Abstract
The broad and potent activity of 4-thiazolidinones has established it as one of the biologically important scaffolds. The synthesis of N-(2-aryl-4-oxothiazolidin-3-yl)isonicotinamide by a novel method of stirring and sonication is described. The conventional method for synthesis of 4-thiazolidinones involves use of a Dean-Stark water separator for the removal of water from the reaction with long reaction times (12–48 h), and the stirring procedure also involves the use of DCC (dicyclohexylcarbodiimide) as a dehydrating agent. We report the synthesis of 4-thiazolidinone analogs of isonicotinic acid hydrazide by novel, green route methods of sonication and stirring using molecular sieves. Results indicate that high yields and shorter reaction times can be achieved by employing novel green route methods of synthesis.
Introduction
Green chemistry involves the use of microwave technology, sonochemistry, stirring, phase transfer catalysis, ionic liquids, and many more techniques. Among the traditional high-energy chemistry areas of principal theoretical and practical importance, such as radiation chemistry, plasma chemistry, photochemistry, and laser chemistry, sonochemistry occupies a special place as it studies the chemical and physicochemical processes induced by acoustic vibrations in a medium Citation1.
The broad and potent activity of 4-thiazolidinones has established it as one of the biologically important scaffolds Citation2. 4-Thiazolidinone analogs possess a wide spectrum of biological activities, such as anti-inflammatory Citation3–6, anticonvulsant Citation7, antibacterial Citation8–10, antifungal Citation11, antihistaminic Citation12 Citation13, FSH receptor agonist Citation14, anticancer Citation15, antiviral Citation16, and anthelmintic Citation17 Citation18 activities.
In the first step Schiff's bases of isoniazid were synthesized, which were further utilized for the synthesis of 4-thiazolidinone analogs by novel green route methods of sonication and stirring.
Results and discussion
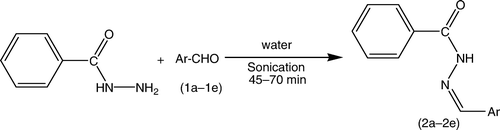
The synthetic strategies adopted to obtain the target compounds are depicted in and . The key intermediates (2a–2e) were prepared with an excellent yield in a one-step reaction () by our previously reported method of sonication Citation19. Furthermore, the intermediates that were synthesized represent versatile building blocks for the synthesis of new heterocycles incorporating a thiazole nucleus. The synthesized intermediates were characterized by the presence of a strong band at 1595–1625 cm−1, and 1H NMR spectra also showed a singlet signal equivalent to one proton for the =CH group between 7.4–8.4, which confirms the formation of Schiff's bases.
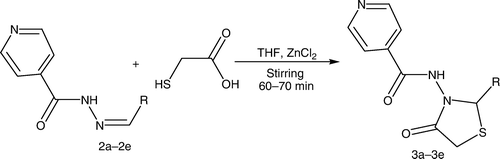
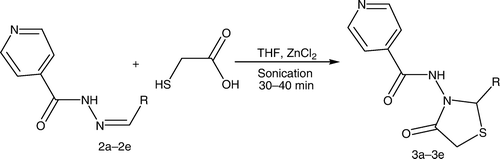
The synthesized intermediates were further utilized for the synthesis of N-(2-aryl-4-oxothiazolidin-3-yl)isonicotinamide derivatives (3a–3e) containing a 4-thiazolidinone nucleus by stirring and sonication ( and ), which were characterized by the presence of a strong band at 1714–1729 cm−1 for the ring carbonyl group, which is considered to be a strong confirmation for the thiazolidinone nucleus formation. Another piece of evidence for cyclization is the appearance of a singlet signal equivalent to one proton in the 1H NMR spectrum between 5.9 and 6.1 ppm (C-2, CH) and doublet of doublet signal equivalent to two protons between 3.3 and 3.8 (C-5, CH2), which represents the formation of the thiazolidinone nucleus.
EIMS of selected compound 3c displayed a molecular ion peak at m/z 344, which confirmed its molecular weight. A base peak in mass spectra was obtained at m/z 298 by the loss of the NO2 + fragment from the molecule.
Experimental
Melting points were determined with the Veego melting point apparatus (VMP PM, 32/1104) and are uncorrected. Thin layer chromatography Citation20 was carried out using silica gel (G-60 mesh). The Rf value for each compound was calculated using chloroform:methanol (8:2) as the solvent, and the spots were located using iodine vapor. UV studies were carried out on a UV visible spectrophotometer (Shimadzu 1700), and the λmax of the respective synthesized compounds were calculated using ethanol as a solvent. IR spectras (KBr) Citation21 were recorded on a FTIR spectrophotometer with a diffuse reflectance attachment (Shimadzu 8400S). 1H NMR spectras Citation22 were obtained from a NMR spectrophotometer (Bruker Avance II 400 NMR) with dimethyl sulfoxide as the solvent. Chemical shifts were expressed in parts per million relative to SiMe4 as an internal standard. A mass spectrum was obtained on an electron impact mass spectrometer at 70eV ionizing beam and using a direct insertion probe. Unless stated otherwise, all materials were obtained from commercial suppliers and used without further purification.
Synthesis of Schiff's bases as intermediates
The synthesis of compounds (2a–2e) was performed according to our previously reported procedure Citation19 (). Isoniazid (0.01 M) and appropriate aromatic aldehydes (0.01 M) (1a–1e) in water were sonicated until the completion of the reaction, which was monitored by TLC (CHCl3:methanol, 8:2). The reaction mixture was filtered, and the residue obtained was washed with water, followed by a sodium thiosulfate solution, and then dried. The crude products obtained on recrystallization from alcohol yielded pure hydrazones of Isoniazid (INH) (2a–2e). The synthesized compounds were characterized by their melting points and spectral data (IR, 1H NMR).
Table 1. Chemical structures and properties of Schiff's bases of isoniazid by sonication (2a–2e).
(2a) Benzylidene isonicotinoyl hydrazone
White crystals; Yield 95.6; mp 194°C–196°C; IR (KBr): ν/cm−1=3197 (NH), 3028 (CH), 1693 (amide C=O), 1600 (imine C=N); 1H NMR (DMSO-d6): δ ppm=9.53–9.44 (d, 2H, pyridine), 8.81–8.78 (d, 2H, pyridine), 8.37 (s, 1H, NH), 7.89–7.57 (s, 5H, aromatic), 7.43 (s, 1H, CH).
(2b) p-Chloro benzylidene isonicotinoyl hydrazone
White crystals; Yield 96.2; mp 198°C–201°C; IR (KBr): ν/cm−1=3190 (NH), 3028 (CH), 1666 (amide C=O), 1596 (imine C=N), 1087 (C–Cl); 1H NMR (DMSO-d6): δ ppm=8.91–8.88 (d, 2H, pyridine), 8.65 (s, 1H, =CH), 8.33 (s, 1H, NH), 8.05–7.99 (d, 2H, pyridine), 7.81–7.7.79 (d, 2H, aromatic), 7.51–7.50 (d, 2H, aromatic).
(2c) p-Nitro benzylidene isonicotinoyl hydrazone
Yellow powder; Yield 98.1; mp 203°C–205°C; IR (KBr): ν/cm−1=3186 (NH), 3001 (CH), 1685 (amide C=O), 1590 (imine C=N), 1512 (NO2); 1H NMR (DMSO-d6): δ ppm=9.35–9.32 (d, 2H, pyridine), 8.55–8.51 (d, 2H, pyridine), 8.34 (s, 1H, =CH), 8.11 (s, 1H, NH), 7.93–7.89 (d, 2H, aromatic), 7.62–7.60 (d, 2H, aromatic).
(2d) p-Hydroxy benzylidene isonicotinoyl hydrazone
Yellow powder; Yield 90.7; mp 262°C–264°C; IR (KBr): ν/cm−1=3440 (OH), 3213 (NH), 3055 (CH), 1662 (amide C=O), 1602 (imine C=N); 1H NMR (DMSO-d6): δ ppm=12–11.5 (s, 1H, OH), 8.8–8.5 (d, 2H, pyridine), 8.4–8.3 (s, 1H, =CH), 8.0 (s, 1H, NH), 7.9–7.7 (d, 2H, pyridine), 7.5 (d, 2H, aromatic), 7.0–6.8 (d, 2H, aromatic).
(2e) Furfurylidene isonicotinoyl hydrazone
Brown crystals; Yield 95.2; mp 165°C–167°C; IR (KBr): ν/cm−1=3271 (NH), 3051 (CH), 1650 (amide C=O), 1620 (imine C=N); 1H NMR (DMSO-d6): δ ppm=8.91–8.89 (d, 2H, pyridine), 8.32 (s, 1H, =CH), 8.18–8.13 (d, 2H, pyridine), 7.84 (s, 1H, NH), 7.54–7.52 (d, 1H, furfural ring CH), 6.85–6.55 (m, 1H, furfural ring CH), 5.97–5.94 (d, 1H, furfural ring CH).
Synthesis of N-(2-aryl-4-oxothiazolidin-3-yl)isonicotinamide analogs using Schiff's bases
Green route procedure
Appropriate Schiff's bases (2.0 mmol) (2a–2e) were dissolved in 10 mL tetrahydrofuran (THF) with addition of thioglycolic acid at low temperature (0°C–5°C) in the presence of molecular sieves and zinc chloride. For the reaction by stirring, the reaction mixture was stirred until the completion of the reaction [TLC; CHCl3:methanol (8:2)]. For the sonication method, the reaction mixture was sonicated until the completion of the reaction [TLC; CHCl3:methanol (8:2)]. The reaction mixture was taken in ethyl acetate. The organic layer was successively washed with water, 5% sodium bicarbonate, and finally brine. The crude products obtained on recrystallization from alcohol yielded 4-thiazolidinones (3a–3e ). The synthesized compounds were characterized by their melting points and by spectral data (IR, 1H NMR, and MS). represents the structures of aldehydes and their respective synthesized Schiff's bases and 4-thiazolidinone analogs.
Table 2. Chemical structures and properties of 4-thiazolidinones (3a–3e).
Table 3. Isonicotinoyl hydrazones and their 4-thiazolidinone analogs.
(3a) N-(2-benzyl-4-oxothiazolidin-3-yl)isonicotinamide
White powder; Yield 92; mp 250°C–253°C; IR (KBr): ν/cm−1=3199 (NH), 3029 (CH), 1729 (ring C=O), 1683 (amide C=O); 1H NMR (DMSO-d6): δ ppm=8.77–8.68 (d, 2H, pyridine), 8.75 (s, 1H, NH), 7.75–7.73 (d, 2H, pyridine), 7.71–7.69 (s, 5H, aromatic), 5.96 (s, 1H, CH), 3.40–3.35 (d, 1H, CH2), 3.29–3.24 (d, 1H, CH2).
(3b) N-(2-p-chlorobenzyl-4-oxothiazolidin-3-yl)isonicotinamide
White crystals; Yield 96.8; mp 239°C–242°C; IR (KBr): ν/cm−1=3193 (NH), 3024 (CH), 1716 (ring C=O), 1670 (amide C=O), 1083 (C–Cl); 1H NMR (DMSO-d6): δ ppm=8.77–8.75 (d, 2H, pyridine), 8.07 (s, 1H, NH), 7.85–7.84 (d, 2H, pyridine), 7.75–7.73 (d, 2H, aromatic), 7.71–7.69 (d, 2H, aromatic), 6.05 (s, 1H, CH), 3.70–3.63 (d, 1H, CH2), 3.59–3.52 (d, 1H, CH2).
(3c) N-(2-p-nitrobenzyl-4-oxothiazolidin-3-yl)isonicotinamide
Yellow powder; Yield 86.4; mp 263°C–266°C; IR (KBr): ν/cm−1=3199 (NH), 3029 (CH), 1720 (ring C=O), 1681 (amide C=O), 1519 (NO2); 1H NMR (DMSO-d6): δ ppm=9.07–9.02 (d, 2H, pyridine), 8.73 (s, 1H, NH), 8.13–8.11 (d, 2H, pyridine), 7.95–7.93 (d, 2H, aromatic), 7.71–7.70 (d, 2H, aromatic), 5.91 (s, 1H, CH), 3.40–3.35 (d, 1H, CH2), 3.31–3.26 (d, 1H, CH2); EIMS (70eV, m/z): 344 (M+), 298 (Base peak).
(3d) N-(2-p-hydroxybenzyl-4-oxothiazolidin-3-yl)isonicotinamide
Yellow powder; Yield 90.2; mp >290°C; IR (KBr): ν/cm−1=3445 (OH), 3217 (NH), 3052 (CH), 1724 (ring C=O), 1668 (amide C=O); 1H NMR (DMSO-d6): δ ppm=8.77–8.75 (d, 2H, pyridine), 8.06 (s, 1H, NH), 7.85–7.84 (d, 2H, pyridine), 7.75–7.73 (d, 2H, aromatic), 7.34–7.29 (d, 2H, aromatic), 6.05 (s, 1H, CH), 5.36 (s, 1H, OH), 3.70–3.63 (d, 1H, CH2), 3.59–3.52 (d, 1H, CH2).
(3e) N-(2-furfuryl-4-oxothiazolidin-3-yl)isonicotinamide
Brown powder; Yield 81; mp 198°C–201°C; IR (KBr): ν/cm−1=3271 (NH), 3112 (CH), 1714 (ring C=O), 1664 (amide C=O); 1H NMR (DMSO-d6): δ ppm=9.08–9.02 (d, 2H, pyridine), 8.62 (s, 1H, NH), 8.21–8.19 (d, 2H, pyridine), 7.94–7.93 (d, 1H, furfural ring CH), 7.06–7.93 (m, 1H, furfural ring CH), 6.27 (s, 1H, furfural ring CH), 5.93 (s, 1H, CH), 3.92–3.90 (d, 1H, CH2), 3.65–3.62 (d, 1H, CH2).
Conclusions
The conventional reaction for the synthesis of 4-thiazolidinones requires longer reaction times (12–48 h reflux) for the completion of the reaction with low yields (less than 65%). The reaction also involves the use of Dean-Stark water separator for the removal of water from the reaction. The synthesis of 4-thiazolidinones with stirring in the presence of dicyclohexylcarbodiimide (DCC) has been reported.
On the other hand, the reaction carried out by us employs novel green route methods of sonication and stirring using molecular sieves. The time span required for the completion of the reaction is less than that for the conventional method. The reaction by the stirring method required 60–70 min while the sonication method required 30–40 min for the completion of the reaction. Furthermore, the present investigation offers rapid and effective new procedures for the synthesis of the 4-thiazolidinone analogs with improved yields (81–96.8%). The results of the physicochemical characterization of the synthesized products by chromatographic and spectroscopic studies suggested that the products obtained by both stirring and sonication methods were comparable in chemical composition.
Acknowledgements
This work was financially supported by the University of Pune, Pune (India), and Padm. Dr. D.Y. Patil Institute of Pharmaceutical Sciences and Research, Pune (India). NMR characterization was carried out by SAIF/CIL, Panjab University, Chandigarh (India).
Related Research Data
References
- Margulis , M.A. High Energy Chem . 2004 , 38 ( 3 ), 135 – 142 .
- Verma , A. ; Saraf , S.K. Eur. J. Med. Chem . 2007 , 20 , 1 – 9 .
- Albanese , C. ; Christin-Maitre , S. ; Sluss , P.M. ; Crowley , W.F. ; Jameson , J.L. Mol. Cell. Endocrinol . 1994 , 101 , 211 – 219 .
- Vane , J. ; Botting , R. FASEBJ Journal 1987 , 1 , 89 – 96 .
- Vigorita , M.G. ; Ottana , R. ; Monforte , F. ; Maccari , R. ; Trovato , A. ; Monforte ; M.T. ; Taviano , M.F. Bioorg. Med. Chem. Lett . 2001 , 11 , 2791 – 2794 .
- Unlu , S. ; Onkol , T. ; Dundar , Y. Arch. Pharm. Pharm. Med. Chem . 2003 , 336 , 353 – 361 .
- Dwivedi , C. ; Gupta , S.S. ; Parmar , S.S. J. Med. Chem . 1972 , 15 , 553 – 554 .
- Kavitha , C.V. Bioorg. Med. Chem . 2006 , 14 , 2290 – 2299 .
- Mohan , L. ; Chadha , V.K. ; Chaudhary H.S. Ind. J. Exp. Biol . 1972 , 10 , 37 – 40 .
- Bonde , C.G. ; Gaikwad , N.J. Bioorg. Med. Chem . 2004 , 12 , 2151 – 2161 .
- Katti , S.B. ARKIVOC 2005 , 2 , 120 – 130 .
- Vittoria , D. ; Orazio , M. ; Eugenio , P. ; Antonio , C. ; Federico , G. ; Adele , B. J. Med. Chem . 1992 , 35 ( 15 ), 2910 – 2912 .
- Agrawal , V.K. ; Sachan , S. ; Khadikar , P.V. Acta Pharm . 2000 , 50 , 281 – 290 .
- Ottana , R. ; Mazzon , E. ; Dugo , L. ; Monforte , F. ; Maccari , R. ; Sautebin , L. ; De Luca , G. ; Vigorita , M.G. ; Alcaro , S. ; Ortuso , F. ; Caputi A.P. ; Cuzzocrea , S. Eur. J. Pharmacol . 2002 , 448 , 71 – 80 .
- Ottana , R. ; Carotti , S. ; Maccari , R. ; Landini , I. ; Chiricosta , G. ; Caciagli , B. ; Vigorita , M.G. ; Mini , E. Bioorg. Med. Chem. Lett . 2005 , 15 , 3930 – 3933 .
- Rawal , R.K. ; Prabhakar , Y.S. ; Katti , S.B. ; Clercq , E. Bioorg. Med. Chem . 2005 , 13 , 6771 – 6776 .
- Aries , R. French Patent 2,186,245 . Chem. Abstr . 1974 , 81 , 140869 .
- Aries , R. French Patent 2,190,431 . Chem. Abstr . 1976 , 84 , 35329 .
- Thomas , A.B. ; Tupe , P.N. ; Badhe , R.V. ; Nanda , R.K. ; Kothapalli , L.P. ; Paradkar , O.D. ; Sharma , P.A. ; Deshpande , A.D. Green Chem. Lett. Rev . 2009 , 2 , 23 – 27 .
- Stahl , E. Thin Layer Chromatography: A Practical Handbook, , 2nd ed. ; New York : Springer-Verlag , 1969 ; pp 96 – 102 .
- Kalsi , P.S. Spectroscopy of Organic Compounds , 5th ed. ; New Delhi : New Age International Publication , 2002 ; pp 364 – 367 .
- Kemp , W. Organic Spectroscopy , 3rd ed. ; Palgrave Macmillan , Basingstoke , 1991 ; pp 285 – 324 .