Abstract
Cyanoacetamides 3a–d were prepared by reacting ethyl cyanoacetate with primary aliphatic amines 2a–d. The formed cyanoacetamides 3a–d were coupled with aromatic diazonium salts to give the corresponding arylhydrazones 4a–i which were used as precursors to title triazoles and pyrazoles by reacting with hydroxylamine and chloroacetonitrile. Yields of products formed by conventional heating are compared with those of microwave and ultrasound irradiation
Introduction
Recently, many papers have been published dealing with 2-arylhydrazononitriles as precursors to heteroaromatics Citation1–6. Elnagdi et al. Citation7–10 have reported efficient synthetic approaches to functionally substituted pyrazoles and 1,2,3-triazoles utilizing arylhydrazononitrile precursors. In the light of our recent interest in adopting green synthetic methodologies for the synthesis of functionally substituted heteroaromatics utilizing microwave (Mw) heating and ultrasound (Us) irradiation Citation11–25, we aim in this work to report on the synthesis and utility of arylhydrazononitrile as precursors to 1,2,3-traizoles 7, 10, and pyrazole derivatives 13.
Results and discussion
Ethyl cyanoacetate Citation1 was reacted with a variety of aliphatic amines 2a–d under Mw heating or Us activation to yield cyanoacetamides 3a–d ().
Scheme 1. Synthesis of cyanoacetamides
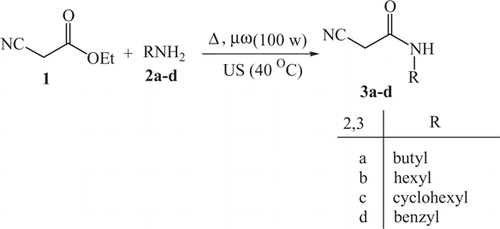
Compounds 3a–d were coupled readily with aromatic diazonium salts to yield the corresponding arylhydrazononitriles 4a–i, in 67–95% yields (). The structures of compounds 4a–i were established on the basis of their elemental analyses and spectral data. 1H NMR spectra of compounds 4a–i showed a singlet signal in region 11.10–14.97 ppm corresponding to the hydrazone (NH) proton. The structure assigned to compounds 4a–i could be unequivocally established by single crystal X-ray diffraction of compound 4c Citation26, as shown in .
Figure 1. X-ray crystal structure of 4c.
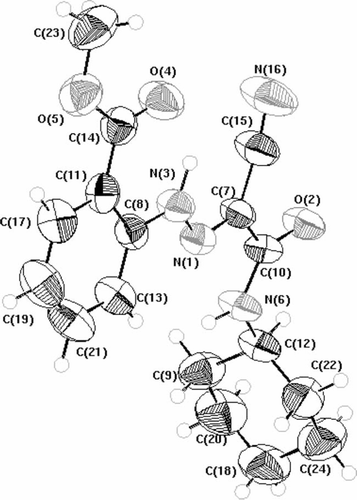
Scheme 2. Synthesis of 2-arylhydrazononitrile derivatives.
Parallel to the recent literature data Citation7 Citation8 Citation27–29, compounds 4a–i reacted with hydroxylamine hydrochloride in the presence of sodium acetate to yield the amidoximes 5a–h. It has been found that the reaction completion time was 1 h in refluxing ethanol and 2–5 min under Mw heating. The structures of the new amidoximes 5a–h have been elucidated by elemental analyses and spectroscopic measurements. For example, the 1H NMR spectra of compound 5a revealed the presence of (NH2) protons at Δ6.52 ppm and a broad singlet signal at Δ14.21 ppm corresponding to (OH) proton. The IR spectra of compound 5a showed absorption bands at ν = 3587, 3456, and 3420 cm−1 due to OH and NH2 groups, respectively.
Upon heating 5 in dimethylformamide (DMF) at reflux temperature for 1 h or under Mw irradiation for 2–5 min or by utilizing Us irradiation for 1 h at 40°C, this compound gave solid products whose structures were assumed to be 6, 7, or 8 ( ).
Scheme 3. Synthesis of 5-amino-1,2,3-triazole and 1,2,3-triazolo[4,5-d]pyrimidin-7-one derivatives
![Scheme 3. Synthesis of 5-amino-1,2,3-triazole and 1,2,3-triazolo[4,5-d]pyrimidin-7-one derivatives](/cms/asset/a57a4e1f-c9fd-4d5f-9f98-086777cb9e6f/tgcl_a_483602_o_f0004g.gif)
The structure of isoxazoles 6 was readily ruled out for the reaction products on the basis of spectral data. Thus, the presence of an amide carbonyl absorption in region ν = 1642–1658 cm−1 in the IR spectra of the reaction products allowed us to discard the possible structure 6. Moreover, 13C NMR spectra of the reaction products confirmed the presence of a CO carbon at Δ≈164 ppm. If the reaction product was the isomer 6, it would be difficult to assign this signal. Elemental analysis and spectral data could not unequivocally differentiate the two isomers 7 and 8. Therefore, the 1,2,3-triazolo[4,5-d]pyrimidines 10 was prepared to chemically verify the structure of 7. Reaction of aminotriazoles 7 with dimethylformamide dimethylacetal (DMF DMA), under different reaction conditions, gave the ring-closed 1,2,3-triazolo[4,5-d]pyrimidines 10, via the intermediate 9 (). It is difficult to obtain these reaction products 10 with the isomer 8.
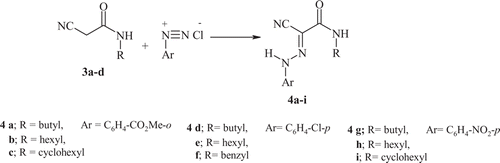
Recently, Elnagdi et al. Citation2 Citation7 Citation30 have reported that refluxing 2-arylhydrazononitriles with functionally substituted alkyl halides afforded 4-aminopyrazole derivatives. Now, compound 4 was next reacted with chloroacetonitrile, under conventional heating, Mw irradiation, and sonication, to afford the 4-aminopyrazoles 13, via the acyclic non-isolable intermediate 12 ( ). The identity of compounds 13 was supported by correct elemental analyses and mass spectra as well as the IR and NMR spectra which were compatible with assigned structures (see Section “Experimental”). The reaction times and yields of the products formed via traditional methods were compared with those of Mw and Us irradiation (see ).
Scheme 4. Synthesis of pyrazole derivatives.
Table 1. Yield as well as reaction times by the three methodologies are compared.
In conclusion, we have shown that the synthesis of 2-aryl-1,2,3-triazoles and 4-aminopyrazoles from arylhydrazononitriles is better conducted by green methodologies through the avoidance of heating and excessive use of solvents. On the other hand, it should be noted that reactions occur at different temperatures with these techniques and therefore strict comparisons will require a balance between effectiveness and energy costs.
Experimental
General
All melting points were measured on a Gallenkamp electrothermal melting point apparatus and are uncorrected. The IR absorption spectra were measured on a Nicolet Magna 520FT IR spectrophotometer. 1H and 13C NMR spectra were recorded in deuterated dimethylsulfoxide (DMSO) or deuterated chloroform (CDCl3) at Bruker DPX 400 MHz spectrometer using tetramethylsilane (TMS) as an internal reference. Mass spectra were performed on a Shimadzu GCMS-QP 1000 EX mass spectrometer at 70 eV. Mw irradiation was carried out using the commercial Mw oven (SGO 1000 W). A thermocouple used to monitor the temperature inside the Mw vessel during the reactions found that the temperature was approximately 105–110°C. Us irradiation was carried out with a microprocessor controlled-2004, high intensity ultrasonic processor with temperature controller (750 W). The ultrasonic frequency of the cleaning bath used was equal to 25 KHz. The reaction temperature was stabilized at 35–40°C even after more than 1 h by addition or removal of water in ultrasonic bath to keep the required temperature. Elemental analyses were measured by means of Perkin Elmer 2400 CHN elemental analyzer flowchart. X-ray crystallography was carried out on a Kappa CCD Enraf Nonius FR 590 diffractometer, at National Research Center, Dokki, Cairo, Egypt.
General procedure for the preparation of N-substituted-2-cyano-acetamide 3a–d
Method I (Δ)
Equimolar amounts (0.1 mol) of both ethyl cyanoacetate and the aliphatic amines 2a–d were stirred at room temperature for 1–4 h and the resulting solid product was re-crystallized from ethanol.
Method II (µω)
A mixture of ethyl cyanoacetate (0.1 mol) and the appropriate amount of aliphatic amines 2a–d (0.1 mol) was placed in the Mw oven and irradiated at 460 W for 1–4 min. Then, the reaction mixture was left to cool to room temperature. The solid product so-formed was filtered and re-crystallized from ethanol.
Method III (Us)
Equimolar amounts (0.1 mol) of both ethyl cyanoacetate and the aliphatic amines 2a–d were mixed and heated under Us irradiation at 40°C for 2–10 min, and then left to cool to room temperature. The solid product so-formed was filtered and re-crystallized from ethanol.
N-Butyl-2-cyanoacetamide (3a)
Orange crystals from ethanol; mp 72°C; IR νmax cm−1: 3299 (NH), 2954 (CH aliphatic), 2258 (CN), and 1653 (C = O); 1H NMR; (DMSO-d 6); Δ = 0.85 (t, 3H, CH3, J=7 Hz), 1.27 (m, 2H, CH2, J=7 Hz), 1.38 (m, 2H, CH2, J=7 Hz), 3.06 (q, 2H, CH2 , J=7 Hz), 3.56 (s, 2H, CH2CN), and 8.18 (br s, 1H, NH) ppm; 13C NMR; (DMSO-d 6); Δ = 14.02, 20.00, 25.76, 31.40 (butyl carbons), 39.27 (CH2CN), 116.71 (CN), and 162.39 (C = O) ppm; MS: 141 [M++1]. Analysis calculated for C7H12N2O (140.19): C, 59.98; H, 8.63; and N, 19.98. found: C, 59.90; H, 8.70; and N, 19.92.
2-Cyano-N-hexyl-acetamide (3b)
Yellow crystals from ethanol; mp 67°C; IR νmax cm−1: 3299 (NH), 2932 (CH aliphatic), 2260 (CN), and 1645 (C = O); 1H NMR; (CDCl3); Δ = 0.81 (t, 3H, CH3, J=6 Hz), 1.24 (m, 6H, 3CH2), 1.45 (m, 2H, CH2, J=7 Hz), 3.17 (q, 2H, CH2, J=7 Hz), 3.42 (s, 2H, CH2CN), and 7.17 (s, 1H, NH) ppm; 13C NMR; (CDCl3); Δ = 13.99, 22.54, 26.04, 26.55, 29.11, 31.43 (hexyl carbons), 40.45 (CH2CN), 115.22 (CN), and 162.03 (C = O) ppm; MS: 169 [M++1]. Analysis calculated for C9H16N2O (168.24): C, 64.25; H, 9.59; and N,16.65. Found: C, 64.20; H, 9.61; and N, 16.69.
2-Cyano-N-cyclohexyl-acetamide (3c)
Colorless crystals from ethanol; mp 136°C; IR νmax cm−1: 3272 (NH), 2933 (CH aliphatic), 2261 (CN), and 1628 (C = O); 1H NMR; (DMSO-d 6); Δ = 1.12 − 1.68 (m, 6H, 3CH2), 1.70–2.03 (m, 4H, 2CH2), 3.46–3.54 (m, 1H, CH), 3.22 (s, 2H, CH2CN), and 8.14 (d, 1H, NH, J=7 Hz) ppm; 13C NMR; (DMSO-d 6); Δ = 24.35, 24.87, 25.62, 25.84, 30.91, 32.60 (cyclohexyl carbons), 48.73 (CH2CN), 116.73 (CN), and 161.51 (C = O) ppm; MS: 166 [M+]. Analysis calculated for C9H14N2O (166.22): C, 65.05; H, 8.49; and N, 16.85. Found: C, 65.10; H, 8.36; and N, 16.89.
N-Benzyl-2-cyano-acetamide (3d)
Brown crystals from ethanol; mp 124°C; IR νmax cm−1: 3295 (NH), 3091 (CH aromatic), 2923 (CH aliphatic), 2220 (CN), and 1640 (C = O); 1H NMR; (DMSO-d 6); Δ = 3.71 (s, 2H, CH2CN), 4.30 (d, 2H, PhCH2, J=5 Hz), 7.29–7.34 (m, 5H, ph–H), and 8.74 (t, 1H, NH, J=7 Hz) ppm; MS: 174 [M+]. Analysis calculated for C10H10N2O (174.20): C, 68.95; H, 5.79; and N, 16.08. Found: C, 68.90; H, 5.67; and N, 16.20.
Preparation of arylhydrazone derivatives 4a–i
A cold solution of aryldiazonium salt (10 mmol) was prepared by adding a solution of sodium nitrite (1g into 10 mL H2O) to a cold solution of arylamine hydrochloride or arylamine nitrate (10 mmol) with stirring. The resulting solution of the aryldiazonium was then added to a cold solution of N-substituted-2-cyanoacetamides 3a–d (0.1mol) in ethanol (50 mL) containing sodium acetate (1 g into 10 mL H2O). The mixture was stirred at room temperature for 1 h and the solid product so-formed was collected by filtration and re-crystallized from ethanol.
2-[N′-(Butylcarbamoyl-cyano-methylene)-hydrazino]-benzoic acid methyl ester (4a)
Yellow crystals from ethanol; yield 70%, mp 166°C; IR νmax cm−1: 3388 (NH), 3026 (CH aromatic), 2953 (CH aliphatic), 2214 (CN), 1696 (C = O ester), and 1668 (C = O amide); 1H NMR; (DMSO-d 6); Δ = 0.89 (t, 3H, CH3, J=7 Hz), 1.31 (m, 2H, CH2, J=7 Hz), 1.49 (m, 2H, CH2, J=7 Hz), 3.23 (q, 2H, CH2, J=7 Hz), 3.91 (s, 3H, ester CH3), 7.20 (t, 1H, Ar H, J=7 Hz), 7.69 (t, 1H, Ar H, J=7 Hz), 7.98 (d, 1H, Ar H, J=8 Hz), 8.16 (d, 1H, Ar H, J=8 Hz), 8.58 (t, 1H, NH, J=5 Hz), and 12.28 (s, 1H, NH) ppm; 13C NMR; (DMSO-d 6); Δ = 14.26, 20.15, 31.92 (butyl carbons), 53.33 (ester CH3), 111.18, 112.99, 113.53, 123.60, 131.23, 135.51 (C6H4COOCH3-o), 116.54 (CN), 143.67 (C = N–NH), and 159.97, 168.15 (2C = O) ppm; MS: 301 [M + –1]. Analysis calculated for C15H18N4O3 (302.34): C, 59.59; H, 6.00; and N, 18.53. Found: C, 59.68; H, 6.35; and N, 18.43.
2-[N′-(Cyano-hexylcarbamoyl-methylene)-hydrazino]-benzoic acid methyl ester (4b)
Yellow crystals from ethanol; yield 67%, mp 140°C; IR νmax cm−1: 3391 (NH), 3023 (CH aromatic), 2951 (CH aliphatic), 2215 (CN), 1697 (C = O ester), and 1670 (C = O amide); 1H NMR; (DMSO-d 6); Δ = 0.83 (t, 3H, CH3CH2, J=6 Hz), 1.24 (m, 6H, 3CH2), 1.48 (m, 2H, CH2, J=7 Hz), 3.17 (q, 2H, CH2, J=7 Hz), 3.88 (s, 3H, ester CH3), 7.16 (t, 1H, Ar H, J=7 Hz), 7.63 (t, 1H, Ar H, J=7 Hz), 7.95 (d, 1H, Ar H, J=8 Hz), 8.12 (d, 1H, Ar H, J=8 Hz), 8.50 (t, 1H, NH, J=5 Hz), and 14.97 (s, 1H, NH) ppm; MS: 330 [M+]. Analysis calculated for C17H22N4O3 (330.39): C, 61.80; H, 6.71; and N, 16.96. Found: C, 61.74; H, 6.98; and N, 16.79.
2-[N′-(Cyano-cyclohexylcarbamoyl-methylene)-hydrazino]-benzoic acid methyl ester (4c)
Orange crystals from ethanol; yield 77%, mp 166°C; IR νmax cm−1: 3290 (NH), 3032 (CH aromatic), 2935 (CH aliphatic), 2210 (CN), 1720 (C = O ester), and 1689 (C = O amide); 1H NMR; (DMSO-d 6); Δ = 1.09–1.62 (m, 6H, 3CH2), 1.78–2.11 (m, 4H, 2CH2), 3.62–3.88 (m, 1H, cyclohexyl CH), 3.91 (s, 3H, COOCH3), 7.01 (d, 1H, NH, J=7 Hz), 7.20 (t, 1H, Ar H, J=7 Hz), 7.70 (t, 1H, Ar H, J=7 Hz), 7.79 (d, 1H, Ar H, J= 8 Hz), 8.13 (d, 1H, Ar H, J=8 Hz), and 12.28 (s, 1H, NH) ppm; 13C NMR; (DMSO-d 6); Δ = 25.58, 25.73, 32.76, 49.01 (cyclohexyl carbons), 53.33 (ester CH3), 111.24, 113.23, 113.54, 123.61, 131.22, 135.48 (C6H4COOCH3-o), 116.66 (CN), 143.62 (C = N–NH), and 159.17, 168.15 (2C = O) ppm; MS: 327[M+–1]. Analysis calculated for C17H20N4O3 (328.37): C, 62.18; H, 6.14; and N, 17.06. Found: C, 62.26; H, 6.32; and N, 17.00.
N′-Butyl-2-[(4-chlorophenyl)-hydrazono]-2-cyano-acetamide (4d)
Brown crystals from ethanol; yield 90%, mp 185°C; IR νmax cm−1: 3380 (2NH), 3086 (CH aromatic), 2927 (CH aliphatic), 2211 (CN), and 1646 (C = O); 1H NMR; (DMSO-d 6); Δ = 0.86 (t, 3H, CH3, J=7 Hz), 1.28 (m, 2H, CH2, J=7 Hz), 1.45 (m, 2H, CH2 , J=7 Hz), 3.19 (q, 2H, CH2, J=7 Hz), 7.35 (d, 2H, Ar H, J=8 Hz), 7.63 (d, 2H, Ar H, J=8 Hz), 8.26 (t, 1H, NH, J=5 Hz), and 13.90 (s, 1H, NH) ppm; MS: 277[M+–1]. Analysis calculated for C13H15ClN4O (278.74): C, 56.02; H, 5.42; and N, 20.10. Found: C, 56.15; H, 5.58; and N, 20.22.
2-[(4-Chlorophenyl)-hydrazono]-2-cyano-N-hexyl-acetamide (4e)
Orange crystals from ethanol; yield 85%, mp 176°C; IR νmax cm−1: 3391 (NH), 3089 (CH aromatic), 2939 (CH aliphatic), 2212 (CN), and 1654 (C = O); 1H NMR; (CDCl3); Δ = 0.60 (t, 3H, CH3, J=6 Hz), 1.30 (m, 6H, 3CH2), 2.29 (m, 2H, CH2, J=7 Hz), 3.05 (q, 2H, CH2, J=7 Hz), 6.90 (t, 1H, NH, J=5 Hz), 6.98 (dd, 2H, Ar H, J=8 Hz), 7.18 (d, 2H, Ar H, J=8 Hz), and 11.10 (s, 1H, NH) ppm; 13C NMR; (CDCl3); Δ = 13.93, 22.36, 26.42, 29.53, 31.30 (hexyl carbons), 108.74, 128.83, 129.02, 140.75 (C6H4–Cl-p), 117.06 (CN), 157.30 (C = N–NH), and 160.56 (C = O) ppm; MS: 306 [M+]. Analysis calculated for C15H19ClN4O (306.80): C, 58.73; H, 6.24; and N, 18.26. Found: C, 58.51; H, 6.15; and N, 18.40.
N′-Benzyl-2-[(4-chlorophenyl)-hydrazono]-2-cyano-acetamide (4f)
Yellow crystals from ethanol; yield 85%, mp 129°C; IR νmax cm−1: 3336 (NH), 3032 (CH aromatic), 2928 (CH aliphatic), 2218 (CN), and 1643 (C = O); 1H NMR; (DMSO-d 6); Δ = 4.29 (d, 2H, CH2ph, J=5 Hz), 7.23–7.36 (m, 5H, Ph–H), 7.41 (d, 2H, Ar–H, J=8 Hz), 7.70 (d, 2H, Ar–H, J=8 Hz), 9.12 (t, 1H, NH, J=5 Hz), and 13.83 (s, 1H, NH) ppm;13C NMR; (DMSO-d 6); Δ = 43.02 (CH2ph), 107.22, 108.91, 111.87, 127.58 (C6H4Cl-p), 116.75 (CN), 127.95, 128.82, 128.91, 139.10 (phenyl carbons), 141.72 (C = N–NH), and 162.73 (C = O) ppm; MS: 311 [M+–1]. Analysis calculated for C16H13ClN4O (312.76): C, 61.45; H, 4.19; and N, 17.91. Found: C, 61.65; H, 4.29; and N, 17.85.
N′-Butyl-2-cyano-2-[(4-nitrophenyl)-hydrazono]-acetamide (4g)
Orange crystals from ethanol; yield 89%, mp 144°C; IR νmax cm−1: 3242 (NH), 3072 (CH aromatic), 2957 (CH aliphatic), 2220 (CN), and 1660 (C = O); 1H NMR; (CDCl3); Δ = 0.77 (t, 3H, CH3, J=7 Hz), 1.22 (m, 2H, CH2, J=7 Hz), 1.41 (m, 2H, CH2, J=7 Hz), 3.22 (q, 2H, CH2, J=7 Hz), 7.06 (t, 1H, NH, J=5 Hz), 7.44 (d, 2H, Ar H, J=8 Hz), 8.03 (d, 2H, Ar H, J=8 Hz), and 11.60 (s, 1H, NH) ppm;13C NMR; (CDCl3); Δ = 13.78, 20.04, 31.11, 31.68 (butyl carbons), 110.57, 112.10, 123.37, 125.32, 134.72, 143.27 (C6H4–NO2-p), 115.58 (CN), 147.42 (C = N–NH), and 160.04 (C = O) ppm; MS: 288 [M+–1]. Analysis calculated for C13H15N5O3 (289.30): C, 53.97; H, 5.23; and N, 24.21. Found: C, 53.83; H, 5.42; and N, 24.22.
2-Cyano-N-hexyl-2-[(4-nitrophenyl)-hydrazono]-acetamide (4h)
Brown crystals from ethanol; yield 92%, mp 186°C; IR νmax cm−1: 3369 (NH), 3091 (CH aromatic), 2929 (CH aliphatic), 2218 (CN), and 1656 (C = O); 1H NMR; (DMSO-d 6); Δ = 0.84 (t, 3H, CH3, J=6 Hz), 1.26 (m, 6H, 3CH2), 1.49 (m, 2H, CH2, J=7 Hz), 3.21 (q, 2H, CH2, J=7 Hz), 7.80 (d, 2H, Ar H, J=8 Hz), 8.19 (d, 2H, Ar H, J=8 Hz), 8.50 (t, 1H, NH, J=5 Hz), and 13.88 (s, 1H, NH) ppm; 13C NMR; (DMSO-d 6); Δ = 14.44, 22.59, 26.65, 29.74, 31.59 (hexyl carbons), 111.33, 112.51, 125.74, 143.13 (C6H4–NO2-p), 116.32 (CN), 148.32 (C = N–NH), and 160.26 (C = O) ppm; MS: 316 [M+–1]. Analysis calculated for C15H19N5O3 (317.35): C, 56.77; H, 6.03; and N, 22.07. Found: C, 56.69; H, 6.15; and N, 22.31.
2-Cyano-N-cyclohexyl-2-[(4-nitrophenyl)-hydrazono]-acetamide (4i)
Brown crystals from ethanol; yield 88%, mp 212°C; IR νmax cm−1: 3313 (NH), 3020 (CH aromatic), 2931 (CH aliphatic), 2214 (CN), and 1651 (C = O); 1H NMR; (DMSO-d 6); Δ = 1.09–410 (m, 11H, cyclohexyl H), 7.58 (d, 2H, Ar H, J=8 Hz), 8.07 (d, 2H, Ar H, J=8 Hz), 8.24 (d, 1H, NH, J=7 Hz), and 13.71 (s, 1H, NH) ppm; MS: 314 [M+–1]. Analysis calculated for C15H17N5O3 (315.33): C, 57.14; H, 5.43; and N, 22.21. Found: C, 57.26; H, 5.18; and N, 22.11.
Preparation of 5a–h
Method I (Δ)
To a solution of hydroxylamine hydrochloride (0.1 mol) and hydrazono-2-cyanoacetamide derivatives 4a–i (0.1 mol) in ethanol (50 mL), anhydrous sodium acetate (0.1 mol) was added and the reaction mixture was refluxed for 1 h. After concentration and cooling to room temperature, the solid product so-formed was filtered and re-crystallized from ethanol.
Method II (µω)
A mixture of hydroxylamine hydrochloride (0.1 mol), hydrazono-2-cyanoacetamide derivatives 4a–i (0.1 mol), anhydrous sodium acetate, and drops of ethanol was irradiated under Mw irradiation at 460 W for 1–5 min, until no starting materials were present (monitored by TLC) in 1-min intervals. The reaction mixture was left to cool to room temperature. The solid product so-formed was filtered and re-crystallized from ethanol.
Method III (Us)
To a solution of hydroxylamine hydrochloride (0.1 mol) and hydrazono-2-cyanoacetamide derivatives 4a–i (0.1 mol) in ethanol (50 mL), anhydrous sodium acetate (0.1 mol) was added and the reaction mixture was irradiated under Us irradiation at 40°C for 30 min, until no starting materials were present (monitored by TLC). The solid product so-formed was filtered and re-crystallized from ethanol.
2-{N-[Hexylcarbamoyl-(N-hydroxycarbamimidoyl)-methylene]-hydrazino}-benzoic acid methyl ester (5a)
Yellow crystals from ethanol; mp 130°C; IR νmax cm−1: 3587 (br OH), 3456, 3420 (NH2), 3379 (br 2NH), 3097 (CH aromatic), 2955 (CH aliphatic), 1701 (C = O ester), and 1651 (C = O amide); 1H NMR; (CDCl3); Δ = 0.87 (t, 3H, CH3, J=6 Hz), 1.35 (m, 6H, 3CH2), 1.59 (m, 2H, CH2, J=7 Hz), 3.34 (q, 2H, CH2, J=7 Hz), 3.87 (s, 3H, ester CH3), 6.52 (s, 2H, NH2), 6.98 (t, 1H, NH, J=5 Hz), 7.16 (t, 1H, Ar H, J=7 Hz), 7.50 (t, 1H, Ar H, J=7 Hz), 7.70 (d, 1H, Ar H, J=8 Hz), 7.96 (d, 1H, Ar H, J=8 Hz), 13.88 (s, 1H, NH), and 14.21 (s, 1H, OH) ppm; 13C NMR; (CDCl3); Δ = 14.10, 22.68, 26.76, 29.68, 31.56, 39.48 (hexyl carbons), 52.33 (ester CH3), 113.98, 114.65, 121.31, 122.23, 131.34, 134.29 (C6H4COOCH3-o), 145.30 (C = N–NH), 151.35 (C = N–OH), 165.70 (HN–C = O), and 167.29 (COOCH3) ppm; MS: 363 [M+]. Analysis calculated for C17H25N5O4 (363.42): C, 56.19; H, 6.93; and N, 19.27. Found: C, 56.31; H, 6.45; and N, 19.55.
2-{N-[Cyclohexylcarbamoyl-(N-hydroxycarbamimidoyl)-methylene]-hydrazino}-benzoic acid methyl ester (5b)
Brown crystals from ethanol; mp 134°C; IR νmax cm−1: 3520 (br OH), 3423, 3401 (NH2), 3279 (NH), 3088 (CH aromatic), 2933 (CH aliphatic), and 1650 (2C = O); MS: 361 [M+]. Analysis calculated for C17H23N5O4 (361.40): C, 56.50; H, 6.41; and N, 19.38. Found: C, 56.36; H, 6.70; and N, 19.25.
N-Butyl-2-[(4-chlorophenyl)-hydrazono]-2-(N-hydroxycarbamimidoyl)-acetamide (5c)
Orange crystals from ethanol; mp 86°C; IR νmax cm−1: 3568 (OH), 3498, 3471 (NH2), 3379, 3356 (NH), 3047 (CH aromatic), 2958 (CH aliphatic), and 1643 (C = O); 1H NMR; (DMSO-d 6); Δ = 0.89 (t, 3H, CH3, J=7 Hz), 1.30 (m, 2H, CH2, J=7 Hz), 1.46 (m, 2H, CH2, J=7 Hz), 3.21 (q, 2H, CH2, J=7 Hz), 6.72 (s, 2H, NH2), 7.29 (d, 2H, Ar H, J=8 Hz), 7.44 (d, 2H, Ar H, J=8 Hz), 8.32 (t, 1H, NH, J=5 Hz), 10.11 (s, 1H, NH), and 13.48 (s, 1H, OH) ppm; 13C NMR; (DMSO-d 6); Δ = 14.20, 19.10, 20.24, 20.29 (butyl carbons), 116.21, 122.76, 126.39, 129.73 (C6H4–Cl-p), 142.50 (C = N–NH), 151.05 (C = N–OH), and 163.85 (C = O) ppm; MS: 310 [M+–1]. Analysis calculated for C13H18 ClN5O2 (311.77): C, 50.08; H, 5.82; and N, 22.46. Found: C, 50.45; H, 5.75; and N, 22.36.
2-[(4-Chlorophenyl)-hydrazono]-N-hexyl-2-(N-hydroxycarbamimidoyl)-acetamide (5d)
Colorless crystals from ethanol; mp 82°C; IR νmax cm−1: 3585 (br OH), 3470, 3434 (NH2), 3392 (NH), 3085 (CH aromatic), 2918 (CH aliphatic), and 1636 (C = O); 1H NMR; (DMSO-d 6); Δ = 0.84 (t, 3H, CH3, J=6 Hz), 1.05 (m, 6H, 3CH2), 1.43 (quintet, 2H, CH2, J=7 Hz), 3.17 (t, 2H, CH2, J=7 Hz), 6.66 (s, 2H, NH2), 7.26 (d, 2H, Ar H, J=8 Hz), 7.39 (d, 2H, Ar H, J=8 Hz), 8.22 (t, 1H, NH, J=5 Hz), 13.36 (s, 1H, NH), and 13.52 (s, 1H, OH) ppm; MS: 338 [M+–1]. Analysis calculated for C15H22N5O2Cl (339.83): C, 53.02; H, 6.53; and N, 20.61. Found: C, 53.31; H, 6.45; and N, 20.55.
N-Benzyl-2-[(4-chlorophenyl)-hydrazono]-2-(N-hydroxycarbamimidoyl)-acetamide (5e)
Yellow crystals from ethanol; mp 135°C; IR νmax cm−1: 3580 (OH), 3483, 3432 (NH2), 3390 (NH), 3091 (CH aromatic), 2923 (CH aliphatic), and 1640 (C = O); 1H NMR; (CDCl3); Δ = 4.51 (d, 2H, CH2ph, J=5 Hz), 6.65 (s, 2H, NH2), 7.31–7.80 (m, 9H, Ar–H), 9.62 (t, 1H, NH, J=5 Hz), 13.10 (s, 1H, NH), and 13.98 (s, 1H, OH) ppm; MS: 344 [M+–1]. Analysis calculated for C16H16N5O2Cl (345.79): C, 55.58; H, 4.66; and N, 20.25. Found: C, 55.70; H, 4.52; and N, 20.16.
N-Butyl-2-(N-hydroxycarbamimidoyl)-2-[(4-nitrophenyl)-hydrazono]-acetamide (5f)
Brown crystals from ethanol; mp185°C; IR νmax cm−1: 3535 (OH), 3471, 3451 (NH2), 3391 (NH), 3052 (CH aromatic), 2918 (CH aliphatic), and 1643 (C = O); 1H NMR; (DMSO-d 6); Δ = 0.87 (t, 3H, CH3, J=7 Hz), 1.32 (m, 2H, CH2, J=7 Hz), 1.45 (m, 2H, CH2, J=7 Hz), 3.20 (q, 2H, CH2, J=7 Hz), 6.62 (s, 2H, NH2), 7.57 (d, 2H, Ar H, J=8 Hz), 8.16 (d, 2H, Ar H, J=8 Hz), 9.41 (t, 1H, NH, J=5 Hz), 13.10 (s, 1H, NH), and 13.65 (s, 1H, OH) ppm; MS: 322 [M+]. Analysis calculated for C13H18N6O4 (322.33): C, 48.44; H, 5.63; and N, 26.07. Found: C, 48.22; H, 5.75; and N, 26.15.
N-Hexyl-2-(N-hydroxycarbamimidoyl)-2-[(4-nitrophenyl)-hydrazono]-acetamide (5g)
Brown crystals from ethanol; mp 195°C; IR νmax cm−1: 3548 (OH), 3491, 3442 (NH2), 3385 (NH), 3049 (CH aromatic), 2922 (CH aliphatic), and 1653 (C = O); MS: 350 [M+]. Analysis calculated for C15H22N6O4 (350.38): C, 51.42; H, 6.33; N, 23.99. Found: C, 51.56; H, 6.29; and N, 23.81.
N-Cyclohexyl-2-(N-hydroxycarbamimidoyl)-2-[(4-nitrophenyl)-hydrazono]-acetamide (5h)
Brown crystals from ethanol; mp 282°C; IR νmax cm−1: 3501 (br OH), 3454, 3398 (NH2), 3242 (NH), 3103 (CH aromatic), 2925 (CH aliphatic), and 1644 (C = O); 1H NMR; (DMSO-d 6); Δ = 1.11–1.65 (m, 6H, 3CH2), 1.78–2.12 (m, 4H, 2CH2), 3.61–3.89 (m, 1H, cyclohexyl CH), 6.62 (s, 2H, NH2), 7.59 (d, 2H, Ar H, J=8 Hz), 8.05 (d, 2H, Ar H, J=8 Hz), 9.52 (d, 1H, NH, J=7 Hz), 13.23 (s, 1H, NH), and 13.56 (s, 1H, OH) ppm; MS: 348 [M+]. Analysis calculated for C15H20N6O4 (348.36): C, 51.72; H, 5.79; and N, 24.12. Found: C, 51.61; H, 5.50; and N, 24.38.
Preparation of triazole compounds 7a–e
Method I (Δ)
To a solution of compounds 5c and 5e–h (0.1 mol) in DMF (10 mL), triethylamine (0.1 mol) was added. The reaction mixture was heated under reflux for 1 h. Then, it was left to cool to room temperature. The solid product so-formed was filtered and re-crystallized from ethanol.
Method II (µω)
A mixture of compounds 5c and 5e–h (0.1 mol) and triethylamine (0.1 mol) was placed in a tightly closed tube and subjected to a Mw irradiation for 1–5 min until completion of the reaction (monitored by TLC). The reaction mixture was left to cool to room temperature. The solid product so-formed was filtered and re-crystallized from ethanol.
Method III (Us)
Triethylamine (0.1 mol) was added to a solution of compounds 5c and 5e–h (0.1 mol) in DMF (10 mL). The reaction mixture was irradiated under Us irradiation at 40°C for 1 h. Then, it was left to cool to room temperature. The solid product so-formed was filtered and re-crystallized from ethanol.
5-Amino-2-(4-chlorophenyl)-2H-[1,2,3]triazole-4-carboxylic acid butyl amide (7a)
Yellow crystals from ethanol; mp120°C; IR νmax cm−1: 3474, 3431 (NH2), 3335 (NH), 3080 (CH aromatic), 2930 (CH aliphatic), and 1648 (C = O); 1H NMR; (CDCl3); Δ = 0.93 (t, 3H, CH3, J=7 Hz), 1.39 (m, 2H, CH2, J=7 Hz), 1.57 (m, 2H, CH2, J=7 Hz), 3.34 (q, 2H, CH2, J= 7 Hz), 6.69 (s, 2H, NH2), 7.08 (d, 2H, Ar H, J=8 Hz), 7.26 (d, 2H, Ar H, J=8 Hz), and 9.28 (t, 1H, NH, J=5 Hz) ppm; 13C NMR; (CDCl3); Δ = 13.87, 20.26, 31.34, 38.81 (butyl carbons), 115.44, 119.34, 128.06, 129.48 (C6H4–Cl-p), 141.46, 153.50 (triazole carbons), and 165.67 (C = O) ppm; MS: 293 [M+]. Analysis calculated for C13H16ClN5O (293.76): C, 53.15; H, 5.49; N, 23.84. Found: C, 53.26; H, 5.35; and N, 23.80.
5-Amino-2-(4-chlorophenyl)-2H-[1,2,3]triazole-4-carboxylic acid benzyl amide (7b)
Brown crystals from ethanol; mp 137°C; IR νmax cm−1: 3461, 3430 (NH2), 3221 (NH), 3021 (CH aromatic), 2920 (CH aliphatic), and 1642 (C = O); 1H NMR; (CDCl3); Δ = 5.21 (d, 2H, CH2ph, J=5 Hz), 6.67 (s, 2H, NH2), 7.31–7.83 (m, 9H, Ar–H), and 7.97 (t, 1H, NH, J=5 Hz) ppm; MS: 327 [M+]. Analysis calculated for C16H14ClN5O (327.78): C, 58.63; H, 4.31; and N, 21.37. Found: C, 58.60; H, 4.47; and N, 21.41.
5-Amino-2-(4-nitrophenyl)-2H-[1,2,3]triazole-4-carboxylic acid butyl amide (7c)
Brown crystals from ethanol; mp103°C; IR νmax cm−1: 3484, 3452 (NH2), 3325 (NH), 3099 (CH aromatic), 2990 (CH aliphatic), and 1658 (C = O); 1H NMR; (DMSO-d 6); Δ= 0.93 (t, 3H, CH3, J=7 Hz), 1.35 (m, 2H, CH2, J=7 Hz), 1.56 (m, 2H, CH2, J=7 Hz), 3.41 (q, 2H, CH2, J=7 Hz), 6.68 (s, 2H, NH2), 7.19 (d, 2H, Ar H, J=8 Hz), 7.51 (d, 2H, Ar H, J=8 Hz), and 9.31 (t, 1H, NH, J=5 Hz) ppm; MS: 304 [M+]. Analysis calculated for C13H16N6O3 (304.31): C, 51.31; H, 5.30; and N, 27.62. Found: C, 51.24; H, 5.42; and N, 27.51.
5-Amino-2-(4-nitrophenyl)-2H-[1,2,3]triazole-4-carboxylic acid hexyl amide (7d)
Brown crystals from ethanol; mp 176°C; IR νmax cm−1: 3492, 3386 (NH2), 3344 (NH), 3053 (CH aromatic), 2920 (CH aliphatic), and 1653 (C = O); 1H NMR; (DMSO-d 6); Δ = 0.84 (t, 3H, CH3, J=6 Hz), 1.27 (m, 6H, 3CH2), 1.48 (m, 2H, CH2, J=7 Hz), 3.19 (t, 2H, CH2, J=7 Hz), 6.18 (s, 2H, NH2), 7.58 (d, 2H, Ar H, J =8 Hz), 8.12 (d, 2H, Ar H, J=8 Hz), and 9.40 (t, 1H, NH, J=5 Hz) ppm; MS: 332 [M+]. Analysis calculated for C15H20N6O3 (332.37): C, 54.21; H, 6.07; and N, 25.29. Found: C, 54.29; H, 6.16; and N, 25.32.
5-Amino-2-(4-nitrophenyl)-2H-[1,2,3]triazole-4-carboxylic acid cyclohexyl amide (7e)
Brown crystals from ethanol; mp167°C; IR νmax cm−1: 3479, 3448 (NH2), 3367 (NH), 3078 (CH aromatic), 2924 (CH aliphatic), and 1651 (C = O); 1H NMR; (DMSO-d 6); Δ = 1.07–1.67 (m, 6H, 3CH2), 1.71–2.86 (m, 4H, 2CH2), 2.93–3.80 (m, 1H, cyclohexyl CH), 7.20–8.28 (m, 4H, Ar–H), 6.70 (s, 2H, NH2), and 9.32 (d, 1H, NH, J=7 Hz) ppm; 13C NMR; (DMSO-d 6); Δ = 24.59, 25.62, 29.75, 32.48 (cyclohexyl carbons), 113.56, 125.01, 125.33, 125.96 (C6H4–NO2-p), 142.38, 147.92 (triazole carbons), and 162.33 (C = O) ppm; MS: 330 [M+]. Analysis calculated for C15H18N6O3 (330.35): C, 54.54; H, 5.49; and N, 25.44. Found: C, 54.39; H, 5.35; and N, 25.20.
General method to reaction of triazole compounds 7a–e with dimethylformamide dimethylacetal (DMF DMA)
Method I (Δ)
To a solution of compounds 7a–e (0.1 mol) in dry xylene (20 mL), DMF DMA (0.1 mol) was added. The reaction mixture was refluxed for 30 min. Then it was left to cool to room temperature, and poured into ice-cold water. The solid product so-formed was filtered and re-crystallized from ethanol.
Method II (µω)
A mixture of compounds 7a–e (0.1 mol) and of DMF DMA (0.1 mol) was placed in a tightly closed tube, and subjected to a Mw irradiation for 2–5 min until completion of the reaction (monitored by TLC). The reaction mixture was left to cool to room temperature, and then poured into ice-cold water. The solid product so-formed was filtered and re-crystallized from ethanol.
2-(4-Chlorophenyl)-6-hexyl-2,6-dihydro-[1,2,3]triazolo-[4,5-d]pyrimidin-7-one (10a)
Yellow crystals from ethanol; mp 78°C; IR νmax cm−1: 3089 (CH aromatic), 2925 (CH aliphatic), and 1645 (C = O); 1H NMR; (CDCl3); Δ = 0.91 (t, 3H, CH3, J=6 Hz), 1.29 (m, 6H, 3CH2), 1.47 (m, 2H, CH2, J=7 Hz), 3.29 (t, 2H, CH2, J=7 Hz), 7.05 (s, 1H, pyrimidine CH), 7.32 (d, 2H, Ar, H, J=8 Hz), and 7.61 (d, 2H, Ar, H, J=8 Hz) ppm; MS: 331 [M+]. Analysis calculated for C16H18ClN5O (331.81): C, 57.92; H, 5.47; and N, 21.11. Found: C, 57.75; H, 5.25; and N, 21.47.
6-Butyl-2-(4-nitrophenyl)-2,6-dihydro-[1,2,3]triazolo-[4,5-d]pyrimidin-7-one (10b)
Brown crystals from ethanol; mp 190°C; IR νmax cm−1: 3092 (CH aromatic), 2939 (CH aliphatic), and 1670 (C = O); 1H NMR; (CDCl3); Δ = 0.89 (t, 3H, CH3, J=6 Hz), 1.40 (m, 2H, CH2, J=7 Hz), 1.66 (m, 2H, CH2, J=7 Hz), 3.49 (q, 2H, CH2, J=7 Hz), 7.15 (s, 1H, pyrimidine CH), 7.31 (d, 2H, Ar, H, J=8 Hz), and 7.65 (d, 2H, Ar, H, J=8 Hz) ppm; MS: 314 [M+]. Analysis calculated for C14H14N6O3 (314.31): C, 53.50; H, 4.49; and N, 26.74. Found: C, 53.72; H, 4.64; and N, 26.59.
Preparation of pyrazole compounds 13a–e
Method I (Δ)
To a solution of compounds 4a–i (0.1 mol) in triethylamine (20 mL) and chloroacetonitrile (0.1 mol) were refluxed for 30 min. The reaction mixture was left to cool to room temperature, and then poured into ice-cold water. The solid product so-formed was filtered and re-crystallized from ethanol.
Method II (µω)
To a mixture of compounds 4a–i (0.1 mol) and chloroacetonitrile (0.1 mol), a few drops from triethylamine were added, then the mixture was placed in a tightly closed tube and subjected to a Mw irradiation for 2 min until completion of the reaction (monitored by TLC). The reaction mixture was left to cool to room temperature, and then poured into ice-cold water. The solid product so-formed was filtered and re-crystallized from ethanol.
Method III (Us)
Chloroacetonitrile (0.1 mol) was added to a solution of compounds 4a–i (0.1 mol) in triethyl amine (20 mL), under Us irradiation at 40°C for 30 min. The reaction mixture was left to cool to room temperature, and then poured into ice-cold water. The solid product so-formed was filtered and re-crystallized from ethanol.
2-(4-Amino-5-cyano-3-cyclohexylcarbamoyl-pyrazol-1-yl)-benzoic acid methyl ester (13a)
Brown crystals from ethanol; mp >300°C; IR νmax cm−1: 3438, 3395, 3277 (NH, NH2), 3088 (CH aromatic), 2933 (CH aliphatic), 2261 (CN), and 1648 (C = O); 1H NMR; (CDCl3); Δ = 1.18–3.77 (m, 11H, cyclohexyl H), 3.93 (s, 3H, ester CH3), 6.67 (s, 2H, NH2), 7.20 (t, 1H, Ar H, J=7 Hz), 7.44 (t, 1H, Ar H, J=7 Hz), 7.63 (d, 1H, Ar H, J=8 Hz), 7.94 (d, 1H, Ar H, J=8 Hz), and 9.75 (t, 1H, NH, J=7 Hz) ppm; MS: 367 [M+]. Analysis calculated for C19H21N5O3 (367.41): C, 62.11; H, 5.76; and N, 19.06. Found: C, 62.26; H, 5.80; and N, 19.17
4-Amino-1-(4-chlorophenyl)-5-cyano-1H-pyrazole-3-cyclohexylic acid butyl amide (13b)
Brown crystals from ethanol; mp 117°C; IR νmax cm−1: 3472, 3378, 3329 (NH, NH2), 3020 (CH aromatic), 2957 (CH aliphatic), 2212 (CN), and 1644 (C = O); 1H NMR; (DMSO-d 6); Δ = 0.91 (t, 3H, CH3, J=7 Hz), 1.37 (m, 2H, CH2, J=7 Hz), 1.54 (m, 2H, CH2, J=7 Hz), 3.38 (q, 2H, CH2, J=7 Hz), 6.71 (s, 2H, NH2), 7.43 (d, 2H, Ar H, J=8 Hz), 7.59 (d, 2H, Ar H, J=8 Hz), and 9.34 (t, 1H, NH, J=5 Hz) ppm; 13C NMR; (DMSO-d 6); Δ = 13.78, 20.14, 31.80, 38.71 (butyl carbons), 116.93 (CN), 110.93, 123.07, 129.66, 132.87 (C6H4–Cl-p), 134.48, 137.01, 143.09 (pyrazole carbons), and 162.25 (C = O) ppm; MS: 317 [M+]. Analysis calculated for C15H16ClN5O (317.78): C, 56.70; H, 5.08; and N, 22.04. Found: C, 56.62; H, 5.20; and N, 22.29.
4-Amino-1-(4-chlorophenyl)-5-cyano-1H-pyrazole-3-cyclohexylic acid hexyl amide (13c)
Brown crystals from ethanol; mp 125°C; IR νmax cm−1: 3450, 3428, 3388 (NH, NH2), 3109 (CH aromatic), 2926 (CH aliphatic), 2210 (CN), and 1645 (C = O); 1H NMR; (DMSO-d 6); Δ = 0.88 (t, 3H, CH3, J=6 Hz), 1.30 (m, 6H, 3CH2), 1.57 (m, 2H, CH2, J=7 Hz), 3.38 (t, 2H, CH2, J=7 Hz), 6.49 (s, 2H, NH2), 7.15 (d, 2H, Ar H, J= 8 Hz), 7.36 (d, 2H, Ar H, J=8 Hz), and 8.96 (t, 1H, NH, J=5 Hz) ppm; MS: 345 [M+]. Analysis calculated for C17H20ClN5O (345.83): C, 59.04; H, 5.83; and N, 20.25. Found: C, 59.17; H, 5.42; and N, 20.54.
4-Amino-1-(4-chlorophenyl)-5-cyano-1H-pyrazole-3-cyclohexylic acid benzyl amide (13d)
Brown crystals from ethanol; mp 130°C; IR νmax cm−1: 3371, 3326, 3299 (NH, NH2), 3064 (CH aromatic), 2923 (CH aliphatic), 2225 (CN), and 1644 (C = O) cm−1; 1H NMR; (DMSO-d 6 ); Δ = 4.28 (d, 2H, phCH2, J=5 Hz), 6.21 (s, 2H, NH2), 7.25–8.10 (m, 9H, Ar–H), and 8.89 (t, 1H, NH, J=5 Hz) ppm; MS: 351 [M+]. Analysis calculated for C18H14ClN5O (351.80): C, 61.46; H, 4.01; and N, 19.91. Found: C, 61.54; H, 4.20; and N, 19.83.
4-Amino-5-cyano-1-(4-nitrophenyl)-1H-pyrazole-3-cyclohexylic acid hexyl amide (13e)
Brown crystals from ethanol; mp 153°C; IR νmax cm−1: 3401, 3358, 3226 (NH, NH2), 3090 (CH aromatic), 2927 (CH aliphatic), 2218 (CN), and 1660 (C = O); 1H NMR; (CDCl3); Δ = 0.88 (t, 3H, CH3, J=6 Hz), 1.32 (m, 6H, 3CH2), 1.58 (m, 2H, CH2, J=7 Hz), 3.40 (t, 2H, CH2, J=7 Hz), 6.59 (s, 2H, NH2), 7.40 (d, 2H, Ar H, J=8 Hz), 7.95 (d, 2H, Ar H, J=8 Hz), and 9.62 (t, 1H, NH, J=5 Hz) ppm; MS: 356 [M+]. Analysis calculated for C17H20N6O3 (356.39): C, 57.29; H, 5.66; and N, 23.58. Found: C, 57.20; H, 5.72; and N, 23.68.
X-ray crystallography
A single crystal of compound 4c was obtained by slow evaporation from a mixture of ethanol:DMF (2:1). The crystal structure was solved and refined using maxus (nonius, Deflt and MacScience, Japan) Citation21 Mo–Kα radiation (λ = 0.71073 Å) and a graphite monochromator were used for data collection. The chemical formula and ring labeling system is shown in . Crystallographic data (, excluding structure factors) for the structure in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 686225. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union, Road, Cambridge CB2 1EZ, UK [fax: 144-(0)1223-336033 or e-mail: [email protected]].
Table 2. Crystal data of compound 4c.
Related Research Data
References
- Al-Mousawi , S.M. ; Moustafa , M.S. Beilstein J. Org. Chem . 2007 , 3 , 12 .
- Ghozlan , S.A.S. ; Badahdah , K.O. ; Abdelhamid , I.A. Beilstein J. Org. Chem . 2007 , 3 , 15 – 17 .
- Gewald , K. ; Rehwald , M. ; Mueller , H. ; Bellmann , P. ; Schaefer , H. Monatsh. Chem . 1995 , 126 , 347 .
- Sotelo , E. ; Mocelo , R. ; Suarez , M. ; Loupy , A. Synth. Commun . 1997 , 27 , 2423 .
- Shawali , A.S. ; Abdelkader , M.H. ; Attalbawy , F.M. Tetrahedron 2002 , 58 , 2875 .
- Al-Mousawi , S. ; Elassar , A. ; El-Apasery , M.A. Phosphours Sulfur Silicon Relat. Elem . 2006 , 181 , 1755 .
- Al-Saleh , B. ; El-Apasery , M.A. ; Elnagdi , M.H. J. Chem. Res . 2004 , 580 .
- El-Dusouqui , O.M.E. ; Abdelkhalik , M.M. ; Al-Awadi , N.A. ; Dib , H.H. ; George , B.G. ; Elnagdi , M.H. J. Chem. Res . 2006 , 302 .
- Riyadh , S.M. ; Abdelhamid , I.A. ; Ibrahim , H.M. ; Al-Matar , H.M. ; Elnagdi , M.H. Heterocycles 2004 , 71 12 , 2586 .
- Elassar , A.A. ; Dib , H.H. ; Al-Awadi , N.A. ; Elnagdi , M.H. ARKIVOC 2007 , ii , 272 – 315 .
- Mason , T.J. Chem. Soc. Rev . 1997 , 26 , 443 – 451 .
- Ahluwalia , V.K. ; Goyal , B. ; Das , U. J. Chem. Res . 1997 , 266 .
- Loupy , A. ; Petit , A. ; Hamelin , J. ; Texier-Boullet , F. ; Jacquanlt , P. ; Mathe , D. Synth. Rev . 1998 , 1213 – 1234 .
- Varma , R.S. Green Chem . 1999 , 1 , 4 – 55 .
- Lidstrom , P. ; Tierney , J. ; Wathey , B. ; Westman , A. Tetrahedron 2001 , 57 , 9225 – 9283 .
- Rehorek , A. ; Tauber , M. ; Gubitz , G. Ultrason. Sonochem . 2004 , 11 , 177 .
- Al-Zaydi , K.M. ; Borik , R.M. ; Elnagdi , M.H. Molecules 2003 , 8 12 , 910 – 923 .
- Al-Zaydi , K.M. ; Elnagdi , M.H. J. Korean Chem. Soc . 2003 , 47 6 , 591 – 596 .
- Al-Zaydi , K.M. Molecules 2003 , 8 , 541 – 555 .
- Bonrath , W. Ultrason. Sonochem . 2004 , 11 , 1 – 4 .
- Bougrin , K. ; Loupy , A. ; Soufiaoui , M. J. Photo Chem. Photobiol. Rev . 2005 , 6 , 139 – 167 .
- Anwar , H.F. ; Metwally , N.H. ; Gaber , H. ; Elnagdi , M.H. J. Chem. Res . 2005 , 29 – 31 .
- Al-Zaydi , K.M. ; Borik , R.M. ; Elnagdi , M.H. Molecules 2007 , 12 , 2061 – 2079 .
- Al-Zaydi , K.M. ; Borik , R.M. ; Elnagdi , M.H. J. Heterocycl. Chem . 2007 , 44 , 1 – 6 .
- Al-Zaydi , K.M. ; Borik , R.M. ; Elnagdi , M.H. Ultrason. Sonochem . 2009 , 16 5 , 660 – 668 .
- Crystallographic data for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publications nos. CCDC . These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK; Fax: _44 1223 336033; E-mail: [email protected]) .
- Abdel-Motaleb , R.M. ; Makloof , A.A. ; Ibrahim , H.M. ; Elnagdi , M.H. J. Heterocycl. chem . 2006 , 43 , 931 – 934 .
- Hassaneen , H.M.E. Synth. Commun . 2007 , 37 , 3579 – 3765 .
- AL-Matar , H.M. ; Riyadh , S.M. ; Elnagdi , M.H. ARKIVOC 2007 , xiii , 53 – 62 .
- Abdallah , S.O. ; Metwally , N.H. ; Anwar , H.F. ; Elnagdi , M.H. J. Heterocycl. chem . 2005 , 42 , 781 – 786 .