Abstract
An efficient synthesis of 1,3-bis(1,4,7-triazacyclonon-1-yl)-2-hydroxypropane is described. In contrast to earlier procedures, an improved synthetic strategy led to the selection of suitable reagents and conditions that afforded the title compound in excellent overall yield with improved process efficiency and scalability. Moreover, the mildness and synthetic efficiency of the chosen conditions were particularly well-suited for the large-scale synthesis of the title compound, which is in increasing demand for a variety of applications, including the preparation of stationary phases for immobilized metal-ion affinity chromatography of recombinant proteins.
Introduction
The synthesis of macrocyclic ligands and their applications have attracted considerable interest across very diverse fields of use, ranging from medical imaging, catalysis through to self-assembling polymers Citation1. As part of our ongoing research efforts into more efficient methods of synthesis of 1,4,7-triazacyclononane (TACN) derivatives and their use in sensing devices, in solid state heterogeneous catalysis and various modes of separation, including immobilized metal-ion affinity chromatography (IMAC), we wished to prepare 1,3-bis(TACN)-2-propanol 1 in significant quantities to enable scale-up of several key process systems, including more efficient purification of biopharmaceutical targets, with reduced waste burden, i.e. with overall improved E factors, decreased water consumption, and with fewer unit operation process steps.
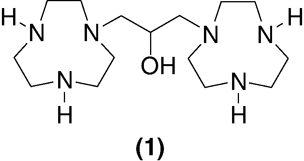
The synthesis of 1,3-bis(1,4,7-triaza-cyclonon-1-yl)-2-hydroxypropane 1 was first reported in 1993 by Sessler et al. Citation2, with confirmatory syntheses subsequently published Citation3, Citation4. Whilst all of these syntheses are amenable to the laboratory scale preparation of 1–2 g of the desired product, they are not practical for the larger scale reactions necessary for the preparation of 100 g–Kg quantities of 1, since they involve a variety of reactants, which is from a green chemistry point of view, are undesirable for multiple reasons ().
Figure 1. Structure of 1,3-bis(TACN)-2-propanol.
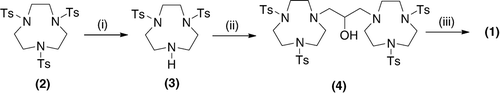
For example, in previously reported small-scale laboratory procedures Citation2–Citation4, the route to 1 involved the selective mono-detosylation of a tri-tosyl TACN 2 to generate the di-tosyl TACN 3. This approach is difficult to control and leads to low overall yields. An alternative approach, involving the conversion of the tri-tosylate 2 to the mono-tosyl TACN followed by selective mono-tosylation to generate the di-tosyl TACN 3 has also been attempted () Citation5, Citation6. Again, this alternative approach is inefficient in terms of yield of the desired product and also reagent utilization. Although conversion of 3–4 proceeds in moderate yield, the reported protocols for detosylation of the tetra-tosyl compound 4 require the use of a large excess of concentrated sulfuric acid at elevated temperatures, e.g. >100°C for 2 days. For the synthesis of the product 1 at larger preparative scales by this route, special handling conditions and equipment are required to accommodate the very large volumes of concentrated sulfuric acid that are employed, with significant amounts of waste generated whilst the product 1 is only obtained in low yields (<30%). Besides the process engineering constraints, as recognized by Sessler et al. Citation2, the use of the harsh acidic conditions required for such detosylations are also not suited for the preparation of 1, mainly due to the presence of the secondary alcohol group. Accordingly, these investigators explored an alternative laboratory scale method for the detosylation of 4 by reduction with 2% Na/Hg and obtained the product 1 in improved yield on a 1.5 mmol scale. However, application of this reductive method on a larger preparative scale would involve the use of prohibitive amounts of Na/Hg amalgam, i.e. even at the relatively modest preparative scale of 100 mmol, in excess of 2.5 Kg of 2% Na/Hg would be required. From OH&S considerations, waste disposal and environmental reasons, use and disposal of large quantities of Hg are unacceptable.
Scheme 1. Route to (1) using tosyl protecting groups.
Note: Reagents and conditions: (1) HBr/HOAc followed by TsCl; (2) BrCH2CH(OH)CH2Br, Et3N, CH3CN, 72 hours, reflux; and (3) concentrated H2SO4, reflux 2 days or 2% Na/Hg, MeOH, reflux 20 hours.
We therefore sought alternative procedures for the synthesis of 1 by taking advantage of modifications to a method described by Iranzo et al. Citation4, whereby the use of the readily removable t-Boc-protecting groups has been reported to allow the synthesis of 1.6 HCl in 25% overall yield from TACN 6. In the present study, it has been found that through the judicious selection of reagents, their stoichiometries and optimized conditions, a much more efficient synthesis of the title compound can be achieved, suitable for large-scale preparations to meet our process demands.
Results and discussion
The title compound 1.6 HCl was prepared as depicted (see ). TACN.3HCl (5) was first converted to the free base 6 in good yield. The free base was then converted to the bis-carbamate 7 as the precursor compound for use in the coupling reaction, (7→9). For the bis-N-protection of TACN 6, Iranzo et al. Citation4 used Boc-anhydride (1.9 equivalents) under standard conditions, to achieve the transformation (6→7) in 62% yield after chromatography Citation4. Using the same conditions, in our hands, we were only able to achieve after repetitive experiments a more modest 43% yield of the pure material (see , Entry 1), with the concomitant formation of the tris-carbamate 8 in 38% yield. Although Boc3TACN 8 could be deprotected to regenerate TACN.3HCl 5 in high yield, this was considered undesirable and uneconomical. Procedures to optimize the synthesis of the bis-carbamate 7 were thus investigated.
Scheme 2. Route to (1) using Boc-protecting groups.
Note: Reagents and conditions: (1) H2O,NaOH; toluene; (2) Boc-ON, Et3N, CH2Cl2, overnight, room temperature; (3) BrCH2CH(OH)CH2Br, Et3N, CH3CN, 72 hours, reflux; and (4) EtOH/concentrated HCl, overnight, room temperature.
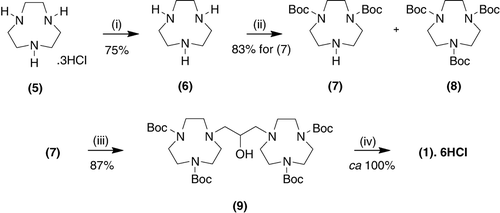
Table 1. Product distribution as a function of Boc-protecting reagent.
Kovacs and Sherry Citation7 have stated in a short note that the use of Boc-ON as a Boc-protecting reagent gave a quantitative yield of Boc2TACN 7, although no experimental conditions were specified. Through the use of Boc-ON, and conditions adapted from a procedure reported by Kimura et al. Citation8 for the synthesis of hexadentate, asymmetric pendant arm macrocycles containing a 1,4,7-triazacyclononane-1,4-diacetate backbone, we were able to readily prepare Boc2TACN 7 in 83% yield (see , Entry 2), in addition to a small amount of the undesired Boc3TACN 8. This reaction was carried out in dichloromethane, a greener substitute than the chloroform used in earlier literature work. In addition, this solvent was readily recycled with very high efficiency in a closed loop system, minimizing waste. Use of aqueous work-up conditions, moreover, circumvented the need for column chromatography, whilst attaining in one step the Boc2TACN 7 in high yield (>83%) and purity. Further adaptation of this procedure by directly using the tri-hydrochloride salt of TACN 5 led to deterioration in yields using either Boc-protecting reagent (see , Entries 3 and 4).
The Boc2TACN 7 was coupled with 1,3-dibromo-2-propanol to generate the bis-TACN derivative, 9. It was found that the use of a small excess (0.1 equivalents) of Boc2TACN 7 gave an excellent yield of the product 9 after separation from the residual Boc2TACN 7 by filtration through a plug of silica. The compound 9 was obtained in high purity and yield (87%). This precursor to the title compound (1) was deprotected using concentrated HCl in absolute ethanol to yield the desired product 1 as the 6 HCl salt in very high yield (ca 100%) and purity at 20-fold larger scales than hitherto achieved. Thus, overall the yield of 1.6 HCl from TACN 6 was >70%. Moreover, it can be noted that in contrast to the method reported by de Leon-Rodriguez et al. Citation9 for the 1,7-bis-Cbz protection/deprotection of cyclen, where the use of dichloromethane gave a lower yield (70%) than the 90% achieved using chloroform, in the case of 1 the reverse was found. The hydrochloride salt, 1.6 HCl, can readily be converted, if required, into the free base 1 in >90% yield.
Experimental
1,4-Bis(tert-butoxycarbonyl)-1,4,7-tri-azacyclononane 7
Triethylamine (81.3 mL, 583.3 mmol) was added to a solution of TACN 6 (25.4 g, 196.6 mmol) in dichloromethane (500 mL). Boc-ON (96.48 g, 391.8 mmol) dissolved in dichloromethane (500 mL) was added with stirring over an hour at room temperature. Stirring was maintained overnight and the mixture concentrated under reduced pressure. The resulting residue was dissolved in ethyl acetate (750 mL) and washed successively with 4% aqueous sodium bicarbonate (750 mL), and saturated aqueous sodium chloride (750 mL), and then extracted with 10% aqueous citric acid (3×750 mL). The citric acid extract was rendered alkaline (pH 10) by the addition of 10 M aqueous sodium hydroxide and was then extracted with dichloromethane (3×500 mL). The organic phase was dried over anhydrous sodium sulfate and concentrated under reduced pressure to yield the title compound 7 as an amber gum (53.5 g, 83%). The ethyl acetate layer was concentrated to yield pure Boc3TACN 8 (8.48 g, 10%). The 1H and 13C NMR and ESI mass spectral data were consistent with literature values Citation4, Citation7, Citation8.
1,3-Bis(4,7-di(tert-butoxycarbonyl)-1,4,7-triazacyclonon-1-yl)-2-hydroxy-propane 9
Triethylamine (27.2 mL, 195.2 mmol) and 1,3-dibromopropan-2-ol (7.89 mL, 77.3 mmol) were added to a solution of Boc2TACN 7 (53.5 g, 162.4 mmol) in acetonitrile (1.8 L). The reaction was stirred for 3 days at reflux. The mixture was concentrated to a dark amber gum which was dissolved in dichloromethane (500 mL) and washed consecutively with 10% aqueous sodium hydroxide (2×200 mL), water (200 mL), and brine (200 mL). The aqueous phases were combined and back-extracted with dichloro-methane (2×50 mL). The combined organic layers were concentrated to an amber gum which was redissolved in dichloromethane (ca 25 mL) and filtered through a plug of silica (100 mm diameter sinter containing 40 mm depth of silica) (ethyl acetate/hexanes, 1:1→2:1) to furnish the title compound 9 as a light yellow gum that expanded to a beige foam under high vacuum (48.3 g, 87%). The 1H and 13C NMR and ESI mass spectral data were consistent with literature values Citation4.
1,3-Bis(1,4,7-triazacyclonon-1-yl)-2-hydroxypropane HCl salt 1, 6HCl
Concentrated HCl (353 mL) was added gradually to a solution of the Boc4 compound 9 (48.26 g, 67.5 mmol) in absolute ethanol (353 mL) maintained at ca 25°C. The mixture was stirred overnight at room temperature then cooled in an ice bath. The white precipitate was collected by filtration and washed with cold ethanol followed by cold acetone and dried under vacuum to yield the pure hydrochloride salt of 1 in almost quantitative yield (37.9 g, 98% for 6 HCl.2.3 H2O). The 1H and 13C NMR and ESI mass spectral data were consistent with literature values Citation4.
Conclusion
In this study, we have demonstrated a greener and more efficient synthesis of the title compound 1.6 HCl, suitable for its large-scale preparation. Through a combination of modified conditions and the judicious choice of reagents, the title compound 1.6 HCl was synthesized in high overall yield and purity.
Acknowledgements
We thank Dr. Kei Saito and Ms Sweta Pant for their useful discussions on TACN synthesis. This work was supported by the Australian Research Council.
References
- Gloe , K. Macrocyclic Chemistry: Current Trends and Future Perspectives ; Springer : Dordrecht , 2005 .
- Sessler , J.L. ; Sibert , J.W. ; Burrell , A.K. ; Lynch , V. ; Markert , J.T. ; Wooten , C.L. Inorg. Chem . 1993 , 32 , 4277 – 4283 .
- Brudenell , S.J. ; Spiccia , L. ; Bond , A.M. ; Comba , P. ; Hockless , D.C.R. Inorg. Chem . 1998 , 37 , 3705 – 3713 .
- Iranzo , O. ; Elmer , T. ; Richard , J.P. ; Morrow , J.R. Inorg. Chem . 2003 , 42 , 7737 – 7746 .
- Sessler , J.L. ; Sibert , J.W. ; Lynch , V. Inorg. Chem . 1990 , 29 , 4143 – 4146 .
- Wieghardt , K. ; Tolksdorf , I. ; Herrmann , W. Inorg. Chem . 1985 , 24 , 1230 – 1235 .
- Kovacs , Z. ; Sherry , A.D. Tetrahed. Lett . 1995 , 36 , 9269 – 9272 .
- Kimura , S. ; Bill , E. ; Bothe , E. ; Weyhermuller , T. ; Wieghardt , K. J. Am. Chem. Soc . 2001 , 123 , 6025 – 6039 .
- De Leon-Rodriguez , L.M. ; Kovacs , Z. ; Esqueda-Oliva , A.C. ; Miranda-Olvera , A.D . Tetrahed. Lett . 2006 , 47 , 6937 – 6940 .