Abstract
A simple, efficient and environmentally friendly process for synthesis of 3,4-dihydropyrimidinones using a one-pot three-component condensation of aromatic aldehydes, β-dicarbonyl compounds, and urea/thiourea is discussed. The cost-effective, new generation ionic liquid (IL), tri-(2-hydroxyethyl) ammonium acetate was used as both solvent and catalyst. The reactions were carried out using both conventional heating and microwave energy. Application of IL technology with microwave energy represents an attractive and rapid alternative to the conventional acid–base catalyzed thermal processes.
Introduction
3, 4-Dihydropyrimidin-2(1H)-ones and their derivatives have been reported to possess remarkable therapeutic and pharmacological activities such as antibacterial, antiviral, anti-inflammatory, and anticancer properties Citation1–4. Because of these diverse pharmacological properties, continuous efforts are being made to improve this reaction in terms of yields, reaction rate, and nature of catalysts being used. The initial synthesis of dihydropyrimidinones via Biginelli condensation gives only 20–45% yield Citation5. This has led to the development of higher yielding multistep synthetic strategies for synthesis of dihydropyrimidinones. However, these lack the efficiency and simplicity of one-step and one-pot synthetic procedures Citation6 Citation7. Thus, the Biginelli reaction has received renewed interest and several improved protocols for synthesis of dihydropyrimidinones and has been reported recently in literature.
Microwave synthesis Citation8 Citation9, ultrasound irradiation Citation10 Citation11, and use of acid catalysts have been tried. Various catalysts such as triflates Citation12–14, metal bromides Citation15, strontium (II) nitrate Citation16, metal triflimides Citation17, polystyrenesulfonic acid Citation18, pyrazolidine dihydrochloride Citation19, ion exchange resins Citation20, polymer based solid acid Citation21, and L-proline Citation22 have been used to replace the strong acid used in the classical Biginelli reaction. However, many of these methods have limitations such as use of fairly high amounts of expensive catalysts, long reaction times, tedious procedures for product isolation, and also some could be environmentally hazardous. Therefore, the search for readily available and environmentally friendly catalysts is still being actively pursued.
Ionic liquids (ILs) now have emerged as powerful alternatives to conventional organic solvents and catalysts due to some of their properties such as negligible vapor pressure, thermal stability, wide range of solubility and ease of recovery and reuse. Imidazole-based ILs such as [bmim]BF4 and [bmim]PF6 have been used as catalysts for the synthesis of dihydropyrimidinones Citation23. However, imidazole-based ILs containing BF4 and PF6 anions are environmentally unsafe as they liberate hazardous HF gas. In addition, their high costs as well as problems in disposability make their use limited. Many cost-effective alkanolamine-based ILs have recently been identified Citation24. The application and catalytic potential of these alkanolamine-based ILs for environmentally friendly organic synthesis has been reported Citation25 Citation26.
Use of microwave energy has become popular in synthetic organic chemistry in order to improve classical organic reactions, shortening reaction times, and improving yields Citation27–30. This can be coupled with solvent free reactions or IL mediated reactions, to make the synthesis more environmentally friendly.
In the present method, an easily synthesizable, cost-effective IL, tri-(2-hydroxyethyl) ammonium acetate was used for the synthesis of dihydropyrimidinones using conventional heating and microwave irradiation.
Results and discussion
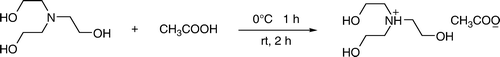
In our research, focusing toward mild, economical, and green route for the synthesis of libraries of novel compounds such as heterocycles of biological importance, we proposed to make use of an easily synthesizable IL (). For the first time, tri-(2-hyroxyethyl) ammonium acetate was utilized to replace imidazole-based ILs for the synthesis of dihydropyrimidinones under solvent free and neutral conditions.
To standardize the requirement of IL in the reaction, several experiments were carried out as depicted in . Increasing the amount of IL from 0.2 to 1 g for a 1 g batch of benzaldehyde led to increase in yields from 60 to 88%. Further increase did not change yields. The reaction did not go to completion in the absence of IL. Variation of temperature from room temperature (25°C) to 120°C resulted in identification of 90°C as the optimum temperature for this reaction.
Table 1. Effect of amount of ionic liquid on the yield of products.a
With these optimized parameters, reactions for synthesis of different dihydropyrimidinones were carried out (). The results of IL catalyzed Biginelli reactions are presented in . Yields obtained using urea as substrates were between 70 and 88% (isolated yields) using conventional heating. In case of thiourea, it was observed that the yields obtained were slightly lower as compared to urea. An important feature of this procedure is that wide variety of functional groups such as hydroxyl, halogen, alkoxy, alkyl, nitro, etc. on the aromatic aldehyde as well as α, β-unsaturated aldehyde (cinnamaldehyde) can be used under these reaction conditions.
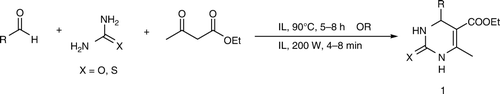
Table 2. Ionic liquid catalyzed Biginelli reactions under conventional heatinga and microwave irradiationb.
The reactions were also studied using the combination of microwave irradiation and IL. Use of microwave energy dramatically decreased reaction times from 5–8 h to 4–8 min as shown in . Isolated yields of all the compounds were slightly improved. Thus, this method offers a simple and easy access to substituted dihydropyrimidinones with a variety of substitution patterns in a time efficient manner.
The IL was recovered by removing water under reduced pressure and could be reused without significant loss of activity. As the reactions were performed on small scale using 1 g IL per reaction, this led to loss of about 5–10% of IL per experiment, which was further compensated by addition of fresh IL. The reusability of IL was quantified by performing a set of experiments () using benzaldehyde, urea, and ethyl acetoacetate. The IL was recovered and reused for three times.
Table 3. Results for recyclability of ionic liquid.
Experimental
Material and methods
The IL was prepared by previously reported method without any modifications and characterized by FTIR, 1H NMR, and 13C NMR spectroscopy. The reagents and solvents were commercially available. All reactions were conducted in parallel synthesizer using similar reaction conditions (Metler Tolledo™). The CEM-Discover Lab-Mate microwave was used for synthesis. All synthesized compounds are known and identified by spectroscopic data. FTIR spectra were obtained on a Perkin–Elmer infrared spectrometer with KBr discs and 1H NMR spectra were recorded in CDCl3 or DMSO-D6 on a JEOL 300 MHz spectrometer with tetramethylsilane as internal standard. Melting points were determined on a Kofler melting point apparatus and uncorrected. Thin-layer chromatography (TLC) was accomplished on 0.2 mm precoated plates of silica gel 60 F-254 (Merck).
Typical procedure for the synthesis of 5-ethoxycarbonyl-4-phenyl-6-methyl-3,4-dihydropyrimidin-2(1H)-one (1a) under conventional heating
Benzaldehyde (1 g, 0.00768 mol), ethyl acetoacetate (0.98 g, 0.00768 mol), urea (0.6 g, 0.00998 mol), and 1g of IL were charged to a 20 ml reactor. Reaction mixture was heated at 90°C for the appropriate time as mentioned in . After the completion of reaction, as indicated by TLC, the reaction mixture was poured onto crushed ice and stirred for 10–15 min. The yellow solid separated was filtered under suction and washed with ice-cold water. A pure product was obtained by washing the product with diethyl ether.
Typical procedure for the synthesis of 5-ethoxycarbonyl-4-phenyl-6-methyl-3,4-dihydropyrimidin-2(1H)-one (1a) under microwave irradiation
Benzaldehyde (1 g, 0.00768 mol), ethyl acetoacetate (0.98 g, 0.00768 mol), and urea (0.6 g, 0.00998 mol) were mixed with IL (1 g) and placed in a microwave reaction vial. Reaction was programmed to 200 W at 90°C. After the completion of reaction, as indicated by TLC, the reaction mixture was poured onto crushed ice and stirred for 10–15 min. The yellow solid separated was filtered under suction and washed with ice-cold water. A pure product was obtained by washing the product with diethyl ether.
Spectroscopic data for selected compounds
5-ethoxycarbonyl-4-phenyl-6-methyl-3,4-dihydropyrimidin-2(1H)-one (Entry 1a)
m.p. 201–202°C (202–204°C Lit.(11)); IR (KBr): ν max (in cm−1) = 3247, 3117, 1721, 1701, 1648; 1H NMR (δ in ppm, DMSO-d6): δ = 9.20 (s, 1H, NH); 7.74 (s, 1H, NH); 7.25 (s, 5H, C6H5); 5.2 (s, 1H, CH); 4.0 (q, J=6.5 Hz, 2H, OCH2CH3); 2.3 (s, 3H, CH3); 1.15 (t, J=6.5 Hz, 3H, OCH2CH3).
5-ethoxycarbonyl-4-(4-methoxyphenyl)-6-methyl-3,4-dihydropyrimidin-2(1H)-one (Entry 1b)
m.p. 200–202°C (200–202°C Lit.(8)); IR (KBr): ν max (in cm−1) = 3241, 3117, 1718, 1636; 1H NMR (δ in ppm, DMSO-d6): δ = 9.15 (s, 1H, NH); 7.76 (s, 1H, NH); 7.15 (d, 2H, CH); 5.12 (s, 1H, CH); 3.98 (q, J=7.1 Hz, 2H, OCH2CH3); 3.76 (s, 3H, OCH3); 2.24 (s, 3H, CH3); 1.16 (t, J=7.1 Hz, 3H, OCH2CH3).
5-ethoxycarbonyl-4-(4-hydroxyphenyl)-6-methyl-3,4-dihydropyrimidin-2(1H)-one (Entry 1c)
m.p. 227–229°C (226–228°C Lit.(11)); IR (KBr): ν max (in cm−1) = 3280, 3113, 1715, 1689, 1647; 1H NMR (δ in ppm, DMSO-d6): δ = 9.34 (s, 1H, OH); 9.10 (s, 1H, NH); 7.64 (s, 1H, NH); 7.03–6.65 (m, 4H, C6H4); 5.02 (s, 1H, CH); 3.95 (q, J=7.0 Hz, 2H, OCH2CH3); 2.21 (s, 3H, CH3); 1.08 (t, J=7.0 Hz, 3H, OCH2CH3).
5-ethoxycarbonyl-4-(3-nitrophenyl)-6-methyl-3,4-dihydropyrimidin-2(1H)-one (Entry 1e)
m.p. 224–226°C (225–227°C Lit.(11)); IR (KBr): ν max (in cm−1) = 3230, 3113, 1694, 1640; 1H NMR (δ in ppm, DMSO-d6): δ = 9.37 (s, 1H, NH); 7.90 (s, 1H, NH); 8.10–7.64 (m, 4H, C6H4); 5.29 (s, 1H, CH); 3.98 (q, J=6.9 Hz, 2H, OCH2CH3); 2.26 (s, 3H, CH3); 1.08 (t, J=6.9 Hz, 3H, OCH2CH3).
5-ethoxycarbonyl-4-(4-hydroxy-3-methoxyphenyl)-6-methyl-3,4-dihydropyrimidin-2(1H)-one (Entry 1f)
m.p. 233–234 (232–233°C Lit.(11)); IR (KBr): ν max (in cm−1) = 3244, 1701, 1645, 1516, 1221; 1H NMR (δ in ppm, DMSO-d6): δ = 8.99 (s, 1H, NH), 8.58 (s, 1H, NH), 7.47 (s, 1H), 6.80 (s, 1H, CH), 6.69–6.63 (m, 2H), 5.1 (s, 1H), 4.02 (q, J=6.57 Hz, 7.37 Hz, 2H, OCH2CH3), 3.7 (s, 3H), 2.26 (s, 3H, CH3), 1.17 (t, J=7.3 Hz, 3H, OCH2CH3).
5-ethoxycarbonyl-4-(4-methylphenyl)-6-methyl-3,4-dihydropyrimidin-2(1H)-one (Entry 1g)
m.p. 213–214°C (214–215°C Lit.(20)); IR (KBr): ν max (in cm−1) = 3244, 3115, 1726, 1706, 1649; 1H NMR (δ in ppm, DMSO-d6): δ = 9.13 (s, 1H, NH); 7.66 (d, J=3.11 Hz, 1H, NH); 7.09 (m, 4H, C6H4); 5.08 (d, J=3.11 Hz, 1H, CH); 3.95 (q, J=7.03 Hz, 2H, OCH2CH3); 2.3 (s, 3H, CH3); 1.15 (t, J=6.5 Hz, 3H, OCH2CH3).
5-ethoxycarbonyl-4-(4-chlorophenyl)-6-methyl-3,4-dihydropyrimidin-2(1H)-one (Entry 1h)
m.p. 211–213°C (210–212°C Lit.(20)); IR (KBr): ν max (in cm−1) = 3240, 1723, 1643; 1H NMR (δ in ppm, DMSO-d6): δ = 9.26 (s, 1, NH), 7.79 (s, 1H, NH), 7.40–7.30 (m, 4H, CH), 5.14 (s, 1H, CH), 3.99 (q, 2H, OCH2CH3), 2.25 (s, 3H, CH3), 1.09 (t, 3H, OCH2CH3).
5-ethoxycarbonyl-4-styryl-6-methyl-3,4-dihydropyrimidin-2(1H)-one (Entry 1i)
m.p. 232–234°C (233–234°C Lit.(20)); IR (KBr): ν max (in cm−1) = 3244, 3103, 1721, 1682, 1654; 1H NMR (δ in ppm, DMSO-d6): δ = 8.95 (s, 1H, NH); 7.45 (d, J=1.7 Hz, 1H, NH); 7.25 (m, 5H, C6H5); 6.2 (d, J=16.4 Hz, 1H, CH); 6.05 (dd, J=16.4 Hz, 1H, CH); 4.25 (d, J=6.0 Hz, 1H); 3.95 (q, J=7.0 Hz, 2H, OCH2CH3); 2.5 (s, 3H, CH3); 1.05 (t, J=7.0 Hz, 3H, OCH2CH3).
5-ethoxycarbonyl-4-phenyl-6-methyl-3,4-dihydropyrimidin-2(1H)-thione (Entry 1j)
m.p. 207–208°C (206–208°C Lit.(20)); IR (KBr): ν max (in cm−1) = 3250, 1651, 1598, 1561; 1H NMR (δ in ppm, DMSO-d6): δ = 10.33 (s, 1H, NH); 9.64 (s, 1H, NH); 7.35–7.19 (m, 5H, C6H5); 5.16 (d, J=3.5 Hz, 1H, CH); 4.0 (q, J=7.0 Hz, 2H, OCH2CH3); 2.28 (s, 3H, CH3); 1.09 (t, J=7.0 Hz, 3H, OCH2CH3).
5-ethoxycarbonyl-4-(4-methylphenyl)-6-methyl-3,4-dihydropyrimidin-2(1H)-thione (Entry 1l)
m.p. 214–215 (214–215°C Lit.(14)); IR (KBr): ν max (in cm−1) = 3313, 3171, 3106, 1667, 1610, 1575; 1H NMR (δ in ppm, DMSO-d6): δ = 10.30 (s, 1H, NH), 9.61 (s, 1H, NH),7.12 (d, 2H, J=8.0 Hz, CH), 6.90 (d, 2H, J=8.0 Hz, CH), 5.12 (s, 1H, CH), 4.00 (q, 2H, J=6.8 Hz, OCH2CH3), 3.72 (s, 3H, CH3), 2.29 (s, 3H, CH3), 1.11 (t, 3H, J=6.8 Hz, OCH2CH3).
Conclusion
In conclusion, we have described for the first time, use of a cost-effective IL, tri-(2-hydroxyethyl)-ammonium acetate as solvent and catalyst for synthesis of 3,4-dihydropyrimidinones under mild and neutral conditions. The simple work up procedure, good yields of products and application of an easily synthesizable and cost-effective IL make this method an attractive and environmentally friendly synthetic protocol for Biginelli reaction.
Acknowledgements
S.S.C. and Y.O.S. are thankful to UGC (SAP) and CSIR New Delhi for financial support.
Related Research Data
References
- Kappe , C.O. 2000 . Acc. Chem. Res. , 33 : 879 – 882 .
- Kappe , C.O. 1993 . Tetrahedron , 49 : 6937 – 6963 .
- Atwal , K.S. , Rovnyak , G.C. , O'Reilly , B.C. and Schwartz , J. 1989 . J. Org. Chem. , 54 : 5898 – 5907 .
- Studer , A. , Jeger , P. , Wipf , P. and Curran , D.P. 1997 . J. Org. Chem. , 62 : 2917 – 2924 .
- Biginellli , P. 1893 . Gazz. Chim. Ital. , 23 : 360 – 413 .
- O’ Reilly , B.C. and Atwal , K.S. 1987 . Heterocycles , 26 : 1185 – 1188 .
- Barluenga , J. , Thomas , M. , Ballesteros , A. and Loez , L.A. 1989 . Tetrahedron Lett. , 30 : 4573 – 4576 .
- Bigi , F. , Carloni , S. , Frullanti , B. , Maggi , R. and Sartori , G. 1999 . Tetrahedron Lett. , 40 : 3465 – 3468 .
- Yadav , J.S. , Reddy , B.V.S. , Reddy , E.J. and Ramalingam , T. 2000 . J. Chem. Res. (S) , 7 : 354 – 355 .
- Zhang , X.L. , Li , Y.P. , Lin , C.J. and Wang , J.D. 2006 . J. Mol. Catal. A: Chem. , 253 : 207 – 211 .
- Li , J.T. , Han , J.F. , Yang , J.H. and Li , T.S. 2003 . Ultrason. Sonochem. , 10 : 119 – 122 .
- Ma , Y. , Qian , C. , Wang , L. and Yang , M. 2000 . J. Org. Chem. , 65 : 3864 – 3868 .
- Yijun , H. , Fengyue , Y. and Chengjian , Z. 2005 . J. Am. Chem. Soc. , 127 : 16386 – 16387 .
- Su , W. , Li , J. , Zheng , Z. and Shen , Y. 2005 . Tetrahedron Lett. , 46 : 6037 – 6040 .
- Hojatollah , S. and Guo , Q. 2004 . Synth. Commun. , 34 : 171
- Liu , C. , Wang , J. and Li , Y. 2006 . J. Mol. Catal. A: Chem. , 258 : 367 – 370 .
- Suzuki , I. , Suzumura , Y. and Takeda , K. 2006 . Tetrahedron Lett. , 47 : 7861 – 7864 .
- Polshettiwar , V. and Varma , R.S. 2007 . Tetrahedron Lett. , 48 : 7343 – 7346 .
- Ichiro , S. , Yukari , I. and Kei , T. 2008 . Tetrahedron Lett. , 49 : 3238 – 3241 .
- Joseph , J.K. , Jain , S.L. and Sain , B. 2006 . J. Mol. Catal. A: Chem. , 247 : 99 – 102 .
- Palaniappan , S. and John , A. 2005 . J. Mol. Catal. A: Chem. , 233 : 9 – 16 .
- Gohain , M. ; Prajapati , D. ; Sandhu , J.S. Synlett 2004 , 235 – 238 .
- Peng , J. and Deng , Y. 2001 . Tetrahedron Lett. , 42 : 5917 – 5919 .
- Yuan , X.L. , Zhang , S.J. and Lu , X.M. 2007 . J. Chem. Eng. Data , 52 : 596 – 599 .
- Sharma , Y.O. and Degani , M.S. 2007 . J. Mol. Cat. A: Chemical , 277 : 215 – 220 .
- Sharma , Y.O. and Degani , M.S. 2009 . Green Chem. , 11 : 526 – 530 .
- Varma , R.S. 1999 . Green Chem. , 1 : 43 – 55 .
- Loupy , A. , Petit , A. , Hamelin , J. , Texier-Boullet , F. , Francoise , P. and Mathe , D. 1998 . Synthesis , 9 : 1213 – 1234 .
- Galema , S.A. 1997 . Chem. Soc. Rev. , 26 : 233 – 238 .
- Strauss , C.R. and Trainor , R.W. 1995 . Aust. J. Chem. , 48 : 1665 – 1692 .