carbon coupling of aryl iodides (37) and terminal acetylenes (38) in water under aerobic conditions using only 1 mol% of PdCl2 and pyrrolidine as base (). No ligand and no co-catalyst were needed. Various aromatic as well as alkyl-terminal alkynes were coupled successfully (65–97% yield). Only trace amounts of the desired product were obtained when using PdCl2(PPh3)2/CuI/Et3N or Pd(PPh3)4/CuI/Et3N).
Benzofuranes synthesis via tandem reaction
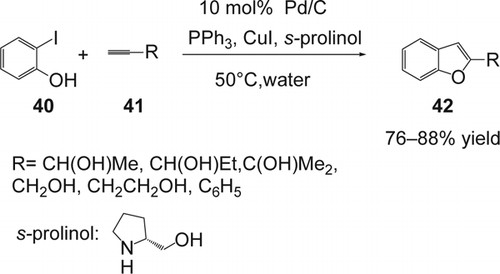
In 2003, Pal and co-workers Citation146–148 reported an efficient Pd/C: PPh3: CuI-catalyzed coupling of 2-iodophenol (40) with terminal alkynes (41) in water (). The reaction proceeds smoothly in neat water under basic conditions. The better solubility of the base (prolinol) in water increased the rate of the coupling reaction and afforded 76–88% yield of the desired benzofurans (42). Various alkynes having non-protecting functional groups gave good yields of the desired product. In addition, the use of a water-soluble phosphine ligands or phase-transfer catalyst was not necessary. Mechanistically, the reaction may proceed via in situ generation of a prolinol-stabilized Pd(0)-complex which perhaps facilitates the reaction in aqueous media due to its interaction with the water molecules (via the hydroxyl group of prolinol). Finally, the tandem reaction allowed an efficient synthesis of various benzofuranes in water which were converted to compounds of potential biological interestFootnote4 Citation149.
Coupling of acid chloride with terminal alkynes
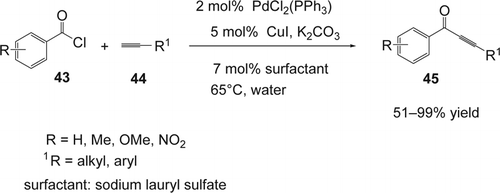
Chen and Li Citation150 reported an efficient coupling of acid chloride (43) with terminal alkynes (44) using PdCl2(PPh3)/CuI as co-catalysts and a catalytic amount of surfactant/phase-transfer reagent under basic conditions in water (). Various aryl acid chlorides were coupled with alkyl and arylalkynes with high yields (51–99%). Among the surfactants used in this study, the sodium laurylsulfate was the most efficient (98% yield) compared to cetyltrimethylammonium bromide (CTAB; 33% yield) and triethylbenzylammonium (27% yield). On the other hand, the yield of the desired ynone (45) dropped from 98 to 9% in the absence of the sodium laurylsulfate.
Platinum
Stereoselective hydrosilylation of terminal alkynes
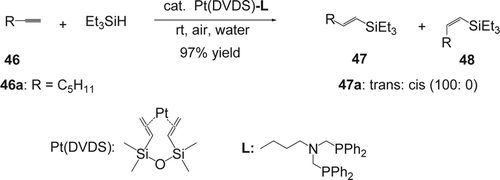
Wu and Li Citation151 reported a highly effective and stereoselective hydrosilylation of terminal alkynes (46) using Pt(DVDS) (DVDS; 1,3-divinyl-1,1,3,3-tetramethyldisiloxane) complex as catalyst and bis(diphenylphosphinomethylene)butylamine as ligand (). The hydrosilylation reaction was effective in water at room temperature under air. The presence of the ligand gave 100% selectivity of the trans-compounds (47a) in the case of the alkylacetylene 46a. Hydroxylated alkynes could be hydrosilylated directly. In all cases except for trimethylsilylacetylene, trans-products were obtained exclusively or selectively. Rhodium-based catalysts such as RhCl(PPh3)3, RhCl3 ·3H2O, Rh(COD)(PPh3)PF6, and Rh(COD)2BF4 gave the desired products with low yields and lower trans/cis selectivity. No reaction was observed with Ru(CO)HCl(PPh3)3 and Ru(Cp)2 catalysts under the same reaction conditions.
N-allylation of aminonaphthalenes with allylic acetates
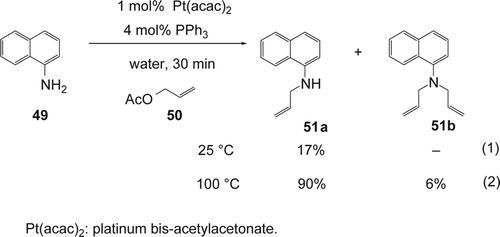
Yang and co-workers reported Citation152 an efficient N-allylation of aminonaphthalenes with allylic acetates using 1 mol% of platinum bis-acetylacetonate [Pt(acac)2] and 4 mol% of PPh3 in water. The reaction at room temperature gave the desired N-allyl-1-naphthylamine 51a selectively with only 17% yield (Equation 1, ). Under reflux, the allylation of 1-aminonaphthalene 49 with allyl acetate 50 afforded N-allyl-1-naphthylamine 51a and N,N-diallyl-1-naphthylamine 51b with 90 and 6% yields, respectively (Equation 2, ). On the other hand, the reaction did not occur in the absence of the platinum species or phosphine ligand. Other Pt catalysts such as Pt[Si(CH3)2C = CH2]2O, and Pt(CH2=CH2)(PPh3)2 were also found to be efficient for this reaction. Most monodentate phosphine ligands afforded the desired product 51a with high yields (51a: 56–90%). PPh3 gave the best yield of 51a. In contrast the bidentate ligands such as dppm, dppb, and dpph gave very low yields (51a: 3–11%). Various aminonaphthalenes and allylic acetates were successfully used.
Nickel
The synthesis of pyrimidine derivatives
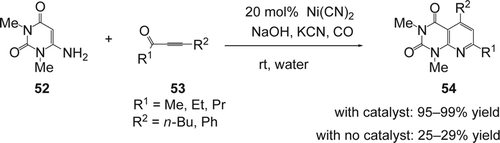
Rosas and co-workers Citation153 reported the reaction of 6-amino-1,3-dimethyluracil (52) with substituted α-ketoalkynes (53) using homogeneous nickel catalyst in aqueous alkaline medium (). Various alkyls and aryles α-ketoalkynes were used successfully. The desired substituted 2,4-dioxopyrido[2,3-d] pyrimidine derivatives (54) were obtained in 95–99% yields while only 25–29% were isolated in the absence of the catalyst.
Cobalt
Intramolecular Pauson–Khand-type reaction
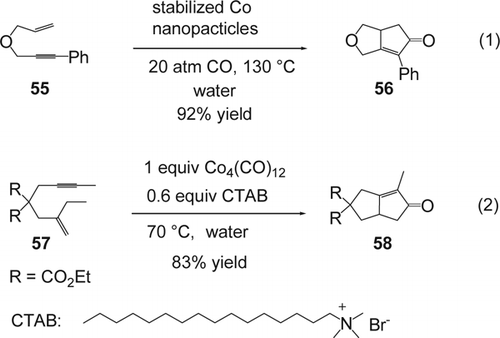
The first effective intramolecular Pauson–Khand reaction using colloidal-cobalt nanoparticles in water was reported by Hyeon and co-workers Citation154 (Equation 1, ). A 20 atm of CO were required in order to obtain high yields of the desired product 56 (92%). Aqueous colloidal-cobalt nanoparticles are simply available and can be recycled, and reused several times without any apparent loss of activity. Krafft and co-workers Citation155 showed that dicobalthexacarbonyl complexes of enynes and alkynes underwent the thermal Pauson–Khand reaction in the presence of surfactants in water. Enyne (57) in the presence of Co4(CO)12, were first converted to the dicobalthexacarbonyl complexes of enynes before adding water and surfactants. Among the investigated surfactants, CTAB and cetyltrimethyl-ammonium hydrogen sulfate were determined to be most effective, although the latter was observed to lead to the hydrolysis of ketals in the substrates. Cyclization in water using Co4(CO)12 with CTAB only provided the desired bicyclic compound (58) with 83% yield (Equation 2, ). Kakiuchi and co-workers Citation156 developed a [RhCl(COD)]2-catalyzed Pauson–Khand-type reaction of enynes in the presence of formaldehyde as a water-soluble source of CO in water and using sodium dodecyl sulfate (SDS) as surfactant.
Rhodium
Enantioselective intramolecular Pauson–Khand-type reaction
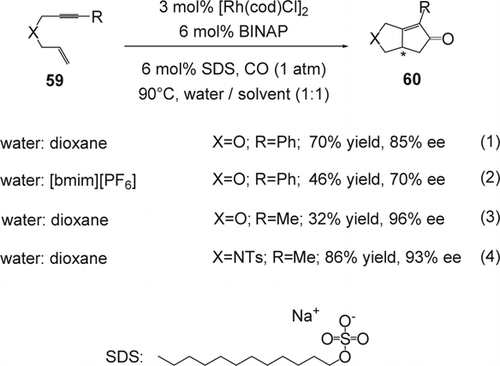
Recently, there has been significant interest in developing enantioselective carbon[sbnd]carbon bonds using Rh transition metal in aqueous media. A successful enantioselective Pauson–Khand reaction with CO in a mixture of 1,4-dioxane and water containing SDS was reported using [Rh(COD)Cl]2-(S)-BINAP as the best catalyst (Equation 1, ). The fused chiral cyclopentenones (60) were prepared in 32–86% yields and 70–93% ee Citation157. Using 1-butyl-3-methyl-imidazolium hexafluorophosphate [bmim][PF6], (Equation 2, ), as co-solvent dropped the ee value as well as the reaction yield to 66% ee and 46% yields, respectively. The study showed that ee's and yields varied depending upon the substrate. When the acetylenic substituent was methyl (Equation 3, ), 96% ee was obtained while the yield of the reaction dropped to 32% yield. On the other hand, in the case of sulfonamide (Equation 4, ), 93% ee and 86% yields were obtained. No reaction was observed when the Pauson–Khand reaction was carried out in neat water solution, presumably due to the insolubility of the substrate and catalyst. Therefore small amounts of 1,4 dioxane were necessary for the efficiency of the reaction. Since surfactants are known to induce reactions in aqueous phase to proceed smoothly, the use of the surfactant as an additive had an influence on the reaction time. For example, when CTAB (a cationic surfactant) was used, the reaction time increased to 24 hours with a slight decrease in enantioselectivity (62% yield; 77% ee). However, when SDS(an anionic surfactant) was used, the reaction time was reduced to 1.5 hours with almost no change in the enantioselectivity. Nevertheless, the reaction rate was rather insensitive to the amount of SDS used.
Addition of phenylboronic acid with various alkenes
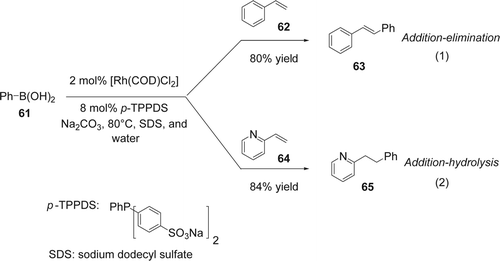
Lautens and co-workers Citation158 reported a highly efficient addition of phenylboronic acid (61) with various alkenes in the presence of a rhodium catalyst and a water soluble ligand (p-TPPDS) under basic conditions in neat water (). In this case, the presence of SDS as surfactant increased slightly the yield of the reaction. In the case of styrene 62, the 1,2-diphenylethylenes (63) was isolated with 80% yield which must arise from a “Hecktype” addition-β H elimination process, an unprecedented mode of reactivity for rhodium (Equation 1, ). In the case of 2-vinylpyridine (64), the compound 65 was isolated with 84% yield which arise from an addition-hydrolysis pathway (Equation 2, ). Later, Genêt and co-workers Citation159 reported also the use of m-TPPTC as a highly reactive water-soluble ligand for the addition of various boronic acids to syrene and 2-vinylpyridine in the presence of Rh catalyst in water under basic conditions. They also showed that it is possible to easily recycle the water-soluble Rh/m-TPPTC without any loss of the selectivity.
Ruthenium
Isomerization of homo-allylic alcohols
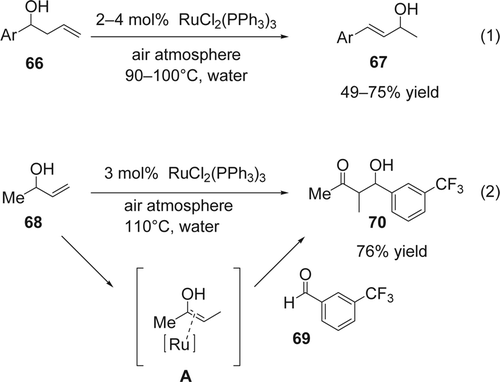
Li and co-workers Citation160 reported the isomerization of homo-allylic alcohols using catalytic amounts of RuC12(PPh3)3 in water under air atmosphere. The homo-allyl alcohols (66) undergo structural reorganization in which both the hydroxyl group and the olefin (67) have been reshuffled with 49–75% yields (Equation 1, ). The corresponding product was not observed in any of the following cases: (1) using aqueous 0.1 N HCl (pH = l) instead of water; (2) using RuCl3 as catalyst in aqueous 0.1 N HCl (pH = l) or aqueous saturated NH4C1 as the reaction solvent; and (3) using other transition metals such as FeCl2, RhCl(PPh3)3, CoC12, and CuC12. Later Wang and Li Citation161 extended their work to the aldol type reaction by coupling efficiently 3-buten-2-ol (68) and 3-fluorobenzaldehyde (69) using RuCl2(PPh3)3 as catalyst in water (Equation 2, ). Mechanistically, the ruthenium complex isomerizes the allyl alcohol to an enol that is co-ordinated with the ruthenium catalyst (A) followed by an in situ reaction between the enol[sbnd]ruthenium complex with the aldehyde generates the aldol product (70; Equation 2, ).
Addition of terminal alkynes to aldehydes and imines
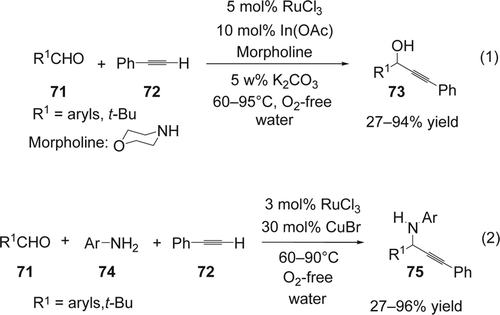
Wei and Li Citation162 developed an efficient Grignard-type reaction of alkynes with aldehydes via C[sbnd]H activation using bi-catalyst systems in aqueous conditions (Equation 1, ). Various catalysts were used for the addition of phenylacetylene 71 to various benzaldehyes 72. The best bi-catalyst was found to be RuCl3/In(OAc)3. On the other hand only trace amounts of the desired product were obtained in the presence of RuCl3/InCl3. Other transition metals such as Rh, Ru, Pd, and Ni gave mixtures of undesired products. Among the organic bases used in this study, morpholine showed higher reactivity. In addition 5 wt% of K2CO3 increased the efficiency of the reaction. Using the best catalyst found, the scope of the reaction was extended to a range of substituted aromatic aldehydes and aliphatic aldehydes without α-hyrdogens. The corresponding propargylalcohols 73 were isolated with modest to high yields (27–94%) (Equation 1, ). In the same year, Wei and Li Citation163 also reported the direct addition of phenylacetylene 71 to imines formed in situ catalyzed by a RuCl3/CuBr co-catalyst in water (Equation 2, ). CuBr and RuCl3 alone gave trace amounts or no desired product, respectively. It is important to mention that the reaction required O2-free media in order to avoid homo-coupling byproducts. Finally the reaction also gave the desired A3 coupling efficiently in free solvent media. This methodology gave a broad range of the desired propargyl-amines 75 in good yields (27–96%).
Copper
Enantioselective addition of terminal alkynes to imines
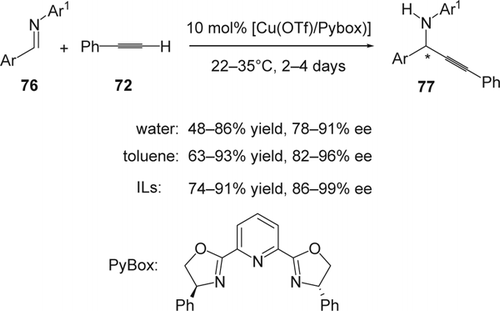
Evans and others showed that chiral copper-bis-(oxazoline) complexes were used effectively as catalysts for enantioselective reactions Citation164–167. In 2002, Wei and Li Citation168 Citation169 reported a highly enantioselective copper(I)-catalyzed direct alkyne (72)-imine (76) addition. The process is simple and provides a diverse range of propargylic amines (77) in high ee and good yield. Among the various bis-oxazoline ligands used, the chiral bis(oxazolinyl) ligand (pybox) was the most effective for this addition in water as well as in toluene (). Various (+)-propargylamines were obtained in high yield and high enantioselectivity although the reaction in toluene provided slightly higher yields and enantioselectivities (63–93% yield, 82–96% ee) than in water (48–86% yield, 78–91% ee). The addition of alkynes to imines enantioselectively was also reported in ILs by Afonso and co-workers Citation170. Using the in situ formed copper(I)[sbnd]bis(oxazoline) complex in [bmim][NTf2], various substrates gave the desired propargylamines with yields between 74 and 91% and ee's between 86 and 99%. In addition, the type of anion was found to have little effect on the ee. Using [BF4] as anion resulted in a significant reduction in yield compared with either [PF6] or [NTf2]. Finally, excellent recyclability, over six times, was shown using [Cu(OTf)/Pybox)] in [bmim][NTf2].
Diastereoselective A3 coupling
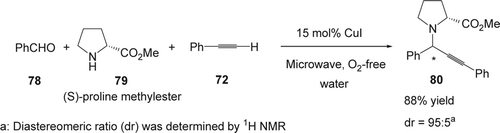
In 2004, Shi and co-workers Citation171 reported Cu(I)-catalyzed three component coupling (A3 coupling) of alkyne (72), aldehyde (77), and amine (79) via C[sbnd]H activation in water under microwave irradiations. In the case of chiral-amine substrate such as (S)-proline methyl ester (79), the chiral propargylamine (80) was obtained with high yield (88%) as well as high diastereoselectivity (dr: 95:5; ).
Silver
Three-component coupling of aldehydes, alkynes, and dialkyl amines
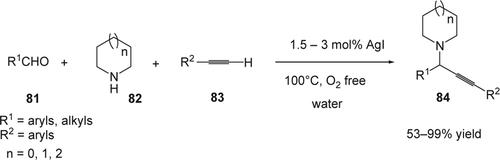
Over the last decade, silver salts were used as catalysts for cross-coupling reactions as well as for the formation of various heterocyclic compoundsFootnote5 Citation172–175. Li and co-workers Citation176 showed that AgI catalyzed efficiently the three-component coupling of aldehydes (81), dialkyl amines (82), and alkynes (83), in neat water under free O2 (). The desired propargylic amines (84) were obtained with modest to high yields (53–99%). AgCl and AgBr also catalyzed the A3 coupling reaction with 55 and 60% yields, respectively. Other silver sources such as AgNO3, Ag2O, AgOAc, Ag2SO4, AgOTf, and AgBF4, which are water-soluble or partially soluble salts, gave the desired product with low conversion (25–45%). In the case of aliphatic aldehydes, the reactions were highly efficient compared to aromatic aldehydes, which provides an effective complement of copper and gold catalyzed A3 coupling.
Alkynylation of aldehydes and ketones
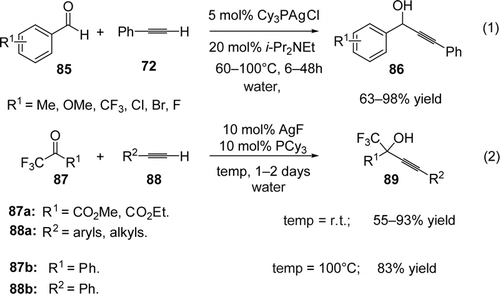
Yao and Li reported the addition of phenylacetylene (72) to aldehydes (85) via C[sbnd]H activation using 5 mol% of Cy3PAgCl complex as catalyst and 20 mol% of iPr2NEt as base in water. Among the mixture of solvent used, water alone gave the best results even though the substrates and the catalyst were not soluble. Both aryl and alkylaldehydes were coupled with phenylacetylene with very high yields of the corresponding propargylalcohools (86; 63–98%). The efficiency of the reaction was related to the presence of tricyclohexylphosphine, iPr2NEt and water (Equation 1, ) Citation177. Very recently, Dong and Li reported an efficient AgF-catalyzed alkynylation of trifluoropyruvate and trifluoroacetophenone in water using PCy3 as ligand Citation178. The addition of various alkynes (88) to trifluoropyruvate (87a) at room temperature gave the desired products with high 55–93% yields (Equation 2, ). In the case of trifluoroacetophenone (87b) (less-reactive ketone), heating the reaction at 100°C was required in order to reach 83% yield of the desired product (Equation 2, ). Other ligands such as PPh3, Pt-Bu3, dppe, and BINAP gave the desired propargylalcohols with very modest yields (up to 24%). Finally, the reaction is also efficient in solventless media.
Alkynylation of arylaldehydes followed by intramolecular cyclization
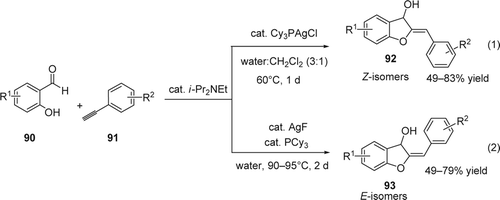
Dihydrobenzofuranol derivatives are an interesting core found in many natural products such as flavanoids Citation179. They are considered great precursors of the aurones which is known to exhibit a variety of biological activities Citation180 Citation181. Very recently, Li and co-workers Citation182 reported an efficient annulation of simple o-hydroxyaldehydes (90) with arylalkynes (91) catalyzed by a silver complex in water. Counter anions in the silver complex proved to be the key factor to Z/E stereoselectivity control. In case of Cy3PAgCl as catalyst, the annulation proceeds efficiently with the Z isomers (92) of the dihydrobenzofuranol derivatives in aqueous media at 60°C with 49–83% yield (Equation 1, ). On the other hand, the E isomers (93) of the dihydrobenzofuranol derivatives were selectively obtained with 49–83% yield using AgF/PCy3 in neat water at 90°C (Equation 2, ).
Gold
Addition of aldehydes, terminal alkynes, and amines
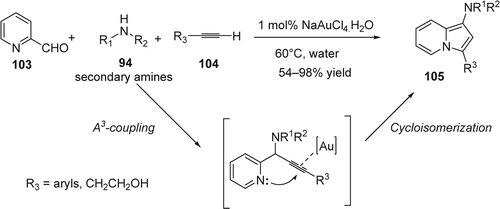
The formation of propargyl amines via the addition of an acetylenic moiety to a C = N bond is a useful method in synthesis Citation183 Citation184. Recently, Li and co-workers Citation185 reported a highly efficient direct coupling of aldehydes (81), secondary amines (94), and phenylacetylene (72), (A3-coupling) catalyzed by gold under such conditions (). The A3-coupling was also catalyzed by Cu and Ag (see Sections “Copper” and “Silver”). In the case of gold as catalyst, no co-catalyst or activator is needed for the reaction. Excellent yields of the corresponding propargyl amine products (95) were obtained. Only <1 mol% of catalyst was necessary. Their study showed that gold-catalyzed A3-coupling tolerates the use of both aromatic and aliphatic aldehydes with alkynes and amines. With aromatic aldehydes, the A3-coupling gave efficiently the desired products with 95–99% yields. While due to the trimerizations of some aliphatic aldehydes, their corresponding propargyl amine products were obtained with modest yields (53–75%). The efficiency of the A3-coupling was only with dialkyl amines. Anilines gave the corresponding products in lower yields and no desired products were observed with N-alkylanilines. The reaction was very clean with almost quantitative yield when water was used as solvent, while the reaction resulted in low conversions and undefined by-products when organic solvents such as THF, toluene, and DMF were used. The reaction mechanism was proposed involving the activation of the C[sbnd]H bond of alkyne by an Au(I) speciesFootnote6 Citation186. The gold acetylide intermediate thus generated the intermediate A which reacted with the iminium ion B generated in situ from aldehydes and secondary amines to give the corresponding propargylamine and regenerate the Au(I) catalyst for further reactions ().
A3-coupling of α-oxyaldehydes, alkynes, and amines
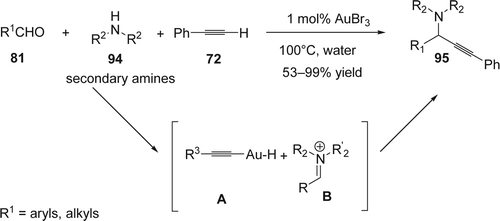
When the three-component coupling was carried with α-oxyaldehyde (96), amine (97), and phenylacetylene (72) in water by using gold, silver, and copper catalysts, gold(I) was found to be the most effective catalyst in this reaction to afford propargylamine (98) in good yield and moderate diastereoselectivity Citation187 (). On the other hand, silver catalysts show the best catalytic activities on non-coordinating α-alkyl-substituted aldehydes.
Gold(III)–salen complexes catalyzed A3-coupling
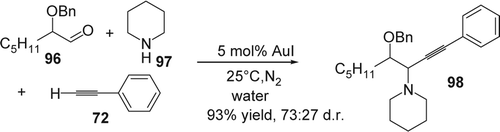
Later Che et al. Citation188 reported that 1 mol% of gold(III)–salen complexes catalyzed the three-component coupling reaction of aldehydes (99), chiral amines (100), and alkynes (101) in water at 40°C (). They prepared and characterized the gold(III)–salen complexes according to a known literature procedure Citation189. The salen ligand was refluxed in CH2Cl2/EtOH with KAu(III)Cl4 in the presence of NH4PF6. The addition of diethyl ether induced the precipitation of the desired gold(III)–salen complexes. The latter afforded a variety of propargylamines (102) with excellent yields and excellent diastereoselectivity (67–89% yield; d.r: up to 99:1). In addition this reaction allowed the synthesis of new propargylamine-modified artemisinin derivatives with modest yield (up to 59%).
Synthesis of 1-substituted 3-aminoindolizines
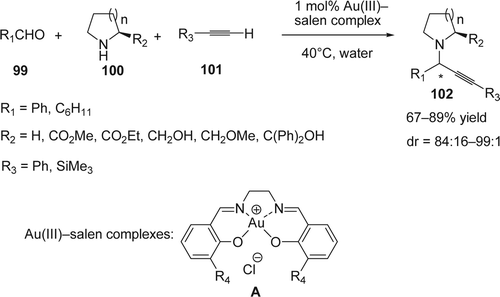
Liu and Yan reported Citation190 the one pot synthesis of 1-substituted 3-aminoindolizines (105) using 1 mol% NaAuCl4-catalyzed three-component reactions of aldehydes (103), secondary amines (94), and alkynes (104) followed by cycloisomerization reaction under solvent free or in H2O at 60°C (). Other gold catalyst such as AuCl3 and AuCl afforded the desired product at the same temperature (60°C) with 86 and 20% yields, respectively. However, at room temperature a longer reaction time was required and lower product yields were observed using NaAuCl4. With respect to amines, secondary amines gave moderate to excellent yields of indolizine derivaritves (68–98%). However, the primary amines such as PhNH2 could not be used for this reaction. On the other hand, no electronic effect was noticed on the aryl alkynes and the reaction gave a high reactivity (88–95%). Alkyl alkynes such as 1-octyne or functionalized alkyne could also be successfully incorporated into the indolizine products (54–66%). The use of pure α-amino acid derivatives with aldehydes and alkynes produces the corresponding indolizines without loss of enantiomeric purity (ee = 99%).
Selected transition metal-catalyzed reactions in ionic liquids (ILs)
In addition to the tremendous interest in exploring chemical reaction in scCO2 and water, ILs were very attractive to use in chemical reactions since the last two decades Citation191–193.
What are ionic liquids (ILs)?
ILs are highly polar salts in which the ions are large and therefore the charges are separated, leading to weaker than normal Coulombic attraction which results in these solvents being liquids below 100°C, or even at room temperature (room temperature ionic liquids (RTILs); ). Contrary to volatile organic compounds (VOCs) ILs are thermally stable, not volatile and have no measurable vapor. They can be recovered and reused. The RTILs (melting point below 22°C) as well as the non-RTILs (melting point below 100°C) can be tuned based on their counter anion. Many researchers consider some ILs to be less toxic than low-boiling points organic solvents. In addition, the dipole characteristics of ILs translate into rapid excitation by microwaves, and consequently faster reactions. Drawbacks of ILs include the amount of time and energy required in their preparation, along with the large amounts of VOCs needed to prepare them Citation194. In 1986, Wilkes and co-workers were the first to report the use of an IL as a catalyst in Friedel–Crafts acylation Citation195.
Figure 4. Examples of ionic liquids (ILs).
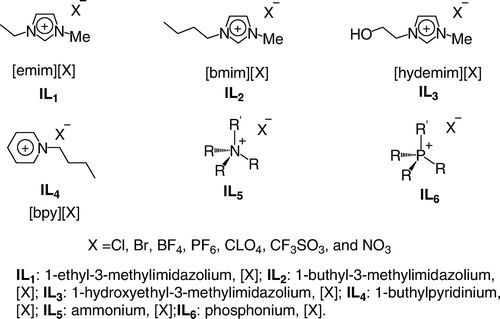
Design of ionic liquids (ILs)
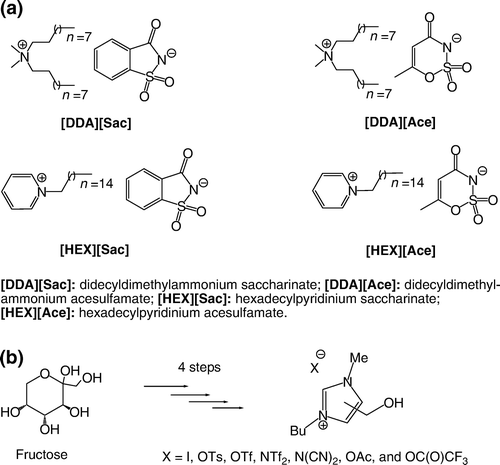
ILs dissolves many organic and organometallic compounds. It is possible to perform a fine-tuning of their miscibility with water and common organic solvents simply by changing the length of the side chains or the nature of the organic cation and the inorganic anion. Such compounds appear very attractive as novel reaction media toward greener processes in organic synthesis. Their unique properties have been reflected by their use in a wide range of stoichiometric, as well as catalytic, reactions Citation196 Citation197. Researchers have focused on ILs formed from dialkylimidazolium halides which are readily prepared from 1-methylimidazole and a slight excess of the desired haloalkane Citation198. The addition of a suitable molar ratio of aluminium chloride to these salts gives rise to ILs containing complex metal anions which have been used in a number of applications such as electroplating, electrochemical devices, and catalysts for organic synthesis Citation199 Citation200. Amines may also be tethered to the IL and used as molecular solvents. Cai et al. have shown that task-specific ILs may be formed by alkylating methyl imidazole with 2-bromo-ethylamine hydrobromide, forming a tethered primary amine Citation201. Very recently, Rogers and co-workers Citation202 prepared a new family of ILs based on quaternary ammonium with dual biological function ((a)). Recently, researchers started exploring new alternative solvent based on biorenewable natural productsFootnote7 Citation203–205. For example, Handy and co-workers Citation206 reported an efficient synthesis of a family of RTILs based on fructose in four steps ((b)).
Figure 5. (a) Examples of ionic liquids with dual biological function; (b) Synthesis of imidazole-based RTILs from biorenewable sources.
Palladium
ILs have been found to be an excellent solvent system and have been used extensively for Heck coupling reactions of a wide range of substrates using both homogeneous and heterogeneous catalysts. Seddon and co-workers Citation207 were among the first groups to study the ILs and show their advantages in terms of recyclability, yield, and control over selectivityFootnote8 , Footnote9 Citation208–218. Other reactions such as Suzuki cross-coupling, Citation219–223 Trost–Tsuji carbon[sbnd]carbon coupling Citation224 Citation225, and hydroarylation Citation226 were also extensively studied in ILs media.
Coupling of aryl bromides with electron-rich olefins
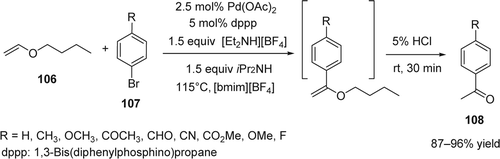
Mo and Xiao reported Citation227 a Pd-catalyzed regioselective Heck reaction of electron-rich olefins (106) and arylbromides (107) in 1-butyl-3-methylimidazolium tetrafluoroborate ([bmim][BF4]) as ILs (). The rates of the reactions were largely accelerated in the presence of an ammonium additive [HNEt3][BF4] and the desired regioselective α-arylated products (108) were obtained with 87–96% yields. On the other hand in the absence of [HNEt3][BF4], the arylation of electron-rich olefins was sluggish and much less selective toward α-olefin products. The acceleration of the reaction rates was also seen in DMF instead of ILs (38% conversion in one hour).The same group reported previously that Pd–dppp catalysis is inhibited by bromide ions in the absence of the ammonium additive Citation228 Citation229. Therefore, it is possible that ammonium additive in this reaction was used to form hydrogen bound with the bromide and enhance the rate of acceleration of the reaction. Recently, the intramolecular Heck reaction of ortho-iodo benzyl allyl ethers to substituted benzofurans was reported with reasonable yields using PdCl2 as the optimal catalyst in [bmim][BF4] at 60°C Citation230.
Coupling of iodoenones with vinyl and phenyl tributyltin
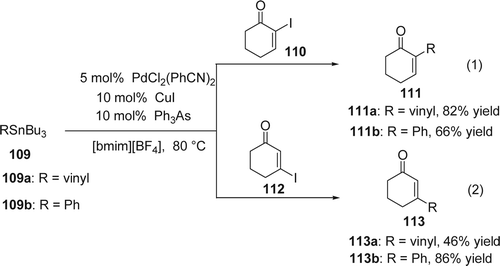
Zhang and Handy Citation231 reported a bis(benzonitrile)palladium(II) chloride/triphenylarsine/copper(I) iodide-catalyzed coupling reaction of α or β-iodoenone and vinyl or phenyl tributyltin in [bmim][BF4]. In the presence of this RTIL, the reaction of vinyltributyltin (109a) and α-iodoenone (110) afforded the desired product 111a with comparable yield than in organic solvent (82% yield) such as N-methyl-2-pyrrolidone (95% yield). The reaction of vinyltributyltin (109a) and β-iodoenone (112) gave only 46% yield of the desired product 113a. On the other hand, the reaction of phenyltributyltin 109b with both α and β-iodocyclohexenones afforded the desired Stille coupling products 111b and 113b in 66% (Equation 1, ) and 86% yields, respectively (Equation 2, ). At the end of the reaction only simple extraction of the product with diethyl ether was required. The recycling studies showed only a slight loss in activity of the catalyst after several uses.
Carbonylation of aryl halides
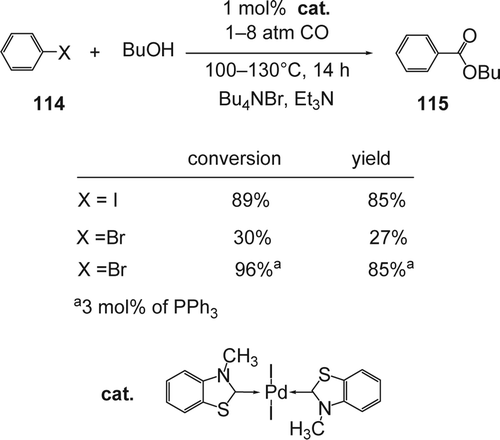
Nacci and co-workers reported Citation232 Pd[sbnd]carbene ligand catalyzed the carbonylation of aryl halides (114) in ILs (). Among the ILs used, tetrabutylammonium bromide (TBAB) was found to give better results. The temperatures and air stable Pd[sbnd]carbene ligands (carbene ligand: benzothiazole carbene) were synthesized and used previously to catalyze C[sbnd]C coupling reactions Citation233–235. In the presence of 1 mol% of the Pd[sbnd]carbene ligand, the carbonylation product of the iodobenzene was obtained with high conversion (89%) and yield (85%) using only 1 atm of CO at 100°C. In the case of bromobenzene, only 30% conversion and 27% yield, even at high temperature and high pressure of CO (130°C, and P CO=8 atm). Under the same conditions using 3 mol% of PPh3 as additive gave the desired carbonylation product (115) with 96% conversion and 85% yield. The presence of triphenylphosphine was required for efficient methoxy-carbonylation. Finally, under these conditions, the catalyst was recovered, reused, and did not show any significant decrease of activity. Recently, Deng and co-workers Citation236 reported the reductive carbonylation of nitrobenzene to the corresponding carbamate using Pd(phen)Cl2 catalyst in a wide range of 1,3-dialkylimidazolium ILs, including those where the alkyl chain is functionalized with a carboxylic and sulfonic acid groups. High catalytic activity was observed with the carboxylic-acid based ILs compared to those based on sulfonic acid. Finally, ILs based on hexafluorophosphate showed higher activity compared to those based on tetrafluoroborate.
Tandem Knoevenagel type-reaction followed by catalytic hydrogenation
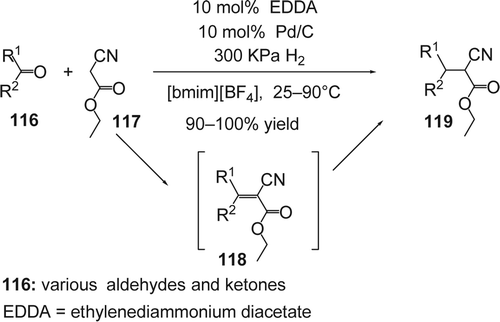
Tandem reactions are highly desirable for increasing the efficiency of a synthetic route by inducing multiple transformations of a substrate in one operation with diminution in the separation and purification of intermediates Citation237 Citation238. Sasson and co-workers reported Citation239 a tandem Knoevenagel type-reaction followed by catalytic hydrogenation in [bmim][BF4] using ethylenediammonium diacetate (EDDA) and heterogeneous Pd-catalyst under hydrogen as to reduce C = C in the condensate products (118) (). This was effective for a range of aromatic aldehydes and ketones as well as methylene compounds such as ethyl acetoacetate, diethyl malonate, and ethyl cyanoacetate. The desired saturated aromatic products (119) were extracted with diethyl ether and the remained catalysts (both Pd/C and EDDA) in the IL were consequently recycled for a second catalytic sequence. The combined IL and catalyst mixture could be used in five consecutive experiments without any loss in activity and selectivity. On the other hand, when the same reaction was carried out in a common organic solvent such as DMA, a considerable amount of benzyl alcohol and toluene was formed by competing direct hydrogenation of the starting aldehyde.
Iridium
Reductive amination of carbonyl compounds with amines
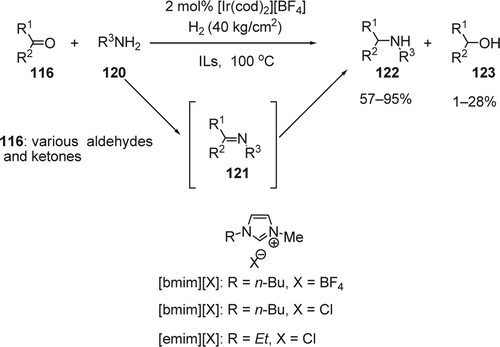
The reductive amination of carbonyl compounds represent a powerful reaction in organic synthesis because ketones and aldehydes can be transformed directly to the corresponding secondary or primary alkylamines without isolation of the intermediary imines or hydroxy amines Citation240–242. Ohta and co-workers reported Citation243 the direct reductive amination (DRA) of carbonyl compounds (116) with amines (120) using the cationic homogeneous iridium catalyst, [Ir(cod)2][BF4], without any other ligands and gaseous hydrogen (). Among the ILs used, [bmim][BF4] was found to be superior to the other organic solvents. While the cation exhibited a small effect, the counteranion of the IL has been found to exert a significant influence on the selectivity. For example, tetraflouroborate gave the best selectivities toward amines. Fonseca et al. Citation244 reported Ir(0) nanoparticles with 2.3±0.4 nm in diameter prepared by simple reduction of [Ir(cod)Cl]2 in 1-n-butyl-3-methylimidazolium hexafluorophosphate IL for the hydrogenation of acetone, 2-pentanone, 3-pentanone, 4-methyl-2-pentanone, cyclopentanone, and benzaldehyde to the corresponding alcohols with high TOFs.
Enantioselective hydrogenation of imines
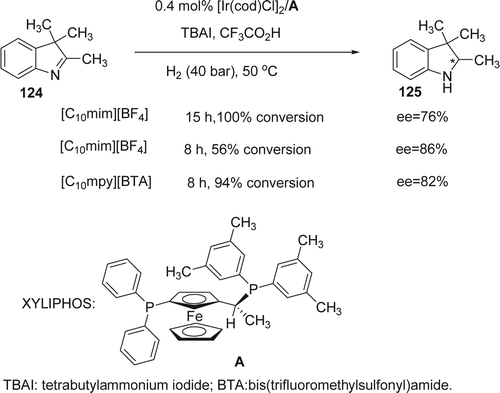
Giernoth and Krumm reported Citation245 an enantioselective hydrogenation of imine (124) using only 0.4 mol% of Ir complexed to chiral XYLIPHOS in ILs such as [C10mim][BF4] and [C10mpy][BTA], (BTA; bis(trifluoromethylsulfonyl)amide) (). The reaction time is reduced from 23 hours for 100% conversion in toluene to less than 15 hours in [C10mim][BF4] with no loss of selectivity, although the ionic media require slightly higher reaction temperatures (50°C), mainly due to their high viscosities. The ee values were sensitive to the time of the reaction as well as the IL used. Decreasing the time of the reaction from 15 to 8 hours decreased the conversion of the reaction (from 100 to 56%), while the ee values incresead slightly (from 72 to 86%). In the case of [C10mpy][BTA] the reaction had reached 94% conversion with 82% ee after only eight hours.
Rhodium
Hydroformylation
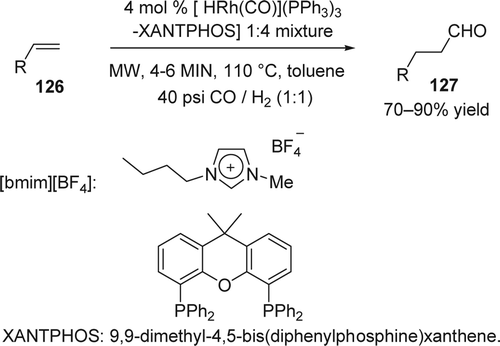
The hydroformylation reaction (oxo process) is one of the most powerful methods for the preparation of aldehydes. Chauvin and co-workers were the first to study the Rh-catalyzed hydroformylation of olefins in ILs employing biphasic conditions Citation246. Wasserscheid and co-workers also showed an efficient biphasic hydroformylation of methyl-3-pentenoate in [bmim][PF6] and [bmim][BF4] using Rh(acac)(CO)2 as catalyst Citation247. The biphasic hydroformylation of pent-1-ene in [bmim][PF6] and [bmim][BF4] with Rh(acac)(CO)2 also showed excellent results Citation248 Citation249. Recently, Taddei and co-workers Citation250 reported that the hydroformylation of alkenes can be carried out in a few minutes under microwave activation at 110°C and at a relatively low pressure (40 psi) using commercially available catalysts and ligands (). The 80 mL vial of a Discover microwave oven was connected to a cylinder of CO and H2, and after filling the reactor at 40 psi, a mixture of alkenes (126), the Wilkinson catalyst HRh(CO)](PPh3)3, and 9,9-dimethyl-4,5-bis(diphenylphosphine)-xanthene (XANTPHOS). XANTPHOS was submitted to microwave irradiation giving, after 4–6 min, high conversion into the corresponding aldehydes (127; 70–90% yield) without formation of the isomerized alkene. The IL [bmim][BF4], was used as co-solvent and an additive recommended to enhance heat transfer from the microwaves toward the reaction mixture Citation251. On the other hand, supported ionic-liquid-phase catalyst systems for Rh-catalyzed olefin hydroformylation was extensivly studied by Fehrmann and Wasserscheid and co-workers Citation252–255. They used the supported IL phase concept to examine the continuous gas-phase hydroformylation of propene in [bmim][C8H17OSO3] as IL and using [Rh(acac)(CO)2: bisphosphine sulfoxantphos] system as catalyst. High degrees of linearity in the aldehyde were observed up to 96%. Yuan and co-workers Citation256 reported the use of TPPTS–Rh complex in an MCM-41-supported IL for the liquid-phase hydroformylation of various alkenes such as hex-1-ene, oct-1-ene, dec-1-ene, and dodec-1-ene. The linear aldehydes were formed in preference to the branched ones (ratio up to 3.8). Hydroformylation of hex-1-ene using CO2 as carbonyl carbon source has also been achieved resulting in the chemoselective formation of heptanols using a ruthenium complex employed in a biphasic 1,3-dialkylimidazolium chloride-toluene system Citation257.
Intramolecular cycloisomerization of dienes with alkenes
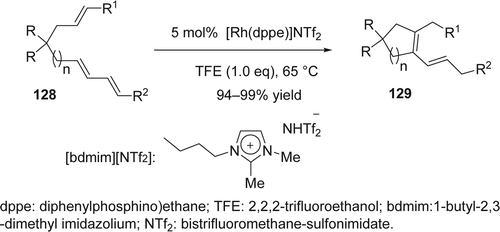
Early this year, Sato and co-workers Citation258 reported an efficient Rh(I)-catalyzed cycloisomerization of dienes with alkenes using ILs as reaction media (). The desired cyclic compounds (129) were obtained with 94–99% yield. The structure of ILs strongly affected the catalyst recycling. For example, 1-butyl-2,3-dimethyl imidazolium [bdmim]+ based IL was more effective than a 1-butyl-3-methyl imidazolium [bmim]+, in which a Rh–NHC complex would be formed from the imidazolium cation part. It has been proven that the addition of 2,2,2 trifluoro-ethanol (TFE) to the reaction mixture prevented the loss of reactivity of the catalyst. The [bdmim][NTf2] containing the Rh(I) catalyst was recovered after the reaction under the present conditions could be reused repeatedly with slight decrease of the desired product yield during the eighth cylcle (82%).
Polymerization of phenylacetylene
Nobile and co-workers Citation259 investigated the use of ILs as reaction media for the polymerization of phenylacetylene (72) catalyzed by (norbornadiene)Rh(acac) and [(norbornadiene)RhCl]2 complexes under basic co-catalysis. Quantitative yields of poly(phenylacetylene) (130) with high cis percentages (96–100%) were obtained within 5 min with (nbd)Rh(acac) in both [bmim]BF4 and [bupy]BF4 (). The molecular weights, determined by gel permeation chromatography (GPC)-technique, were in the range between 110,000 and 199,000 Da. A considerably higher molecular weight was obtained in [bmim]BF4 with respect to [bupy]BF4. The catalyst solution in IL could be recycled without significant loss in activity provided that PPA (poly(phenylacetylene)) separation was performed either by filtration or by toluene extraction. Isolated yield in both [bmim]BF4 and [bupy]BF4 were between 63 and 99% yields. However, the poly-dispersities (Mw/Mn) were between 3.9 and 8.5 in both ILs.
Table 2. Polymerization of phenylacetylene in various ILs.
Tandem cyclization-1,3-dipolar cycloaddition reaction of α-diazo ketones
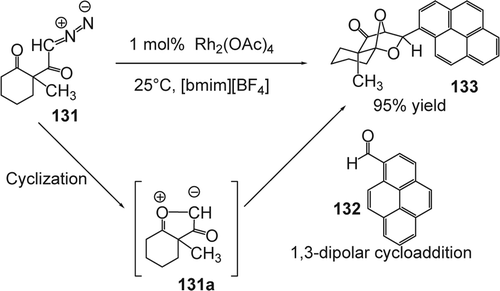
Muthusamy and Gnanaprakasam Citation260 reported the synthesis of dioxa-bridged polycyclic systems using the cyclization–cycloaddition reaction of transient five-membered-ring carbonyl ylides with a-diazo ketones in broad range of ILs. The reaction was efficiently catalyzed by Rh(II) dissolved in [bmim][BF4], [bbim][BF4], [mmim][BF4], and [bmim][PF6]. For example, the rhodium(II) acetate catalyzed reaction of diazo ketone (131) and pyrene-1-carboxaldehyde (132) in [bmim]BF4 IL at room temperature for an hour afforded the dioxa-bridged compound (133) in 95% yield with high regio and exo-selectivity (). The exo-selectivity in (133) was confirmed based on a characteristic singlet resonance around 4.5 ppm for the bridgehead proton in the 1H NMR spectrum. The product (133) confirms the successful generation of fused five-membered-ring carbonyl ylide (131a) in the IL medium ().535 The reaction was found to proceed with high stereoselectivity in all ILs and molecular solvents. However, in the case of [bmim][BF4], a significant increase in the yield of the cycloaddition product (95%) was also observed compared with organic solvents (dichloromethane). Copper (II) also catalyzed this reaction but higher yields were obtained using the rhodium-based catalysts. The ILs based on [BF4] rather than [PF6] also lead to increased yields.
Ruthenium
Enantioselective hydrogenation of ketone and derivatives
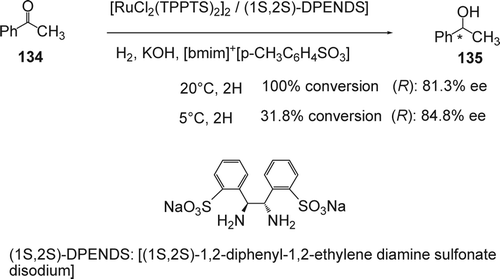
Many researchers were studying Ru-catalyzed homogeneous hydrogenation of ketones. Chan and co-workers reported the asymmetric hydrogenation of α-keto esters in ILs using Ru–BINAP as a catalyst. The enantioselectivity was found to be strongly dependent on the anion of the IL used. For example, for the hydrogenation of methyl pyruvate, 81% ee was obtained in [bmim][BF4] while only 55% ee was obtained in [bmim][PF6] Citation261. They also studied the enantioselective hydrogenation of α-alkyl ketoesters and α-aryl ketoesters. With the α-alkyl ketoesters showing high rates in ILs. Li and co-workers reported Citation262 [RuCl2(TPPTS)2]2-(1S,2S)-DPENDS–KOH] (DPENDS = 1,2-diphenyl-1,2-ethylenediamine sulfonate disodium)-catalyzed asymmetric hydrogenation of acetophenone (134) in a series of hydrophilic ILs [Cnmim][p-CH3C6H4SO3] (n=2, 4, 8, 12; ). The hydrogenation occurred with 100% conversion and 81.3% ee at 20°C during 2 hours. Decreasing the temperature to 5°C gave only 31.8% conversion, but with slightly higher ee (84.8%). A synergistic effect between (1S,2S)-DPENDS and KOH in IL solution significantly accelerates the reaction and enhances the enantioselectivity. The IL phase containing the active catalyst and chiral ligand can be recycled and reused for several times without significant decrease in activity and ee value. An ee of 79.2% was obtained in the asymmetric hydrogenation of acetophenone; however, using 2-fluoroacetophenone, 2-chloroacetophenone, 2-bromoacetophenone, 2-(trifluoromethyl) acetophenone, 4-(trifluoromethyl) acetophenone, 2-methoxyacetophenone, and 4-methoxyacetophenone led to only moderate ee's. Hydrogenation of ethyl acetoacetate with the ammonium salt of 4,4′- and 5,5'-RuBINAP in ILs based on 1,3-dialkyl imidazolium, N-alkylpyridinium, and tetralkylphosphonium cations occurred with moderate ee's was also reported Citation263. Asymmetric hydrogenation of α-aryl ketoesters, using Ru modified by 4,4'-substituted BINAP derivatives, has been carried out in both methanol and the monophasic mixture of [bmim][BF4] and methanol Citation264 Citation265. In general, the differences in ee obtained in the IL/methanol mixture compared with results in methanol were very small and the IL only played a role to aid the recycling of the catalyst. The Rh complexed with optically active (R)-BINAP ligand showed also a good enantioselective catalytic hydrogenation in IL based on (N-n-butylpyridinium (BP) ions) Citation266.
Ring-closing metathesis (RCM) of dienes
Since ILs are immiscible with many organic solvents, it was very attractive to perform a ring-closing metathesis (RCM) and attempt to recover and recycle the IL layer containing the Ru catalyst after a simple extraction of the product. Yao and co-workers reported Citation267 Citation268 recently the RCM of diene and enyne using Grubbs and Hoveyda-Grubbs ruthenium carbene complex bearing an IL tag in mixed solvent systems containing [bmim][PF6] and CH2Cl2 or toluene. Buijsman and co-workers Citation269, have also reported that the Grubbs's Ru catalyst dissolved in [bmim][PF6] promoted the RCM of several dienes for at least three cycles. However, low conversion was obtained at the last cycle caused by extraction of the catalyst to the organic phase. More recently, Dixneuf and co-workers Citation270 have described RCM performed by ruthenium allenylidene salts in [bmim][PF6], but the catalyst proved to be efficient only for the first two cycles due to the slow decomposition of the catalytic system. Even though in many cases the IL protects the catalyst and enables reuse, when the catalyst is modified with the IL tag the recyclability is improved significantly. Mauduit and co-workers reported Citation271 the synthesis of an alkyl imidazolium salt supported ruthenium catalyst A (). The catalyst was used in RCM of N,N-diallyltosylamide (136) in [bmim][PF6]. The cyclic olefin (137) was selectively obtained with 98% conversion after 45 min at 60°C and no cross-metathesis products were observed. The comparative recycling and reuse of the catalyst A, B, and C in [bmim][PF6] for the RCM of the diene (136; ) showed high efficiency of the alkyl imidazolium salt supported ruthenium catalyst A (up to nine times recyclable without loss of activity). Similarly, imidazolium-tagged ruthenium catalysts have also been reported to perform di or trisubstituted diene metathesis including substrates containing oxygen in [bmim][PF6] and [bmim][NTf2]. Citation271
Table 3. Comparative recycling and reuse of Ru-based catalysts for RCM in ILs.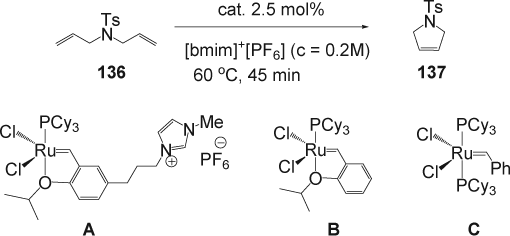
Aldol and Mannich-type reactions
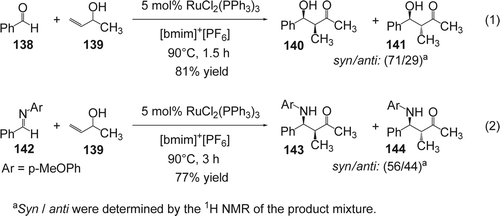
Li and co-workers reported Citation272 a RuCl2(PPh3)3 catalyzed aldol condensation between allylic alcohols and various aldehydes in [bmim][PF6]. For example, the condensation between formaldehyde (138) and but-3-en-2-ol (139) gave the desired aldol-type products (140/141) in 81% yield with 71/29: syn/anti ratio (Equation 1, ). The desired products were obtained efficiently even with aldehydes with low reactivity such as 2-naphthaldehyde or p-anisaldehyde. Interestingly, in the IL, the electron-deficient aldehydes showed the highest activity which is the opposite trend to that found in molecular solvents, including water. This was attributed to the increased solvation by the IL of the more polarized electron-deficient aldehydes. In contrast, neither aromatic nor aliphatic ketones gave good yields under these conditions. The IL containing the catalyst system was recovered and reused at least five times without loss of activity. Mechanistically, the ruthenium complex isomerizes the allyl alcohol to an enol that is coordinated with the ruthenium catalyst followed by an in situ coupling between the enol–ruthenium complex with the aldehyde generates the aldol-type product. On the other hand, the reaction between the allylic alcohol (139) and imine (142) was also catalyzed by RuCl2(PPh3)3 in [bmim][PF6]. The desired products (143/144) were obtained in 77% yield with 56/44: syn/anti ratio (Equation 2, ). The corresponding reaction in water was found to form the aldol product due to hydrolysis of the imine; however, in the IL only Mannich products were obtained with increased yields over molecular solvent systems.
Cyclooligomerization of arylethynes
Conte and co-workers Citation273 reported the cyclooligomerization of arylethynes (145) catalyzed by ruthenium(II) porphyrins in [bmim][PF6] and 1-octyl-3-methylimidazolium hexafluorophosphate [omim][PF6] (). In the case of phenylacetylene as substrate, 1-phenylnaphthalene (146) was isolated as the main product with 32% in [bmim][PF6] and with 60% in [omim][PF6]. While 1,3,5 and 1,2,4-triphenylbenzenes (147/148) were obtained as minor products. Once the p-methoxyphenylacetylene was used, only the compound (146) was isolated as ratio of two isomers. The use of [omim][PF6] gave high yield (60–67%) of naphthalene (146) compared to [bmim][PF6] (21–32% yield).
Table 4. Cyclooligomerization of arylethynes in ILs.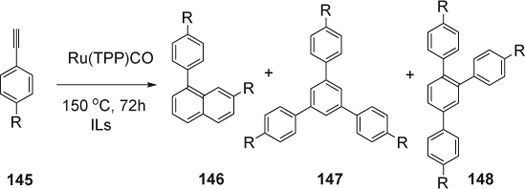
Copper
Selective oxidation of alcohols
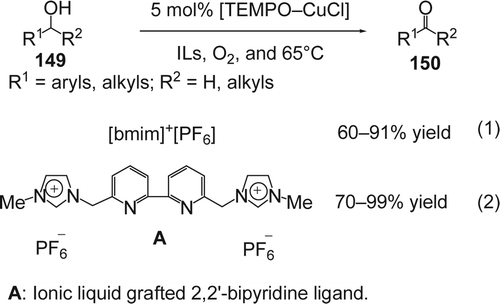
Selective oxidation of alcohols to the corresponding aldehydes or ketones in an oxygen atmosphere has been studied in ILs using the TEMPO–CuCl system. Using this catalytic system, Ansari and Gree reported Citation274 an aerobic oxidation of primary and secondary alcohols (149) to the corresponding aldehydes and ketones (150) in [bmim][PF6] with 60–91% yields (Equation 1, ). In the case of primary alcohols, no over oxidation to the corresponding carboxylic acids were observed. The catalytic activity of a TEMPO 2,2,6,6-tetramethylpiperidine-1-oxyl derivative bearing an IL-type appendage has also been examined for oxidation of alcohols under IL-aqueous biphasic conditions in the presence of NaOCl and KBr. Similar results were obtained under these conditions in the single-phase medium with the functionalized IL. The latter was easily recycled and reused without loss of activity and selectivity Citation275. In order to increase the recyclability of the catalyst, Gao and co-workers Citation275 synthesized the IL (A) incorporating a tethered 2,2'-bipyridine ligand which can complex with CuCl during the TEMPO-catalyzed oxidation reaction, thus immobilizing it more efficiently. In this case the yields of the desired products were between 70 and 99% (Equation 2, ).
Click chemistry
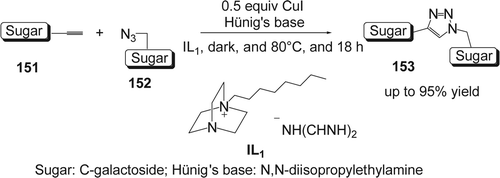
Since Sharpless and co-workers discovered the click chemistry concept, there was a tremendous interest in using the prototypical click reaction constituting the copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC). The later gave efficiently 1,4 disubstituted 1,2,3-triazoles under mild conditions Citation276 Citation277. Marra and co-workers reported Citation278 the first study of a copper(I)-catalyzed azide-alkyne click reaction in ILs (). The cycloaddition of a sugar azide (152) with a sugar acetylene (151) (CuI, i-Pr2EtN, 80°C) was carried out in 10 different ILs as well as in standard molecular solvents (toluene and DMF) to give the 1,4-disubstituted triazole-linked C-disaccharide (153). The highest yields (84 and 95%) were registered in N-octyl dabco-cation-based dicyanamide ([C8dabco] [N(CN)2]) as ILs. The latter IL was recycled in four subsequent reactions without loss of the reaction efficiency. Reactions carried out in the absence of the Hünig's base afforded mixtures of 1,4 and 1,5-disubstituted triazole regioisomers. The same research group also extended their work to the synthesis of glycoclusters on a calix[4]arene platform in ILs using the CuAAC methodologie Citation279 Citation280.
Diels–Alder reaction
Imidazolium-tagged bis(oxazolines) L1 have been prepared and used as chiral ligands in the copper(II)-catalyzed Diels–Alder reaction of N-crotonoyloxazolidinones (154) with cyclopentadiene (154) in the IL 1-ethyl-3-methylimidazolium bis[(trifluoromethyl)sulfonyl]-imide, [emim][NTf2] Citation281. A significant and substantial enhancement in the rate and enantioselectivity was achieved in [emim] [NTf2] compared with dichloromethane. For example, in the presence of the imidazolium-tagged catalyst L 1 complete conversion with 84% ee was obtained for the reaction between N-acryloyloxazolidinone (154) and cyclopentadiene within 2 min in [emim] [NTf2], whereas the corresponding reaction in dichloromethane gave only 43% conversion and 12% ee even with longer reaction time (60 min; ). The enhanced rates obtained in the IL enabled a catalyst loading as low as 0.5 mol% to give complete conversion within 2 min while retaining the same level of enantioselectivity. In the presence of Box ligand L 2 , complete conversion with 95% ee were obtained within 2 min reaction in [emim][NTf2]. On the other hand, the reaction was slow in dichloromethane/L2 and only 78% conversion and 78% ee were obtained. Finally, the imidazolium-tagged catalyst L 1 can be recycled 10 times, compared to L 2 , without any loss in activity or enantioselectivity and showed much higher affinity for the IL phase during the recycling procedure than the analogous uncharged ligand L 2 .
Table 5. Comparative study of Diels–Alder reaction in ILs and dichloromethane.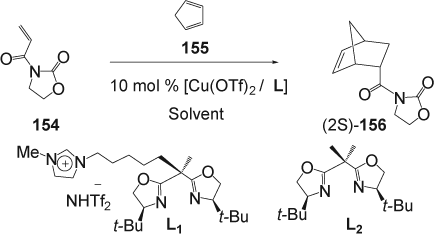
Concluding remarks
Over the last two decades, alternative solvents have received an increase in interest as better replacements for conventional solvents in metal-catalyzed reactions. Most examples shown in the literature were to substitute toxic organic solvents and improve well known processes. As a conclusion from this review, catalysis in alternative solvents (e.g. scCO2, water, and ILs) is hugely varied and in many cases those sustainable solvents enable more efficient reactions to take place compared to traditional solvents. Switching from an organic solvent to scCO2, water, or an ionic media can result in: (1) recycling and reusing the catalyst; (2) increasing the solubility and the stability of the catalyst; (3) increasing the reactivity and the selectivity of some specific reactions; (4) increasing the efficiency of the reaction (e.g. E-factor Citation282), synthesis without protections Citation283 Citation284, and with minimum reaction steps); (5) exploring new reactivities; and finally (6) improving the design to decrease toxic wastes and byproducts.
The unusual chemical and physical properties of scCO2 such as low toxicity, easy separations, variable density, low viscosity, low surface tension, non-flammability, and naturally abundancy is well established. It is clear from the selected examples in this review that scCO2 became very attractive as a sustainable and “green” solvent. However the use of scCO2 is still limited for only certain cases in industry because of the costs associated to the high-pressure equipment and the energy requirements. Nevertheless, it is widely accepted that the advantages associated with scCO2 are likely to lead to a number of new CO2-based processes in the future.
ILs have been found to be effective as catalysts, bases, or solvents and represent a clean industrial technology with significant cost and environmental benefits. Drawbacks still remain, including the expense to manufacture, difficulty found in purification and recycling. However, researchers from Queen's University of Belfast have shown that many ILs can be purified by distillation at 200–300°C at low pressure without decomposition Citation285. Therefore it is possible to recycle and reuse them efficiently. This discovery will certainly open the door to many applications of ILs in the near future. However there is still a lack of information about their toxicity Citation286–292.
Water represents one of the cheapest solvents, abundantly available, non-toxic, and non-flammable. The use of aqueous biphasic catalysis provides an ideal basis for recovery and recycling of the (water-soluble) catalyst. Very recently, Chai and Lautens showed that water has a dramatic effect on both the reactivity of the substrate and the reduction of the byproduct in the tandem Suzuki–Miyaura coupling Citation293. Water is also the ideal solvent for many processes catalyzed by nature's catalysts enzymes. For these reasons, the use of water as a reaction medium co-ordinates well with the current trend toward a sustainable chemical industry based on the utilization of renewable raw materials rather than fossil fuels as the basic feedstock.
Taken together, the current status of this field convincingly substantiates the potential of scCO2, water, and ILs as alternative solvents for green chemistry. It is clear from this review that this field of chemistry will continue to be challenging and an attractive topic.
Acknowledgements
The author is deeply grateful to Professor Chao-Jun Li for guidance, advice, and support during the PhD thesis in the field of green chemistry. The author is also deeply grateful to Professor David N. Harpp as a source of inspiration in teaching. Thanks are also given to McGill University and FQRNT for the fellowships as well as to Professor Ronald Breslow for his encouragement.
Notes
1. For representative reviews in aqueous media, see (Citation7).
2. For an intramolecular Heck reactions catalyzed by [Pd(OCOCF3)2:P(2-furyl)3:EtN i-Pr2] see (Citation34).
3. For reviews on hydrophobic interactions and chemical reactivity, see (Citation119).
4. For the study of benzofuro[3,2-c]pyridine as a potential antidepressant, see (Citation149).
5. For recent reviews on silver-catalyzed reactions, see (Citation172).
6. For synthesizing alkynyl gold complexes, see (Citation186).
7. For examples, see (Citation203).
8. For recent studies on homogeneous Heck reaction using ILs, see (Citation208).
9. For recent studies on heterogeneous Heck reaction using ILs, see (Citation215).
Related Research Data
References
- Anastas , P.T. and Warner , J.C. 1998 . Green Chemistry: Theory and Practice , Oxford : Oxford University Press .
- Poliakoff , M. and Anastas , P. 2001 . Nature , 413 : 257
- Poliakoff , M. and Licence , P. 2007 . Nature , 450 : 810 – 812 .
- Wai , C.M. , Hunt , F. , Ji , M. and Chen , X. 1998 . J. Chem. Edu 1641 – 1645 .
- Oakes , R.S. , Clifford , A.A. and Rayner , C.M. 2001 . J. Chem. Soc., Perkin Trans 917 – 941 .
- Raveendran , P. Ikushima , Y. Wallen , S.L. Acc. Chem. Res . 2005 , 38 , 478 – 485 .
- Li , C-J. Chem. Rev . 1993 , 93 , 2023 – 2035 .
- Lindstrom , U.M. Chem. Rev . 2002 , 102 , 2751 – 2772 .
- Li , C-J. Chem. Rev . 2005 , 105 , 3095 – 3165 .
- Li , C-J. ; Chen , L. Chem. Soc. Rev . 2006 , 35 , 68 – 82 .
- Lindstrom , U.M. 2007 . Organic Reactions in Water: Principles, Strategies and Applications , Oxford, , UK : Blackwell Publishing .
- Herrerías , C.I. ; Yao , X. ; Li , Z. ; Li , C-J. Chem. Rev . 2007 , 107 , 2546 – 2562 .
- Li , C-J. ; Trost , B.M. Proc. Natl. Acad. Sci. USA 2008 , 105 , 13197 – 13202 .
- Scammells , P.J. ; Scott , J.L. ; Singer , R.D. Aust. J. Chem . 2005 , 58 , 155 – 169 .
- Pârvulescu , V.I. ; Hardacre , C. Chem. Rev . 2007 , 107 , 2615 – 2665 .
- Hough , W.L. ; Rogers , R.D. Bull. Chem. Soc. Jpn . 2007 , 80 , 2262 – 2269 .
- Hough , W.L. Smiglak , M. Rodriguez , H. Swatloski , R.P. Spear , S.K. Daly , D.T. Pernak , J. Grisel , J.E. Carliss , R.D. Soutullo , M.D. Davis , J.H. Jr. ; Rogers , R.D. New J. Chem . 2007 , 31 , 1429 – 1436 .
- Ipat'ev , V. ; Rutala , O. Berichte der Deutschen Chemischen Gesellschaft 1913 , 46 , 1748 – 1755 .
- Jessop , P.G. ; Ikariya , T. ; Noyori , R. Science 1995 , 269 , 1065 – 1069 ; Jessop , P.G. ; Ikariya , T. ; Noyori , R. Chem. Rev 1999 , 99 , 475 – 493 .
- Jessop , P.G. and Leitner , W. 1999 . Chemical Synthesis using Supercritical Fluids , Weinheim, , Germany : VCH/Wiley .
- Leitner , W. Science 1999 , 284 , 1780 – 1781 ; Leitner , W. Nature 2000 , 405 , 129 – 130 ; Leitner , W. Nature 2000 , 407 , 295 – 296 ; Leitner , W. Nature 2003 , 423 , 930 – 931 ; Leitner , W. Acc. Chem. Res . 2002 , 35 , 746 – 756 .
- Darr , J.A. ; Poliakoff , M. Chem. Rev . 1999 , 99 , 495 – 541 .
- Poliakoff , M. and King , P. 2001 . Nature , 412 : 125
- Jessop , P. G. In Supercritical Fluid Technology for Drug Product Development , York , P. ; Kompella , U.B. ; Shekunov , B.Y. New York : Marcel Dekker , 2004 ; pp 461 – 496 .
- Prajapati , D. ; Gohain , M. Tetrahedron 2004 , 60 , 815 – 833 .
- Campestrini , S. ; Tonellato , U. Curr. Org. Chem . 2005 , 9 , 31 – 47 .
- Jiang , H-F. Curr. Org. Chem . 2005 , 9 , 289 – 297 .
- Jessop , P.G. J. Supercrit. Fluids 2006 , 38 , 211 – 231
- McHugh , M. ; Krukonis , V. Supercritical Fluid Extraction ; Boston, MA : Butterworths , 1986 .
- Jessop , P.G. ; Hsiao , Y. ; Ikariya , T. ; Noyori , R. J. Am. Chem. Soc. 1996 , 118 , 344 – 355 .
- Kainz , S. ; Koch , D. ; Baumann , W. ; Leitner , W. Angew. Chem., Int. Ed. 1997 , 36 , 1628 – 1630 .
- Early , T.R. ; Gordon , R.S. ; Carroll , M.A. ; Holmes , A.B. ; Shute , R.E. ; McConvey , I.F. Chem. Commun . 2001 , 19 , 1966 – 1967 .
- Saffarzadeh-Matin , S. ; Chuck , C.J. ; Kerton , F.M. ; Rayner , C.M. Organometallics 2004 , 23 , 5176 – 5181 .
- Shezad , N. ; Clifford , A.A. ; Rayner , C.M. Tetrahedron Lett . 2001 , 42 , 323 – 325 .
- Carroll , M.A. Holmes , A.B. Chem. Commun . 1998 , 13 , 1395 – 1396 .
- Morita , D.K. Pesiri , D.R. David , S.A. Glaze , W.H. Tumas , W. Chem. Commun . 1998 , 13 , 1397 – 1398 .
- Shezad , N. Oakes , R.S. Clifford , A.A. Rayner , C.M. Tetrahedron Lett . 1999 , 40 , 2221 – 2224 .
- Smith , C.J. Early , T.R. Holmes , A.B. Shute , R.E. Chem. Commun . 2004 , 17 , 1976 – 1977 .
- Shezad , N. Clifford , A.A. Rayner , C.T. Green Chem . 2002 , 4 , 64 – 67 .
- Fujita , S. Yuzawa , K. Bhanage , B.M. Ikushima , Y. Arai , M. J. Mol. Catal. A: Chem . 2002 , 180 , 35 – 42 .
- Osswald , T. Schneider , S. Wang , S. ; Bannwarth , W. Tetrahedron Lett . 2001 , 42 , 2965 – 2967 .
- Cheng , J-S. Jiang , H-F. Eur. J. Org. Chem . 2004 , 3 , 643 – 646 .
- Jiang , H-F. Curr. Org. Chem . 2005 , 9 , 289 – 297 .
- Li , J.H. ; Jiang , H.F. ; Chen , M.C. Green Chem . 2001 , 3 , 137 – 139 .
- Ikariya , T. Kayaki , Y. Kishimoto , Y. Noguchi , Y. Prog. Nucl. Energy 2000 , 37 , 429 – 434 .
- Kayaki , Y. ; Noguchi , Y. Iwasa , S. Ikariya , T. Noyori , R. Chem. Commun . 1999 , 13 , 1235 – 1236 .
- Jia , L.Q. ; Jiang , H.F. Li , J.H. Chem. Commun . 1999 , 11 , 985 – 986 .
- Jiang , H.F. Jia , L.Q. Li , J.H. Green Chem . 2000 , 2 , 161 – 164 .
- Ran , X-G. Jiang , H-F. Zhu , X-H. Chin. J. Chem . 2004 , 22 , 1384 – 1386 .
- de Vries , T.J. Kemmere , M.F. Keurentjes , J.T.F. Macromolecules 2004 , 37 , 4241 – 4246 .
- de Vries , T.J. Duchateau , R. Vorstman , M.A.G. Keurentjes , J.T.F. Chem. Commun . 2000 , 4 , 263 – 264 .
- Kemmere , M. de Vries , T.J. Vorstman , M.A.G. Keurentjes , J.T.F. Chem. Eng. Sci . 2001 , 56 , 4197 – 4204 .
- Wittmann , K. Wisniewski , W. Mynott , R. Leitner , W. Kranemann , C.L. Rische , T. Eilbracht , P. Kluwer , S. Ernsting , J.M. Elsevier , C.L. Chem. Eur. J. 2001 , 7 , 4584 – 4589 .
- Martin , F. Roelen , O. Feisst , W. US Patent 2127127, August 16, 1938 .
- Hu , Y.L. Chen , W.P. Osuna , A.M.B. Stuart , A.M. Hope , E.G. Xiao , J.L. Chem. Commun . 2001 , 8 , 725 – 726 .
- Yonker , C.R. ; Linehan , J.C. J. Organomet. Chem . 2002 , 650 , 249 – 257 .
- Haji , S. Erkey , C. Tetrahedron 2002 , 58 , 3929 – 3941 .
- Hu , Y. Chen , W. Xu , L.J. Xiao , J.L. Organometallics 2001 20 , 3206 – 3208 .
- Chen , W. Xu , L. Hu , Y. Osuna , A.M.B. Xiao , J. Tetrahedron 2002 , 58 , 3889 – 3899 .
- Kani , I. ; Flores , R. ; Fackler , J.P. ; Akgerman , A. J. Supercrit. Fluids 2004 , 31 , 287 – 294 .
- Erkey , C. Diz , E.L. Suss-Fink , G. Dong , X. Catal. Commun . 2002 , 3 , 213 – 219 .
- Guo , Y. Akgerman , A. Ind. Eng. Chem. Res . 1997 , 36 , 4581 – 4585 ; Guo , Y. ; Akgerman , A. J. Supercrit. Fluids 1999 , 15 , 63 – 71 .
- Chen , M.J. Klingler , R.J. Rathke , J.W. Kramarz , K.W. Organometallics 2004 , 23 , 2701 – 2707 .
- Rathke , J.W. Klingler , R.J. Krause , T.R. Organometallics 1991 , 10 , 1350 – 1355 .
- Jessop , P.G. Ikariya , T. Noyori R. Nature 1994 , 368 , 231 – 233 .
- Kayaki , Y. Suzuki , T. Noguchi , Y. Sakurai , S. Imanari , M. Ikariya , T. Chem. Lett . 2002 , 4 , 424 – 425 .
- Bach , I. Chem. Commun . 1998 , 14 , 1463 – 1464 .
- Sellin , M.F. Bach , I. Webster , J.M. Montilla , F. Rosa , V. Aviles , T. Poliakoff , M. Cole-Hamilton , D.J. J. Chem. Soc., Dalton Trans . 2002 , 24 , 4569 – 4576 .
- Fiddy , S.G. Evans , J. Neisius , T. Sun , X-Z. Jie , Z. George , M.W. Chem. Commun . 2004 , 6 , 676 – 677 .
- Estorach , C.T. ; Orejón , A. ; Masdeu-Bultó , A.M. Green Chem . 2008 , 10 , 545 – 552 .
- Trzeciak , A.M. , Ziólkowski , J.J. and Choukroun , R. 1996 . J. Organomet. Chem. , 525 : 145 – 149 .
- Estorach , C.T. , Orejón , A. and Masdeu-Bultó , A.M. 2008 . Eur. J. Inorg. Chem. , 17 : 2659 – 2663 .
- Monflier , E. , Fremy , G. , Castanet , Y. and Mortreux , A. 1995 . Angew. Chem., Int. Ed. , 34 : 2269 – 2271 .
- Shimizu , S. , Shirakawa , S. , Sasaki , Y. and Hirai , C. 2000 . Angew. Chem., Int. Ed. , 39 : 1256 – 1259 .
- Shirakawa , S. , Shimizu , S. and Sasaki , Y. 2001 . New J. Chem. , 25 : 777 – 779 .
- Palo , D. R. and Erkey , C. 1998 . Ind. Eng. Chem. Res. , 37 : 4203 – 4206 .
- Haar , C.M. , Huang , J. and Nolan , S.P. 1998 . Organometallics , 17 : 5018 – 5024 .
- Smith , D.C. Jr. , Stevens , E.D. and Nolan , S.P. 1999 . Inorg. Chem. , 38 : 5277 – 5281 .
- Bhattacharyya , P. , Gudmunsen , D. , Hope , E.G. , Kemmitt , R.D.W. , Paige , D.R. and Stuart , A.M. 1997 . J. Chem. Soc. Perkin Trans. , 1 : 3609 – 3612 .
- Carter , C.A.G. , Baker , R.T. , Nolan , S.P. and Tumas , W. 2000 . Chem. Commun. , 5 : 347 – 348 .
- Lyubimov , S.E. , Tyutyunov , A.A. , Kalinin , V.N. , Said-Galiev , E.E. , Khokhlov , A.R. , Petrovskii , P.V. and Davankov , V.A. 2007 . Tetrahedron Lett. , 48 : 8217 – 8219 .
- Lyubimov , S.E. , Tyutyunov , A.A. , Kalinin , V.N. , Said-Galiev , E.E. , Khokhlov , A.R. , Petrovskii , P.V. and Davankov , V.A. 2008 . J. Supercrit. Fluids , 45 : 70 – 73 .
- Lyubimov , S.E. , Davankov , V.A. , Said-Galiev , E.E. and Khokhlov , A.R. 2008 . Cat. Comm. , 9 : 1851 – 1852 .
- Beckman , E.J. 2004 . J. Supercrit. Fluids , 28 : 121 – 191 .
- Chen , D-J. , Schmitkamp , M. , Franciò , G. , Klankermayer , J. and Leitner , W. 2008 . Angew. Chem., Int. Ed. , 47 : 7339 – 7341 .
- Kainz , S. ; Koch , D. ; Leitner , W. In Selective Reactions of Metal Activated Molecules ; Werner , H. , Schreier W. Vieweg , Wiesban, , Germany , 1998 ; pp 151 – 156 .
- Kani , I. , Omary , M.A. , Rawashdeh-Omary , M.A. , Lopez-Castillo , Z.K. , Flores , R. , Akgerman , A. and Fackler , J.P. 2002 . Tetrahedron , 58 : 3923 – 3928 .
- Zhao , F. , Fujita , S-I. , Sun , J. , Ikushima , Y. and Arai , M. 2004 . Chem. Commun. , 20 : 2326 – 2327 .
- Lange , S. , Brinkmann , A. , Trautner , P. , Woelk , K. , Bargon , J. and Leitner , W. 2000 . Chirality , 12 : 450 – 457 .
- Hu , Y. , Birdsall , D.J. , Stuart , A.M. , Hope , E.G. and Xiao , J. 2004 . J. Mol. Catal. A: Chem. , 219 : 57 – 60 .
- Wang , S.N. and Kienzle , F. 2000 . Ind. Eng. Chem. Res. , 39 : 4487 – 4490 .
- Jessop , P.G. , Hsiao , Y. , Ikariya , T. and Noyori , R. 1994 . J. Am. Chem. Soc. , 116 : 8851 – 8852 .
- Munshi , P. , Heldebrant , D. , McKoon , E. , Kelly , P.A. , Tai , C-C. and Jessop , P.G. 2003 . Tetrahedron Lett. , 44 : 2725 – 2727 .
- Kröcher , O. , Köppel , R.A. and Baiker , A. 1997 . Chem. Commun. , 5 : 453 – 454 .
- Schmid , L. , Schneider , M.S. , Engel , D. and Baiker , A. 2003 . Catal. Lett. , 88 : 105 – 113 .
- Schneider , M.S. , Grunwaldt , J-D. , Burgi , T. and Baiker , A. 2003 . Rev. Sci. Instrum. , 74 : 4121 – 4128 .
- Ting , S.S.T. , Macnaughton , S.J. , Tomasko , D.L. and Foster , N.R. 1993 . Ind. Eng. Chem. Res. , 32 : 1471 – 1481 .
- Dobbs , J.M. , Wong , J.M. , Lahiere , R.J. and Johnston , K.P. 1987 . Ind. Eng. Chem. Res. , 26 : 56 – 65 .
- Xiao , J-L. , Nefkens , S.C.A. , Jessop , P.G. , Ikariya , T. and Noyori , R. 1996 . Tetrahedron Lett. , 37 : 2813 – 2816 .
- Jessop , P.G. , Stanley , R.R. , Brown , R.A. , Eckert , C.A. , Liotta , C.L. , Ngo , T.T. and Pollet , P. 2003 . Green Chem. , 5 : 123 – 128 .
- Dong , X. and Erkey , C. 2004 . J. Mol. Catal. A: Chem. , 211 : 73 – 81 .
- Berthod , M. , Mignani , G. and Lemaire , M. 2004 . Tetrahedron Asym. , 15 : 1121 – 1126 .
- Adams , D.J. , Chen , W. , Hope , E.G. , Lange , S. , Stuart , A.M. , West , A. and Xiao , J. 2003 . Green Chem. , 5 : 118 – 122 .
- Noyori , R. Asymmetric Catalysis in Organic Synthesis ; Wiley : New York , 1994 ; chapter 2 , pp 16 – 94
- Johansson , A. 1995 . Contemp. Org. Synth. , 2 : 393 – 407 .
- Margitfalvi , J.L. . In Catalysis of Organic Reactions, Chemical Industries Series ; Scaros M.G. , Prunier M.L. Dekker , New York , 1995 62 189 – 200 .
- Kainz , S. , Brinkmann , A. , Leitner , W. and Pfaltz , A. 1999 . J. Am. Chem. Soc. , 121 : 6421 – 6429 .
- Breslow , R . Acc. Chem. Res . 2004 , 37 , 471 – 478 ; Breslow , R. Acc. Chem. Res . 1991 , 24 , 159 – 164 .
- Grieco , P.A. Aldrichimica Acta 1991 , 24 , 59 – 66 ; Grieco , P.A. Organic Synthesis in Water ; Thomson Science : Glasgow, , UK , 1998 .
- Grubbs , R.H. 2003 . Handbook of Metathesis Catalysts , Weinheim, , Germany : Wiley-VCH .
- Novak , B.M. and Grubbs , R.H. 1988 . J. Am. Chem. Soc. , 110 : 7542 – 7543 .
- Nguyen , S.B.T. , Johnson , L.K. , Grubbs , R.H. and Ziller , J.W. 1992 . J. Am. Chem. Soc. , 114 : 3974 – 3975 .
- Narayan , S. , Muldoon , J. , Finn , M.G. , Fokin , V.V. , Kolb , H.C. and Sharpless , K.B. 2005 . Angew. Chem., Int. Ed. , 44 : 3275 – 3279 .
- Kobayashi , S. ; Manabe , K. Acc. Chem. Res . 2002 , 35 , 209 – 217 ; Kobayashi , S. Org. Syn. Water 1998 , 262 – 305 .
- Li , C-J. and Chan , T.H. 1997 . Organic Reactions in Aqueous Media , New York : John Wiley & Sons .
- Li , C-J. Angew. Chem., Int. Ed . 2003 , 42 , 4856 – 4858 ; Li , C-J. Acc. Chem. Res . 2002 , 35 , 533 – 538 .
- Skouta , R. , Varma , R.S. and Li , C-J. 2005 . Green Chem. , 7 : 571 – 575 .
- Cornils , B. , Herrmann , W.A. . Aqueous-Phase Organometallic Catalysis, Concepts and Applications ; Weinheim, , Germany : Wiley-VCH , 1996 .
- Sijbren , O. and Engberts , J.B.F.N. 2003 . Org. Biomol. Chem. , 1 : 2809 – 2820 .
- Okuhara , T. 2002 . Chem. Rev. , 102 : 3641 – 3665 .
- Kobayashi , S. and Manabe , K. 2002 . Acc. Chem. Res. , 35 : 209 – 217 .
- Uozumi , Y. and Shibatomi , K. 2001 . J. Am. Chem. Soc. , 123 : 2919 – 2920 .
- Lubineau , A. , Auge , J. and Queneau , Y. 1994 . Synthesis , 8 : 741 – 760 .
- Reissig , H.U. Org. Synth. Highlights 1991 , 71 – 76 .
- Engberts , J.B.F.N. 1982 . Pure Appl. Chem. , 54 : 1797 – 1808 .
- Pinault , N. and Bruce , D.W. 2003 . Coord. Chem. Rev. , 241 : 1 – 25 .
- Katti , K.V. , Gali , H. , Smith , C.J. and Berning , D.E. 1999 . Acc. Chem. Res. , 32 : 9 – 17 .
- Amengual , R. , Genin , E. , Michelet , V. , Savignac , M. and Genêt , J-P. 2002 . Adv. Synth. Catal. , 344 : 393 – 398 .
- Lopez-Deber , M.P. , Castedo , L. and Granja , J.R. 2001 . Org. Lett. , 3 : 2823 – 2826 .
- Genêt , J-P. and Savignac , M. 1999 . J. Organomet. Chem. , 576 : 305 – 317 .
- Mori , A. , Ahmed , M.S.M. , Sekiguchi , A. , Masui , K. and Koike , T. 2002 . Chem. Lett. , 7 : 756 – 757 .
- Bumagin , N.A. , Sukhomlinova , L.I. , Luzikova , E.V. , Tolstaya , T.P. and Beletskaya , I.P. 1996 . Tetrahedron Lett. , 37 : 897 – 900 .
- Bleicher , L. and Cosford , N.D.P. 1995 . Synlett , 11 : 1115 – 1116 .
- Amatore , C. , Blart , E. , Genêt , J-P. , Jutant , A. , Lemaire-Audoire , S. and Savignac , M. 1995 . J. Org. Chem. , 60 : 6829 – 6839 .
- Casalnuovo , A.L. and Calabrese , J.C. 1990 . J. Am. Chem. Soc. , 112 : 4324 – 4330 .
- Dibowski , H. and Schmidtchen , F.P. 1998 . Tetrahedron Lett. , 39 : 525 – 528 .
- Yokoyama , Y. , Hikawa , H. , Mitsuhashi , M. , Uyama , A. and Murakami , Y. 1999 . Tetrahedron Lett. , 40 : 7803 – 7806 .
- Sonogashira , K. , Tohda , Y. and Hagihara , N. 1975 . Tetrahedron Lett. , 16 : 4467 – 4470 .
- Sonogashira , K. In Metal Catalyzed Cross-Coupling Reactions ; Diederich , F. , Stang , P.J. Wiley-VCH : Weinheim, , Germany , 1998 ; Chapter 5 .
- Chanteau , S.H. and Tour , J.M. 2001 . Tetrahedron Lett. , 42 : 3057 – 3060 .
- Pugh , V.J. , Hu , Q-S. , Zuo , X. , Lewis , F.D. and Pu , L. 2001 . J. Org. Chem. , 66 : 6136 – 6140 .
- Arterburn , J.B. , Venkateswara , K. , Perry , R. and Perry , M.C. 2000 . Tetrahedron Lett. , 41 : 839 – 842 .
- Nicolaou , K.C. and Dai , W-M. 1991 . Angew. Chem. Int. Ed. , 30 : 1387 – 1416 .
- Inouye , M. , Takahashi , K. and Nakazumi , H. 1999 . J. Am. Chem. Soc. , 121 : 341 – 345 .
- Liang , B. , Dai , M. , Chen , J. and Yang , Z. 2005 . J. Org. Chem. , 70 : 391 – 393 .
- Pal , M. , Subramanian , V. and Yeleswarapu , K.R. 2003 . Tetrahedron Lett. , 44 : 8221 – 8225 .
- Uozumi , Y. and Kobayashi , Y. 2003 . Heterocycles , 59 : 71 – 74 .
- Kundu , N.G. , Pal , M. , Mahanty , J.S. and De , M. 1997 . J. Chem. Soc. Perkin Trans. , 1 : 2815 – 2820 .
- Kennis , L.E.J. , Bischoff , F.P. , Mertens , C.J. , Love , C.J. , Van den Keybus , F.A.F. , Pieters , S. , Braeken , M. , Megens , A.A.H.P. and Leysen , J.E. 2000 . Bioorg. Med. Chem. Lett. , 10 : 71 – 74 .
- Chen , L. and Li , C-J. 2004 . Org. Lett. , 6 : 3151 – 3153 .
- Wu , W. and Li , C-J. 2003 . Chem. Commun. , 14 : 1668 – 1669 .
- Gan , K-H. , Jhong , C-J. , Shue , Y-J. and Yang , S-C. 2008 . Tetrahedron Lett. , 64 : 9625 – 9629 .
- Rosas , N. , Sharma , P. , Alvarez , C. , Cabrera , A. , Ramirez , R. , Delgado , A. and Arzoumanian , H. 2001 . J. Chem. Soc. Perkin , 1 : 2341 – 2343 .
- Son , S.U. , Lee , S.I. , Chung , Y.K. , Kim , S-W. and Hyeon , T. 2002 . Org. Lett. , 4 : 277 – 279 .
- Krafft , M.E. , Wright , J.A. and Bonaga , L.V.R. 2003 . Tetrahedron Lett. , 44 : 3417 – 3422 .
- Fuji , K. , Morimoto , T. , Tsutsumi , K. and Kakiuchi , K. 2003 . Angew. Chem., Int. Ed. , 42 : 2409 – 2411 .
- Suh , W.H. , Choi , M. , Lee , S.I. and Chung , Y.K. 2003 . Synthesis , 14 : 2169 – 2172 .
- Lautens , M. , Roy , A. , Fukuoka , K. , Fagnou , K. and Martn-Matute , B. 2001 . J. Am. Chem. Soc. , 123 : 5358 – 5359 .
- Amengual , R. , Michelet , V. and Genêt , J-P. 2002 . Tetrahedron Lett. , 43 : 5905 – 5908 .
- Li , C.J. , Wang , D. and Chen , D.L. 1995 . J. Am. Chem. Soc. , 117 : 12867 – 12868 .
- Wang , M.W. and Li , C-J. 2002 . Tetrahedron Lett. , 43 : 3589 – 3591 .
- Wei , C.M. and Li , C-J. 2002 . Green Chem. , 4 : 39 – 41 .
- Li , C.J. and Wei , C.M. 2002 . Chem. Commun. , 3 : 268 – 269 .
- Evans , D.A. , Kozlowski , M.C. , Murry , J.A. , Burgey , C.S. , Campos , K.R. , Connell , B.T. and Staples , R.J. 1999 . J. Am. Chem. Soc. , 121 : 669 – 685 .
- Fache , F. , Schulz , E. , Tommasino , M.L. and Lemaire , M. 2000 . Chem. Rev. , 100 : 2159 – 2231 .
- Ghosh , A.K. , Mathivanan , P. and Cappiello , J. 1998 . Tetrahedron: Asymm. , 9 : 1 – 45 .
- Nishiyama , H. 1991 . Organometallics , 10 : 500 – 508 .
- Wei , C.M. and Li , C-J. 2002 . J. Am. Chem. Soc. , 124 : 5638 – 5639 .
- Wei , C.M. , Mague , J.T. and Li , C-J. 2004 . Proc. Natl. Acad. Sci. USA , 101 : 5749 – 5754 .
- Rosa , J.N. , Santos , A.G. and Afonso , C.A.M. 2004 . J. Mol. Catal. A , 214 : 161 – 165 .
- Shi , L. , Tu , Y-Q. , Wang , M. , Zhang , F-M. and Fan , C-A. 2004 . Org. Lett. , 6 : 1001 – 1003 .
- Yamamoto , Y. 2008 . Chem. Rev. , 108 : 3199 – 3222 .
- Weibel , J-M. , Blanc , A. and Pale , P. 2008 . Chem. Rev. , 108 : 3149 – 3173 .
- Halbes-Letinois , U. , Weibel , J-M. and Pale , P. 2007 . Chem. Soc. Rev. , 36 : 759 – 769 .
- Li , Z. and He , C. 2006 . Eur. J. Org. Chem. , 19 : 4313 – 4322 .
- Wei , C.M. , Li , Z.G. and Li , C-J. 2003 . Org. Lett. , 5 : 4473 – 4475 .
- Yao , X. and Li , C-J. 2005 . Org. Lett. , 7 : 4395 – 4398 .
- Deng , G-J. and Li , C-J. 2008 . Synlett , 10 : 1571 – 1573 .
- Boumendjel , A. 2003 . Curr. Med. Chem. , 10 : 2621 – 2630 .
- Morimoto , M. , Fukumoto , H. , Nozoe , T. , Hagiwara , A. and Komai , K. 2007 . J. Agric. Food Chem. , 55 : 700 – 705 .
- Okombi , S. , Rival , D. , Bonnet , S. , Mariotte , A-M. , Perrier , E. and Boumendjel , A. 2006 . J. Med. Chem. , 49 : 329 – 333 .
- Yu , M. , Skouta , R. , Zhou , L. , Jiang , H-F. , Yao , X. and Li , C-J. 2009 . J. Org. Chem. , 74 : 3378 – 3383 .
- Cozzi , P.G. , Hilgraf , N. and Zimmermann , N. 2004 . Eur. J. Org. Chem. , 20 : 4095 – 4105 .
- Prajapati , D. , Laskar , D.D. , Gogoi , B.J. and Devi , G. 2003 . Tetrahedron Lett. , 44 : 6755 – 6757 .
- Wei , C. and Li , C-J. 2003 . J. Am. Chem. Soc. , 125 : 9584 – 9585 .
- Vicente , J. , Chicote , M.T. and Abrisqueta , M.D. 1995 . J. Chem. Soc. Dalton Trans. , 3 : 497 – 498 .
- Huang , B. , Yao , X. and Li , C-J. 2006 . Adv. Synth. Catal. , 348 : 1528 – 1532 .
- Lo , V.K-Y. , Liu , Y. , Wong , M-K. and Che , C-M. 2006 . Org. Lett. , 8 : 1529 – 1532 .
- Barnholtz , S.L. , Lydon , J.D. , Huang , G. , Venkatesh , M. , Barnes , C.L. , Ketring , A.R. and Jurisson , S.S. 2001 . Inorg. Chem. , 40 : 972 – 976 .
- Yan , B. and Liu , Y. 2007 . Org. Lett. , 9 : 4323 – 4326 .
- Welton , T. Chem. Rev . 1999 , 99 , 2071 – 2083 ; Welton , T. Coord. Chem. Rev . 2004 , 248 , 2459 – 2477 .
- Bradley , D. , Dyson , P. and Welton , T. 2000 . Chem. Rev. , 9 : 18 – 21 .
- Visser , A.E. , Swatloski , R.P. , Reichert , W.M. , Griffin , S.T. and Rogers , R.D. 2000 . Ind. Eng. Chem. Res. , 39 : 3596 – 3604 .
- Zhang , Y. , Bakshi , B. and Demessie , E.S. 2008 . Env. Sci. Tech. , 42 : 1724 – 1730 .
- Boon , J.A. , Levinsky , J.A. , Pflug , J.I. and Wilkes , J.S. 1986 . J. Org. Chem. , 51 : 480 – 483 .
- Wasserscheid , P. and Keim , W. 2000 . Angew. Chem., Int. Ed. , 39 : 3772 – 3789 .
- Sheldon , R.A. Chem. Commun . 2001 , 2399 – 2407 and references therein .
- Wilkes , J.S. , Levisky , J.A. , Wilson , R.A. and Hussey , C.L. 1982 . Inorg. Chem. , 21 : 1263 – 1264 .
- Fannin , A.A. Jr. , King , L.A. , Levisky , J.A. and Wilkes , J.S. 1984 . J. Phys. Chem. , 88 : 2609 – 2614 .
- Laher , T.M. and Hussey , C.L. 1983 . Inorg. Chem. , 22 : 1279 – 1283 .
- Cai , Y. , Peng , Y. and Song , G. 2006 . Catal. Lett. , 109 : 61 – 64 .
- Hough-Troutman , W.L. , Smiglak , M. , Griffin , S. , Reichert , W.M. , Mirska , I. , Jodynis Liebert , J. , Adamska , T. , Nawrot , J. , Stasiewicz , M. , Rogers , R.D. and Pernak , J. 2009 . New J. Chem. , 33 : 26 – 33 .
- Abbott , A.P. , Capper , G. , Davies , D.L. , Munro , H.L. , Rasheed , R.K. and Tambyrajah , V. 2001 . Chem. Commun. , 19 : 2010 – 2011 .
- Abbott , A.P. ; Capper , G. ; Davies , D.L. ; Rasheed , R.K. ; Tambyrajah , V. Green Chem . 2002 , 4 , 24 – 26 ; Abbott , A.P. ; Capper , G. ; Davies , D.L. ; Rasheed , R.K. ; Tambyrajah , V. Chem. Commun . 2003 , 1 , 70 – 71 .
- Wasserscheid , P. , Boesmann , A. and Bolm , C. 2002 . Chem. Commun. , 3 : 200 – 201 .
- Handy , S.T. , Okello , M. and Dickenson , G. 2003 . Org. Lett. , 5 : 2513 – 2515 .
- Carmichael , A.J. , Earle , M.J. , Holbrey , J.D. , McCormac , P.B. and Seddon , K.R. 1999 . Org. Lett. , 1 : 997 – 1000 .
- Reetz , M.T. and Westermann , E. 2000 . Angew. Chem., Int. Ed. , 39 : 165 – 168 .
- Hamill , N.A. , Hardacre , C. and McMath , S.E. 2002 . Green Chem. , 4 : 139 – 142 .
- Deshmukh , R.R. , Rajagopal , R. and Srinivasan , K.V. 2001 . Chem. Commun. , 17 : 1544 – 1545 .
- Böhm , V.P.W. and Herrmann , W.A. 2000 . Chem. Eur. J. , 6 : 1017 – 1025 .
- Selvakumar , K. , Zapf , A. and Beller , M. 2002 . Org. Lett. , 4 : 3031 – 3033 .
- Park , S.B. and Alper , H. 2003 . Org. Lett. , 5 : 3209 – 3212 .
- Li , S. , Lin , Y. , Xie , H. , Zhang , S. and Xu , J. 2006 . Org. Lett. , 8 : 391 – 394 .
- Xie , X.G. , Lu , J.P. , Chen , B. , Han , J.J. , She , X.G. and Pan , X.F. 2004 . Tetrahedron Lett. , 45 : 809 – 811 .
- Hagiwara , H. , Shimizu , Y. , Hoshi , T. , Suzuki , T. , Ando , M. , Ohkubo , K. and Yokoyama , C. 2001 . Tetrahedron Lett. , 42 : 4349 – 4351 .
- Okubo , K. , Shirai , M. and Yokoyama , C. 2002 . Tetrahedron Lett. , 43 : 7115 – 7118 .
- Forsyth , S.A. , Gunaratne , H.Q.N. , Hardacre , C. , McKeown , A. , Rooney , D.W. and Seddon , K.R. 2005 . J. Mol. Catal. A , 231 : 61 – 66 .
- Mathews , C.J. ; Smith , P.J. ; Welton , T. J. Mol. Catal. A 2003 , 206 , 77 – 82 ; Mathews , C.J. ; Smith , P.J. ; Welton , T. J. Mol. Catal. A 2004 , 214 , 27 – 32 .
- McLachlan , F. , Mathews , C.J. , Smith , P.J. and Welton , T. 2003 . Organometallics , 22 : 5350 – 5357 .
- Wang , R. , Twamley , B. and Shreeve , J.M. 2006 . J. Org. Chem. , 71 : 426 – 429 .
- Jin , C-M. , Twamley , B. and Shreeve , J.M. 2005 . Organometallics , 24 : 3020 – 3023 .
- Xiao , J-C. , Twamley , B. and Shreeve , J.M. 2004 . Org. Lett. , 6 : 3845 – 3847 .
- Chen , W. , Xu , L. , Chatterton , C. and Xiao , J. 1999 . Chem. Commun. , 13 : 1247 – 1248 .
- de Bellefon , C. , Pollet , E. and Grenouillet , P. 1999 . J. Mol. Catal. A , 145 : 121 – 126 .
- Cacchi , S. , Fabrizi , G. and Goggiamani , A. 2004 . J. Mol. Catal. A , 214 : 57 – 64 .
- Mo , J. and Xiao , J-L. 2006 . Angew. Chem., Int. Ed. , 45 : 4152 – 4157 .
- Mo , J. , Xu , J-L. and Xiao , L-J. 2005 . J. Am. Chem. Soc. , 127 : 751 – 760 .
- Xu , L-J. , Chen , W-P. , Ross , J. and Xiao , J-L. 2001 . Org. Lett. , 3 : 295 – 297 .
- Xie , X. , Chen , B. , Lu , J. , Han , J. , She , X. and Pan , X. 2004 . Tetrahedron Lett. , 45 : 6235 – 6237 .
- Handy , S.T. and Zhang , X. 2001 . Org. Lett. , 3 : 233 – 236 .
- Calò , V. , Giannoccaro , P. , Nacci , A. and Monopoli , A. 2002 . J. Organomet. Chem. , 645 : 152 – 157 .
- Calò , V. , Nacci , A. , Lopez , L. and Mannarini , N. 2000 . Tetrahedron Lett. , 41 : 8973 – 8976 .
- Calò , V. , Nacci , A. , Monopoli , A. , Lopez , L. and di Cosmo , A. 2001 . Tetrahedron , 57 : 6071 – 6077 .
- Calò , V. , Nacci , A. , Lopez , L. and Napola , A. 2001 . Tetrahedron Lett. , 42 : 4701 – 4703 .
- Shi , F. , He , Y. , Li , D. , Ma , Y. , Zhang , Q. and Deng , Y. 2006 . J. Mol. Catal. A , 244 : 64 – 67 .
- Tietze , L.F. 1996 . Chem. Rev. , 96 : 115 – 136 .
- Linghu , X. , Nicewicz , D.A. and Johnson , J.S. 2002 . Org. Lett. , 4 : 2957 – 2960 .
- Baidossi , M. , Joshi , A.V. , Mukhopadhyay , S. and Sasson , Y. 2005 . Tetrahedron Lett. , 46 : 1885 – 1887 .
- Trost , B.M. ; Verhoeven , T.R. In Comprehensive Organometallic Chemistry ; Willkinson , G. , Stone , F.G.A. , Abel , E.W. Oxford : Pergamon , 1982 ; 8 789 – 938 .
- March , J. 1992 . Advanced Organic Chemistry , 889 – 890 . New York : Wiley .
- Hutchins , R.O. ; Hutchins , M.K. In Comprehensive Organic Synthesis ; Trost , B.M. , Fleming , I. Oxford : Pergamon , 1991 ; 8 25 – 78 .
- Imao , D. , Fujihara , S. , Yamamoto , T. , Ohta , T. and Ito , Y. 2005 . Tetrahedron , 61 : 6988 – 6992 .
- Fonseca , G.S. , Scholten , J.D. and Dupont , J. 2004 . Synlett , 9 : 1525 – 1528 .
- Giernoth , R. and Krumm , M.S. 2004 . Adv. Synth. Catal. , 346 : 989 – 992 .
- Chauvin , Y. , Mussmann , L. and Olivier , H. 1995 . Angew. Chem., Int. Ed. , 34 : 2698 – 2700 .
- Keim , W. , Vogt , D. , Waffenschmidt , H. and Wasserscheid , P. 1999 . J. Catal. , 186 : 481 – 484 .
- Wasserscheid , P. , Waffenschmidt , H. , Machnizti , P. , Kottsieper , K.W. and Stelzer , O. 2001 . Chem. Commun. , 5 : 451 – 452 .
- Brasse , C.C. , Englert , U. , Salzer , A. , Waffenschmidt , H. and Wasserscheid , P. 2000 . Organometallics , 19 : 3818 – 3823 .
- Petricci , E. , Mann , A. , Schoenfelder , A. , Rota , A. and Taddei , M. 2006 . Org. Lett. , 8 : 3725 – 3727 .
- Leadbeater , N.E. and Torenius , H.M. 2002 . J. Org. Chem. , 67 : 3145 – 3148 .
- Mehnert , C.P. , Cook , R.A. , Dispenziere , N.C. and Afeworki , M. 2002 . J. Am. Chem. Soc. , 124 : 12932 – 12933 .
- Riisager , A. , Fehrmann , R. , Flicker , S. , van Hal , R. , Haumann , M. and Wasserscheid , P. 2005 . Angew. Chem., Int. Ed. , 44 : 815 – 819 .
- Riisager , A. , Eriksen , K.M. , Wasserscheid , P. and Fehrmann , R. 2003 . Catal. Lett. , 90 : 149 – 153 .
- Riisager , A. , Wasserscheid , P. , van Hal , R. and Fehrmann , R. 2003 . J. Catal. , 219 : 452 – 455 .
- Yang , Y. , Deng , C. and Yuan , Y. 2005 . J. Catal. , 232 : 108 – 116 .
- Tominaga , K-I. and Sasaki , Y. 2004 . Chem. Lett. , 33 : 14 – 15 .
- Oonishi , Y. , Saito , A. and Sato , Y. 2009 . Green Chem. , 11 : 330 – 333 .
- Mastrorilli , P. , Nobile , C.F. , Gallo , V. , Suranna , G.P. and Farinola , G. 2002 . J. Mol. Catal. A , 184 : 73 – 78 .
- Muthusamy , S. and Gnanaprakasam , B. 2005 . Tetrahedron , 61 : 1309 – 1315 .
- Lam , K.H. , Xu , L. , Feng , L. , Ruan , J. , Fan , Q. and Chan , A.S.C. 2005 . Can. J. Chem. , 83 : 903 – 908 .
- Xiong , W. , Lin , Q. , Ma , H. , Zheng , H. , Chena , H. and Li , X. 2005 . Tetrahedron: Asymm. , 16 : 1959 – 1962 .
- Berthod , M. , Joerger , J-M. , Mignani , G. , Vaultier , M. and Lemaire , M. 2004 . Tetrahedron: Asymm. , 15 : 2219 – 2221 .
- Hu , A. , Ngo , H.L. and Lin , W. 2004 . Angew. Chem., Int. Ed. , 43 : 2501 – 2504 .
- Ngo , H.L. , Hu , A. and Lin , W. 2003 . Chem. Commun. , 15 : 1912 – 1913 .
- Yinghuai , Z. , Carpenter , K. , Bun , C-C. , Bahnmueller , S. , Ke , C-P. , Shanmugham Srid , V. , Kee , L-W. and Hawthorne , M.F. 2003 . Angew. Chem., Int. Ed. , 42 : 3792 – 3795 .
- Yao , Q. and Sheets , M. 2005 . J. Organomet. Chem. , 690 : 3577 – 3584 .
- Yao , Q. and Zhang , Y. 2003 . Angew. Chem., Int. Ed. , 42 : 3395 – 3398 .
- Buijsman , R.C. , van Vuuren , E. and Sterrenburg , J.G. 2001 . Org. Lett. , 3 : 3785 – 3787 .
- Semeril , D. , Olivier-Bourgbigou , H. , Bruneau , C. and Dixneuf , P.H. 2002 . Chem. Commun. , 2 : 146 – 147 .
- Clavier , H. ; Audic , N. ; Guillemin , J-C. ; Mauduit , M. J. Am. Chem. Soc . 2003 , 125 , 9248 – 9249 ; Clavier , H. ; Audic , N. ; Guillemin , J-C. ; Mauduit , M. J. Organomet.Chem . 2005 , 690 , 3585 – 3599 .
- Yang , X-F. , Wang , M. , Varma , R.S. and Li , C-J. 2004 . J. Mol. Catal. A , 214 : 147 – 154 .
- Conte , V. , Elakkari , E. , Floris , B. , Mirruzzo , V. and Tagliatesta , P. 2005 . Chem. Commun. , 12 : 1587 – 1588 .
- Ansari , I.A. and Gree , R. 2002 . Org. Lett. , 4 : 1507 – 1509 .
- Wu , X-E. ; Ma , L. ; Ding , M-X. ; Gao , L-X. Synlett 2005 , 4 , 607 – 610 ; Wu , X-E. ; Ma , L. ; Ding , M-X. ; Gao , L-X. Chem. Lett . 2005 , 34 , 312 – 313 .
- Rostovtsev , V.V. , Green , L.G. , Fokin , V.V. and Sharpless , K.B. 2002 . Angew. Chem., Int. Ed. , 41 : 2596 – 2599 .
- Torn⊘e , C.W. , Christensen , C. and Meldal , M. 2002 . J. Org. Chem. , 67 : 3057 – 3062 .
- Marra , A. , Vecchi , A. , Chiappe , C. , Melai , B. and Dondoni , A. 2008 . J. Org. Chem. , 73 : 2458 – 2461 .
- Vecchi , A. , Melai , B. , Marra , A. , Chiappe , C. and Dondoni , A. 2008 . J. Org. Chem. , 73 : 6437 – 6440 .
- Dondoni , A. and Marra , A. 2007 . Tetrahedron Lett. , 63 : 6339 – 6345 .
- Doherty , S. , Goodrich , P. , Hardacre , C. , Knight , J.G. , Nguyen , M.T. , Pârvulescu , V.I. and Paun , C. 2007 . Adv. Synth. Catal. , 349 : 951 – 963 .
- Sheldon , R.A. 1994 . Chem. Tech. , 24 : 38 – 47 .
- Chan , T.H. and Li , C-J. 1992 . J. Chem. Soc. Chem. Commun. , 10 : 747 – 748 .
- Baran , P.S. , Maimone , T.J. and Richter , J.M. 2007 . Nature , 446 : 404 – 408 .
- Earle , M.J. , Esperanca , J.M.S.S. , Gilea , M.A. , Canongia Lopes , J.N. , Rebelo , L.P.N. , Magee , J.W. , Seddon , K.R. and Widegren , J.A. 2006 . Nature , 439 : 831 – 834 .
- Torrecilla , J.S. , Garcia , J. , Rojo , E. and Rodriguez , F. 2009 . J. Hazard. Mater. , 164 : 182 – 194 .
- Studzinska , S. and Buszewski , B. 2009 . Anal. Bioanal. Chem. , 393 : 983 – 990 .
- Bailey , M.M. , Townsend , M.B. , Jernigan , P.L. , Sturdivant , J. , Hough-Troutman , W.L. , Rasco , J.F. , Swatloski , R.P. , Rogers , R.D. and Hood , R.D. 2008 . Green Chem. , 10 : 1213 – 1217 .
- Stolte , S. , Matzke , M. , Arning , J. , Boeschen , A. , Pitner , W-R. , Welz-Biermann , U. , Jastorff , B. and Ranke , J. 2007 . Green Chem. , 9 : 1170 – 1179 .
- Zhao , D. , Liao , Y. and Zhang , Z. 2007 . Clean , 35 : 42 – 48 .
- Pretti , C. , Chiappe , C. , Pieraccini , D. , Gregori , M. , Abramo , F. , Monni , G. and Intorre , L. 2006 . Green Chem. , 8 : 238 – 240 .
- Matsumoto , M. , Mochiduki , K. and Kondo , K.J. 2004 . Biosci. Bioeng. , 98 : 344 – 347 .
- Chai , D.I. and Lautens , M. 2009 . J. Org. Chem. , 74 : 3054 – 3061 .