Abstract
2-Azetidinones possess broad and potent activity due to presence of β-lactam ring and has been established as one of the biologically important scaffolds. The synthesis of N-(4-aryl-2-oxoazetidinone)-isonicotinamide by novel methods of stirring and sonication are described. The conventional method for synthesis of 2-azetidinones involves use of Dean–Stark water separator for the removal of water from the reaction with long reaction time (12–16 h reflux) at a very low temperature (−70 to −90°C). The microwave method reported requires inert atmosphere of nitrogen gas for the synthesis of 2-azetidinones. We report herein the synthesis of 2-azetidinone analogs of isonicotinic acid hydrazide by novel green route methods of sonication and stirring using molecular sieves. Results indicate that higher yields and shorter reaction times can be achieved by employing novel green route methods of synthesis.
Introduction
Green Chemistry is designing chemical products and processes that reduce or eliminate the use and/or the generation of hazardous substances. Various green chemistry methods like microwave technology, sonochemistry, stirring, phase transfer catalyst, ionic liquid, and many more techniques include approaches for the creation of “benign-by-design” synthetic methods which are now accepted worldwide Citation1 Citation2. Processes designed by green routes help in the promotion of resource and energy utilization efficiently. It involves low level of waste, and is inherently safe making the processes economically and environmentally beneficial.
The broad and potent activity of 2-azetidinone has been established as one of the biologically important scaffolds Citation3. 2-Azetidinone possesses a broad spectrum of activities, such as antibacterial Citation4–8, antihyperglycemic Citation9, antihyperlipidemic Citation10–15, CNS activity Citation16, tryptase inhibitory Citation17 Citation18, human leukocyte elastase inhibitory Citation19–21, anti-inflammatory Citation22 Citation23, vasopressin v1a antagonist Citation24, and anticancer activity Citation25 Citation26. The Staudinger's ketene–imine cycloaddition reaction [2 + 2] is the most common method for the synthesis of azetidinones and has been reviewed till date by several researchers. The reactions are carried out thermally Citation27 or photochemically Citation28–30 using acid chlorides in the presence of triethylamine or α-diazoketones as ketene precursors. However, the conventional methods reported for the synthesis of 2-azetidinones requires longer reaction time (12–16 h reflux) Citation31–37 at very low temperature (−70 to −90°C) Citation38–44 with low yields (less than 70%) Citation45–47. The reactions also involve the use of a Dean–Stark water separator for the removal of water from the reaction. The previous decade has also seen the use of microwave radiation in the synthesis of azetidinones. However, the reported microwave methods also required inert atmosphere of nitrogen gas to enable the completion of the reaction Citation48 Citation49.
Herein, we report the synthesis of Schiff's bases of isoniazid, which were further utilized for the synthesis of 2-azetidinone analogs by novel green routes of sonication and stirring.
Results and discussion
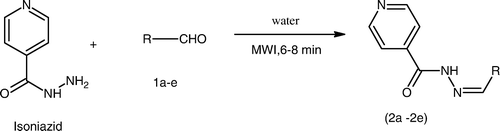
The synthetic strategies adopted to obtain the target compounds are outlined in 2 and 3. The key intermediate Schiff's bases (2a–2e) were prepared in excellent yields in a one step reaction () by our previously reported synthesis using microwave Citation50.
Scheme 1. Microwave assisted synthesis of Schiff's bases of isoniazid.
Furthermore, the intermediates which were synthesized represent versatile building blocks for the synthesis of new heterocycles incorporating azetidinone nucleus. The synthesized intermediates were characterized by the presence of a strong IR band at 1595–1625 cm–1, 1H NMR spectra also showed singlet signal equivalent to one proton for =CH group between 7.4 and 8.4, which confirms the formation of Schiff's bases.
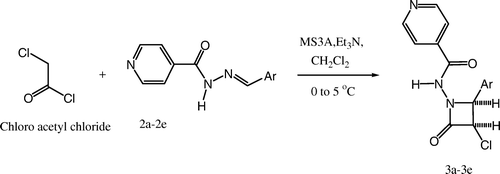
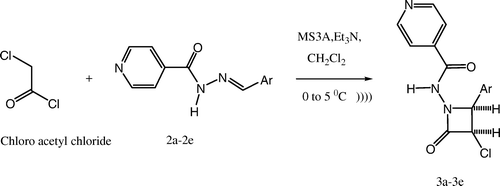
The synthesized intermediates were further utilized for the synthesis of N-(4-aryl-2-oxoazetidinone)-isonicotinamide derivatives (3a–3e) containing 2-azetidinone nucleus by stirring and sonication ( and ), which were characterized by the presence of a strong band at 1715–1780 cm–1 for the ring carbonyl group, which is considered a strong confirmation for the azetidinone nucleus formation. Another piece of evidence for cyclization is the appearance of a doublet signal equivalent to one proton in 1H NMR spectrum between 5.2 and 5.6 ppm (C-3, CH) and doublet signal equivalent to 1 proton spectrum between 5.7 and 5.9 ppm (C-4, CH), which represents the formation of azetidinone nucleus.
Scheme 2. Synthesis of 2-azetidinones analogs using Schiff's bases by stirring.
Scheme 3. Synthesis of 2-azetidinones analogs using Schiff's bases by sonication.
The Electron Impact Mass Spectrometry of selected compound 3b displayed a molecular ion peak at m/z of 317 which confirmed its molecular weight. A base peak in the mass spectrum was obtained at m/z =241 and is contributed to the loss of the (O = C–C–Cl) fragment from the molecule.
Experimental
Melting points (mp) were determined with Veego melting point apparatus (VMP PM, 32/1104) and are uncorrected. Thin layer chromatography (TLC) Citation51 was carried out using silica gel (G-60 mesh). R f value for each compound was calculated using toulene:ethanol (4.5:0.5) as solvent and spots were located using iodine vapor. UV studies were carried out on UV–Visible Spectrophotometer (Shimadzu 1700) and the λmax of the respective synthesized compounds were calculated using ethanol as the solvent. IR spectras (KBr) Citation52 were recorded on a FTIR Spectrophotometer with diffuse reflectance attachment (Shimadzu 8400S). 1H NMR spectra Citation53 were obtained from NMR Spectrophotometer (Bruker Avance II 400 NMR) using dimethyl sulphoxide as the solvent. Chemical shifts were expressed in parts per million relative to SiMe4 as internal standard.
Unless stated otherwise, all the materials were obtained from commercial suppliers and used without further purification.
Synthesis of Schiff's bases as intermediate
The synthesis of compounds (2a–2e) was performed according to our previously reported procedure Citation50 (). Isoniazid (0.01 M) and appropriate aromatic aldehydes (0.01 M) (1a–1e) in water were irradiated under microwave using a microwave synthesizer (Make-Raga's Scientific) at power level 3 (240 W, 35% irradiation) until the completion of the reaction. The reaction was monitored by TLC (acetone:ethanol = 4:1). The reaction mixture was filtered. The residue obtained was washed with water, followed by sodium thiosulphate (Na2S2O3) solution, and then dried. The crude product was obtained upon recrystallization from alcohol to give the pure hydrazones of INH (2a–2e), . The synthesized compounds were characterized by their mp and spectral data (UV, IR, and 1H NMR).
Table 1. Chemical structures and properties of Schiff's bases of isoniazide by microwave (2a–2e).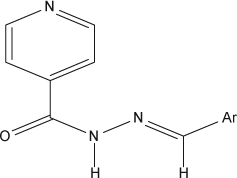
Salicylidene isonicotinyl hydrazone (2a)
White crystals; yield 95.6%; mp 262–264°C; IR (KBr): v cm−1=3344 (–OH), 3178 (–NH), 3004 (–CH), 1685 (amide C = O), and 1616 (imine C = N); 1H NMR (DMSO-d 6): δ ppm = 11 (s, OH), 8.8–8.9 (d, pyridine 2H), 8.6–8.8 (d, pyridine 2H), 8.7 (s, CH), 7.5–7.6 (d, aromatic 1H), 8.6 (s, NH), 6.8–7.0 (d, aromatic 2H), and 7.3–7.4 (m, aromatic 1H).
4-Hydroxy benzylidene isonicotinyl hydrazone (2b)
Yellow powder; yield 94.5%; mp 264–268°C; IR (KBr): v cm–1=3340 (–OH), 3213 (–NH), 3055 (–CH), 1662 (amide C = O), and 1608 (imine C = N); 1H NMR (DMSO-d 6): δ ppm = 11.5 (s, OH), 8.8–8.5 (d, pyridine 2H), 8.4–8.3 (d, pyridine 2H), 7.8 (s, CH), 8.0 (s, NH) 7.5–7.3 (d, aromatic 2H), and 7.0–6.8 (d, aromatic 2H).
p-Cl benzylidene isonicotinyl hydrazone (2c)
White crystals; yield 98.1%; mp 218–221°C; IR (KBr): v cm−1=3166 (–NH), 3020 (–CH), 1674 (amide C = O), 1593 (imine C = N), and 784 (–Cl); 1H NMR (DMSO-d 6): δ ppm = 8.88–8.91 (d, pyridine 2H), 7.99–8.05 (d, pyridine 2H), 8.32 (s, CH), 8.65 (s, NH) 7.79–7.80 (d, aromatic 2H), and 7.50–7.51 (d, aromatic 2H).
p-NO2 benzylidene isonicotinyl hydrazone (2d)
Yellow powder; yield 90.7%; mp 255–257°C; IR (KBr): v cm−1=3186 (–NH), 3001 (–CH), 1685 (amide C = O), 1558 (imine C = N), and 1280 (–NO2); 1H NMR (DMSO-d 6): δ ppm = 9.32–9.35 (d, pyridine 2H), 8.51–8.55 (d, pyridine 2H), 8.10 (s, CH), 8.34 (s, NH), 7.89–7.93 (d, aromatic 2H), and 7.60–7.62 (d, aromatic 2H).
2,5-Di MeO benzylidene isonicotinyl hydrazone (2e)
Yellow crystals; yield 95.2%; mp 185–188°C; IR (KBr): v cm–1=3190 (–NH), 3061 (–CH), 1654 (amide C = O), 1600 (imine C = N), and 1218 (O–CH3); 1H NMR (DMSO-d 6): δ ppm = 8.89–8.92 (d, pyridine 2H), 8.11–8.15 (d, pyridine 2H), 8.22 (s, CH), 8.48 (s, NH), 7.5–7.8 (d of d, aromatic 2H), 6.92 (s, aromatic 1H), and 3.92–4.12 (m, O–CH3).
Synthesis of N-(4-aryl-2-oxoazetidinone)-isonicotinamide analogs using Schiff's bases
The appropriate Schiff's base (0.0025 M) was dissolved in 10 ml of dichloromethane. Molecular sieves [MS (1–2 g, 3A×1.5 mm)] were added to the reaction mixture. The reaction mixture was then stirred in an ice bath at 0–5°C. Chloroacetyl chloride (0.0037 M) followed by triethyl amine (0.0075 M) were then added drop wise to the reaction mixture with constant stirring at low temperature (0–5°C). The reaction mixture was further stirred at room temperature until the completion of reaction [TLC (toulene:ethanol = 4.5:0.5)]. For the sonication method, the reaction mixture was sonicated on an ultrasonic bath (local make) until the completion of reaction [TLC (toulene:ethanol = 4.5:0.5)].
The reaction mixture was added into crushed ice and stirred to obtain the crude product. The product obtained was washed with concentrated brine and sodium bicarbonate solution and then dried. The crude product was obtained upon recrystallization from alcohol to give the pure 2-azetidinones of isonicotinoyl hydrazones (3a–3e) (). The synthesized compounds were characterized by their mp and spectral data (UV, IR, 1H NMR, and MS).
Table 2. Chemical structures and properties of 2-azetidinones (3a–3e).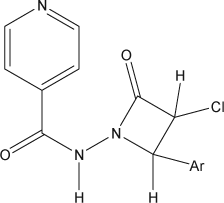
represents structures of aldehydes and their respective synthesized Schiff's bases and 2-azetidinone analogs.
Table 3. Isonicotinyl hydrazones and their 2-azetidinone analogs.
N-(4-salicylidene-2-oxoazetidinone)-isonicotinamide (3a)
Yellow powder; yield 91.1%; mp 98–105°C; IR (KBr): v cm−1=3467 (–OH), 3193 (–NH), 3043 (–CH), 1774 (ring C = O), and 1677 (amide C = O); 1H NMR (DMSO-d 6): δ ppm = 8.89–8.83 (d, pyridine 2H), 7.77–7.75 (d, pyridine 2H), 4.91 (s, –NH), 7.36–6.97 (m, aromatic, 4H), 4.51–4.42 (d, CH Aze ring (C-4), 1H), 5.75–5.71 (d, CH Aze ring (C-3), 1H), and 8.22 (s, OH).
N-[4-(4-hydroxy benzylidene)-2-oxoazetidinone]-isonicotinamide (3b)
Yellow powder; yield 86%; mp 102–110°C; IR (KBr): v cm–1=3556 (–OH), 3186 (–NH), 3012 (–CH), 1762 (ring C = O), and 1662 (amide C = O); 1H NMR (DMSO-d 6): δ ppm = 8.77–8.79 (d, pyridine 2H), 7.96–7.93 (d, pyridine 2H), 4.21 (s, –NH), 7.24–7.23 (d, aromatic, 2H), 6.86–6.84 (d, aromatic, 2H), 4.56–4.54 (d, CH Aze ring (C-4), 1H), and 5.71–5.68 (d, CH Aze ring (C-3), 1H); EIMS (70 eV, m/z): 317 (M+), 241 (base peak).
N-(4-p-Cl benzylidene-2-oxoazetidinone)-isonicotinamide (3c)
Brown crystals; yield 81.9%; mp 92–100°C; IR (KBr): v cm–1=3193 (–NH), 3024 (–CH), 1720 (ring C = O), 1666 (amide C = O), and 829 (–Cl); 1H NMR (DMSO-d 6): δ ppm = 9.38–9.36 (d, pyridine 2H), 8.30–8.28 (d, pyridine 2H), 4.78 (s, –NH), 7.70–7.76 (d, aromatic, 2H), 7.35–7.33 (d, aromatic, 2H), 5.43–5.42 (d, CH Aze ring (C-4), 1H), and 5.81–5.80 (d, CH Aze ring (C-3), 1H).
N-(4-p-NO2 benzylidene-2-oxoazetidinone)-isonicotinamide (3d)
Brown crystals; yield 85.8%; mp 112–116°C; IR (KBr): v cm–1=3035 (–NH), 2974 (–CH), 1735 (ring C = O), 1685 (amide C = O), and 1172 (–NO2); 1H NMR (DMSO-d 6): δ ppm = 9.38–9.36 (d, pyridine 2H), 7.97–7.95 (d, pyridine 2H), 4.91 (s, –NH), 8.30–8.28 (d, aromatic, 2H), 7.19–7.17 (d, aromatic, 2H), 5.43–5.42 (d, CH Aze ring (C-4), 1H), and 5.81–5.80 (d, CH Aze ring (C-3), 1H).
N-(4-2,5-di MeO benzylidene-2-oxoazetidinone)-isonicotinamide (3e)
Brown crystals; yield 92.2%; mp 150–152°C; IR (KBr): v cm–1=3195 (–NH), 2974 (–CH), 1735 (ring C = O), 1681 (amide C = O), and 1073 (O–CH3); 1H NMR (DMSO-d 6): δ ppm = 9.38–9.36 (d, pyridine 2H), 7.97–7.84 (d, pyridine 2H), 4.42 (s, –NH), 6.83–6.86 (d, aromatic 1H), 6.19 (s, aromatic 1H), 6.21 (doublet, aromatic, 1H), 5.42–5.43 (d, CH Aze ring (C-4), 1H), 4.78–4.90 (d, CH Aze ring (C-3), 1H), and 3.77–3.60 (m, o–CH3, 6H).
Conclusion
The synthesis of 2-azetidinones by novel green route methods of sonication and stirring are described. The reactions were carried out in the presence of molecular sieves for removal of water generated during the reaction. The time span required for the completion of reaction is less than that of conventional methods. The reaction by stirring method requires 110–130 min, while the sonication method requires 20–30 min for the completion of the reaction. Furthermore, the present investigation offers rapid and effective new procedures for the synthesis of 2-azetidinone analogs with improved yield (81–93%).
The results of physiochemical characterization of the synthesized products by chromatographic and spectroscopic studies suggested that the product obtained by both stirring and sonication methods were comparable in chemical composition.
Acknowledgements
This work was financially supported by University of Pune, Pune, India. The authors wish to thank the Principal, Padm. Dr. D.Y. Patil Institute of Pharmaceutical Science and Research, Pune, India, for providing the necessary infrastructural facilities to carry out this work. NMR characterization was carried out by SAIF/CIL, Punjab University, Chandigarh, India.
References
- Afonso , C.A.M. Crespo , J.G. Green Separation Processes ; Weinheim : Wiley-VCH Verlag GmbH & Co. KgaA , 2005 ; pp 3 11 .
- Cintas P. Luche J-L. Green Chem 1999 115 125
- Staudinger H.L. Ann. Chem. 1907 365 51 123
- Singh , G.S. Mmolotsi , B.J. Il Farmaco . 2005 , 60 , 727 730 .
- Chavan , A.A. Pai , N.R. Molecules 2007 , 12 , 2467 2477 .
- Gunner , V. Il Farmaco . 2000 , 55 , 147 150 .
- Desai , K.G. Desai , K.R. Bioorg. Med. Chem . 2006 , 14 , 8271 8279 .
- Gradelski , E. Kolek , B. Bonner , D.P. Int. J. Antimicrob. Agents 2001 , 17 , 103 107 .
- Goel , R.K. Kulkarni , S.K. J. Pharm. Pharm. Sci . 2004 , 7 , 80 83 .
- Leach , C.A. Deirdre , M.B. Il Farmaco . 2001 , 56 , 45 50 .
- Vaccaro , W.D. Sher , R. Davis , H.R. Bioorg. Med. Chem. 1998 , 6 , 1429 1437 .
- Wu , G. Wong , Y. Chen , X. Ding , Z. J. Org. Chem. 1999 , 64 , 3714 3718 .
- Clader , J.W. Burnett , D.A. J. Med. Chem . 1996 , 39 , 3684 3693 .
- Xu , X. Bioorg. Med. Chem. Lett . 2007 , 17 , 101 104 .
- Vaccaro , W.D. Bioorg. Med. Chem . 1998 , 6 , 1429 1437 .
- Goelab , R.K. Singha , A. Naidu , P.S. J. Pharm. Pharm. Sci . 2005 , 8 2 , 182 189 .
- Qian , X. Zheng , B. J. Org. Chem . 2002 , 67 , 3595 3600 .
- Bisacchi , G.S. Slusarchyk , W.A. Bioorg. Med. Chem. Lett . 2004 , 14 , 2227 2231 .
- Hagmannt , W.K. Bioorg. Med. Chem. Lett . 1992 , 2 , 681 684 .
- Moreno , G. Bioorg. Med. Chem . 2004 , 12 , 129 138 .
- Hagmannt , W.K. Shah , S.K. Dornt , C.P. Bioorg. Med. Chem. Lett . 1991 , 1 10 , 545 550 .
- Kumar , A. Rajput , C.S. Eur. J. Med. Chem . 2009 , 44 , 83 90 .
- Kumar , A. Rajput , C.S. Bhati , S.K. Bioorg. Med. Chem . 2007 , 15 , 3089 3096 .
- Guillon , C.D. Koppel , G.A. Bioorg. Med. Chem . 2004 , 15 , 2054 2080 .
- Banik , I. Becker , F.F. Banik , B.K. J. Med. Chem . 2003 , 46 , 12 15 .
- Banik , I. Becker , F.F. Banik , B.K. Bioorg. Med. Chem . 2005 , 13 , 3611 3622
- Alcaide , B. ; Almendros , P. ; Salgado , N.R. ; Rodriguez Vicente , A. J. Org. Chem . 2000 , 65 , 4453 4455 .
- Lindler , M.R. ; Frey , W.U. ; Podlech , J. J. Chem. Soc. Perkin Trans . 2001 , 1 , 2566 2577 .
- Sierra , M.A. ; Mancheno , M.J. ; Vicente , R. ; Gomez-Gallego , M. 2001 J. Org. Chem 66 , 8920 8925 .
- Griesbeck , A.G. ; Heckroth , H. Synlett . 2002 , 131 133 .
- Thiagrajan , K. ; Puranik , V.G. ; Deshmukh , A.R.A.S. Bhawal B.M. Tetrahedron 2000 56 7811 7816 .
- Karupaiyan , K. , Puranik , V.G. , Deshmukh , A.R.A.S. and Bhawal , B.M. 2000 . Tetrahedron , 56 : 8555 – 8560 .
- Hassan , H. and Soliman , R. 2000 . Synth Commun. , 30 : 2465 – 2478 .
- Escalante , J. , Gonzalez-Tototzin , M.A. , Avina , J. , Munoz Muniz , O. and Juaristi , E. 2001 . Tetrahedron , 57 : 1883 – 1890 .
- Barrett , A.G.M. , Ahmed , M. , Baker , S.P. , Baugh , S.P.D. , Braddock , D.C. , Procopiou , P.A. , White , A.J.P. and Williams , D.J. 2000 . J. Org Chem. , 65 : 3716 – 3721 .
- Mori , M. , Kozawa , Y. , Nishida , M. , Kanamura , M. , Onozuka , K. and Takimoto , M. 2000 . Org Lett. , 2 : 3245 – 3247 .
- Kozawa , Y. and Mori , M. 2001 . Tetrahedron Lett. , 42 : 4869 – 4873 .
- Banik , B.K. and Becker , F.F. 2000 . Tetrahedron Lett. , 41 : 6551 – 6554 .
- Shindo , M. , Oya , S. , Murakami , R. , Sato , Y. and Shishido , K. 2000 . Tetrahedron Lett. , 41 : 5943 – 5946 .
- Deng , B.L. , Demillequand , M. , Laurent , M. , Toillaux , R. , Belmans , M. , Kemps , L. , Ceresiat , M. and Merchand-Brynaert , J. 2000 . Tetrahedron , 56 : 3209 – 3217 .
- Cainelli , G. , Galletti , P. , Gazzano , M. , Giacomini , D. and Quintavalla , A. 2002 . Tetrahedron Lett. , 43 : 233 – 235 .
- Gerona-Navarro , G. , Bonache , M.A. , Heranz , R. , Garcia Lopez , M.T. and Gonzalez-Muniz , R. 2001 . J. Org Chem. , 66 : 3538 – 3547 .
- Alcaide , B. , Almendros , P. and Aragoncillo , C.J. 2001 . Org Chem. , 66 : 1612 – 1620 .
- Desai , P. and Aube , J. 2000 . Org Lett. , 2 : 1657 – 1659 .
- Abbiati , G. and Rossi , E. 2001 . Tetrahedron , 57 : 7205 – 7212 .
- Ceric , H. , Kovacevic , M. and Sindler-Kulyk , M. 2000 . Tetrahedron , 56 : 3985 – 3993 .
- Penfold , D.J. , Pike , K. , Genge , A. , Anson , M. , Kitteringham , J. and Kilburn , J.D. 2000 . Tetrahedron Lett. , 41 : 10347 – 10351 .
- Bose A.K. Banik B.K. Manhas M.S. Tetrahedron Lett 1995 36 213 216
- Bose , A.K. , Jayaraman , M. , Okawa , A. , Bari , S.S. , Robe , E.W. and Manhas , M.S. 1996 . Tetrahedron Lett. , 37 : 6989 – 6992 .
- Thomas , A.B. Tupe , P.N ; Badhe , R.V. Nanda , R.K. Kothapalli L.P. , ; Paradkar , O.D. Sharma , P.A. Deshpande , A.D. Green Chem. Lett. Rev . 2009 , 2 , 23 27 .
- Stahl , E. Thin Layer Chromatography – A Practical Handbook , 2nd ed Springer-Verlag : New York ; 1969 96 102 .
- Kalsi , P.S. 2002 . Spectroscopy of Organic Compounds , 5th ed , 364 – 367 . New Delhi : New Age International Publication .
- Kemp , W. Organic Spectroscopy , 3rd ed .; ELBS funded by British Government, Palgrave, New York , 1991 285 324 .