Abstract
An efficient and rapid synthesis of an array of naphthyl-substituted cyclohexenone carboxylates, indazolonols, and nitrohydrazones by using activated fly ash as catalyst in dry media is described. All the synthesized compounds were characterized by their melting point, elemental analysis, mass spectrometry, Fourier transform infrared, one-dimensional nuclear magnetic resonance (1H and D2O exchanged 1H and 13C), and two-dimensional homonuclear correlation (HOMOCOR) and heteronuclear single quantum coherence (HSQC) spectroscopic data. Compared to classical conditions, this new protocol had the advantage of excellent yields, mild reaction conditions, simple workup, and short reaction times. Also, the catalyst could be reused up to four runs without any reduction in efficiency, making the process greener and environmentally benign. 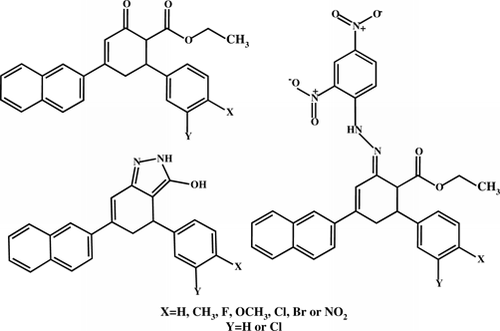
Introduction
A wide range of structurally diverse indazole nuclei have aroused enormous interest in the recent years due to their wide variety of pharmacological activities such as antimicrobial activity Citation1, inhibitors of protein kinase B/Akt Citation2, leishmanocidal activity Citation3, antiprotozoal agents Citation3, antichagasic activity Citation3, trypanocidal activity Citation3, inhibitors of S-adenosylhomocysteine/methylthio adenosine nucleosides Citation4, inhibition of platelet aggregation Citation5, and potent activator of the nitric oxide receptor Citation5. The indazole nucleus is a widely studied biological scaffold that has emerged as a core structural unit of a variety of bioactive molecules Citation6–10.
Adjudin is currently under phase II human trials as a potential nonhormonal male contraceptive drug comprising of the indazole nucleus, which acts by blocking the production of sperm in the testes but without affecting testosterone production Citation6. It is an analog of the chemotherapy drug lonidamine, an indazole–carboxylic acid, which, for a long time, has been known to inhibit aerobic glycolysis in cancer cells Citation7. 7-Nitroindazole is a heterocyclic small molecule containing an indazole ring that has been nitrated at position 7. Nitroindazole acts as a selective inhibitor for neuronal nitric oxide synthase, a hemoprotein enzyme, which in neuronal tissue converts arginine to citrulline and nitric oxide Citation8. YM-348, (S)-1-(7-ethyl-1H-furo[2,3-g]indazol-1-yl)propan-2-amine, is an indazole derivative drug that acts as a potent and selective 5-HT2C receptor agonist. It shows thermogenic and anorectic effects in animal studies Citation9. AL-38022A, (S)-1-(8,9-dihydropyrano[2,3-g]indazol-1(7H)-yl)-propan-2-amine, is an indazole derivative drug that acts as a potent and selective agonist for the 5-HT2 family of serotonin receptors.
The interesting starting material 2-acetyl naphthalene was widely used in perfume formulations Citation11. Hydrazones have been demonstrated to possess different biological activities. For example, isonicotinoyl hydrazones are antitubercular and nifuroxazide is an intestinal antiseptic. Acetylhydrazones provided good protection against convulsions Citation12. 3-Phenyl-5-sulfonamidoindole-2-carboxylic acid 3,4-methylenedioxy/4-methyl/4-nitrobenzylidene-hydrazide showed antidepressant activity Citation13. The most important anti-inflammatory derivative 2-(2-formylfuryl)pyridylhydrazone presented a 79% inhibition of pleurisy Citation14. [(4′-N,N-Dimethylaminobenzylidene-3-(3′,4′-ethylenedioxyphenyl)propionylhydrazine] was used as a standard antinociceptive drug Citation15. The antiplatelet activity of novel tricyclic acylhydrazone derivatives was evaluated by their ability to inhibit platelet aggregation of rabbit platelet-rich plasma Citation16. The arylhydrazone chelator 2-hydroxy-1-naphthylaldehyde isonicotinoyl hydrazone showed greater antimalarial agent activity than did desferrioxamine against chloroquine-resistant and -sensitive parasites Citation17. Triazolohydrazones possess good antibacterial activity Citation18. A new bioactive compound of the N-acylhydrazone class, 3,4-methylenedioxybenzoyl-2-thienyl hydrazone, was shown to have inotropic and vasodilatory effects Citation19. Triaryl hydrazones exhibited antitumoral activity against MCF-7 and ZR-75-1 human malignant breast cell lines Citation20.
Fly ash, an environmental pollutant generated during the combustion of coal for energy production, is an industrial by-product Citation21. Fly ash was utilized in large quantities under three categories: (a) low-value-added utilization (as a fill material for land or mines), (b) medium-value-added utilization (bricks, cement, concrete, lightweight aggregates, and other building construction materials), and (c) high-value-added utilization (mineral recovery, ceramics, and cenospheres). Thus, during the initial stages, attention was paid only to its pozzolanic activity Citation22. Many researchers have devoted themselves to the research of the potential activity of fly ash and the hydration process of fly ash cement Citation23. The chemical composition of fly ash is as follows: SiO2 (64.03%), Al2O3 (15.50%), Fe2O3 (6.50%), CaO (4.62%), MgO (3.00%), loss on ignition (4.35%), and insoluble residue (2.00%).
Activated fly ash was used to catalyze a variety of organic reactions including Knoevenagel condensation; “one-pot” conversions of ketones to amides via Beckmann rearrangement, Schiff bases formation, and Biginelli and Hantzsch reactions Citation24; “one-pot” synthesis of 1,2,4,5-tetrazinanes Citation25; microwave-assisted, solvent-free alkylation and acylation of 2-mercaptobenzothiazole Citation26; different ether and ester derivatives of carvacrol Citation27; liquid-phase esterification of salicylic acid Citation28 with acetic anhydride and methanol; grindstone chemistry solid-state reaction procedure for the synthesis of heterocyclic oximes Citation29; and one-pot synthesis of pyrimidino thiazolidin-4-ones Citation30. Moreover, in several adsorption processes, fly ash was used as a cost-effective adsorbent for removal of pollutants in air and water systems Citation31 Citation32. The presence of active components such as aluminosilicates and Fe2O3 made fly ash a good heterogeneous catalyst or catalyst support in a variety of catalytic reactions Citation33. Also, fly ash was used as a solid support for the preparation of a Ru-based catalyst that produces highest catalytic ammonia decomposition Citation34.
Taking these factors into account and as part of our research program aimed at the synthesis of bioactive novel structurally diverse heterocycles in dry media Citation35–37, herein is reported the molecular conjugation of the naphthyl-substituted chalcones moiety with two or more active counterparts designed and synthesized with the hope of producing novel ethyl 4-(naphthalen-2-yl)-2-oxo-6-arylcyclohex-3-enecarboxylates 9–16, intermediates with three versatile functional groups, i.e. ketone, olefin, and ester, for the synthesis of 4,5-dihydro-6-(naphthalen-2-yl)-4-aryl-2H-indazol-3-ols 17–24, novel fused indazole derivatives; and (2E)-ethyl-2-(2-(2,4-dinitrophenyl)hydrazono)-4-(naphthalen-2-yl)-6-arylcyclohex-3-enecarboxylates 25–32 in dry media catalyzed by activated fly ash under microwave irradiation.
Results and discussion
Chemistry
The conventional approach for the synthesis of 4,5-dihydro-6-(naphthalen-2-yl)-4-aryl-2H-indazol-3-ols 17–24 is as follows: (E)-1-(naphthalen-2-yl)-3-arylprop-2-ene-1-ones 1–8 are synthesized by the Claisen–Schmidt condensation of 2-acetyl naphthalene and substituted benzaldehydes in the presence of alcoholic sodium hydroxide. Treatment of (E)-1-(naphthalen-2-yl)-3-arylprop-2-ene-1-ones 1–8 with ethyl acetoacetate in the presence of sodium ethoxide in refluxing ethanol ( and ) affords ethyl 4-(naphthalen-2-yl)-2-oxo-6-arylcyclohex-3-enecarboxylates 9–16. Compounds 9–16 on reaction with hydrazine hydrate in the presence of anhydrous sodium acetate in refluxing ethanol yield the respective indazole derivatives: 4,5-dihydro-6-(naphthalen-2-yl)-4-aryl-2H-indazol-3-ols 17–24. Compounds 9–16 on reaction with 2,4-dinitrophenylhydrazine in the presence of two to four drops of sulfuric acid in refluxing ethanol yields the respective nitrohydrazone derivatives: (2E)-ethyl-2-(2-(2,4-dinitrophenyl)hydrazono)-4-(naphthalen-2-yl)-6-arylcyclohex-3-enecarboxylates 25–32.
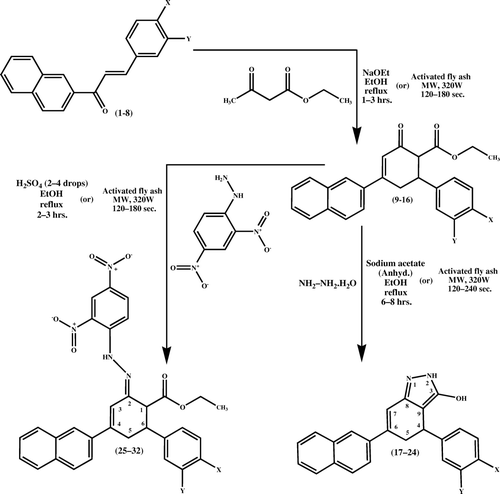
Table 1. Physical and analytical data for compounds 9–32.
There are some problems associated with the above synthesis, such as severe conditions, low yields for the reaction, use of corrosive bases such as sodium ethoxide, sulfuric acid, usage of solvents such as ethanol and longer reaction times. Hence there is a need to develop a better method. By using microwave heating it is often possible to enhance the rate of reactions and improve product yields Citation38–40.
In the present investigation, as-received fly ash is activated by the physical method followed by the microwave method () and to study the influence of activated fly ash to catalyze the synthesis of an array of naphthyl-substituted cyclohexenone carboxylates, indazolonols, and nitrohydrazones in “dry media”.
Figure 1. Flowchart for the preparation of activated fly ash.

In the present reaction procedure, treatment of various substituted 0.005 mol of (E)-1-(naphthalen-2-yl)-3-arylprop-2-ene-1-ones 1–8 with 0.005 mol of ethyl acetoacetate along with a catalytic amount of activated fly ash (20 mg) affords the corresponding ethyl 4-(naphthalen-2-yl)-2-oxo-6-arylcyclohex-3-enecarboxylates 9–16 in a short reaction time ( and ) in high yields in dry media under microwave (MW) irradiation at a power level of 320 W.
Activated fly ash catalyst was shown to be one of the most efficient MW absorbers with a very high specificity to MW heating. It was able to reach a temperature of 110°C after 120 s of irradiation in a microwave oven (320 W). Mere 20 mg of activated fly ash catalyst and 0.005 mol of substrates is the most acceptable ratio in terms of efficiency and safety; a power level of 320 W is the most suitable one.
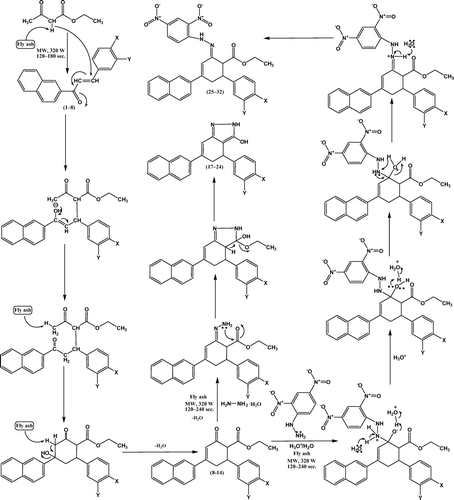
Ethyl acetoacetate undergoes Michael addition with (E)-1-(naphthalen-2-yl)-3-arylprop-2-ene-1-ones 1–8 in the presence of activated fly ash catalyst () to give an addition product. Two major constituents of fly ash – namely, silica and alumina – drive the reaction to proceed smoothly Citation24. Later this addition product undergoes an intramolecular aldol reaction in the presence of activated fly ash, which serves as a dehydrating agent to furnish ethyl 4-(naphthalen-2-yl)-2-oxo-6-arylcyclohex-3-enecarboxylates 9–16. Compounds 9–16 (0.005 mol) containing a β-dicarbonyl system with hydrazine hydrate (0.005 mol) along with a catalytic amount of fly ash (20 mg) initially form Schiff bases. Fly ash assists in the removal of water molecules to form Schiff base intermediate. During the course of reaction, the formed Schiff bases underwent cyclization to afford 4,5-dihydro-6-(naphthalen-2-yl)-4-aryl-2H-indazol-3-ols 17–24 in quantitative yields. Similarly, compounds 9–16 (0.005 mol) with 2,4-dinitrophenylhydrazine (0.005 mol) along with a catalytic amount of fly ash (20 mg) did not undergo usual cyclization reaction to yield the respective 4,5-dihydro-6-(naphthalen-2-yl)-2-(2,4-dinitrophenyl)-4-aryl-2H-indazol-3-ols. Instead, compounds 9–16 underwent condensation reaction, with two molecules joining together with loss of water. It is also called addition–elimination reaction: nucleophilic addition of the –NH2 group to the C = O carbonyl group, followed by the removal of a water molecule. In the present reaction, 2,4-dinitrophenylhydrazine does not react with other carbonyl-containing functional groups such as ester. Fly ash assists for dehydration to form compounds 25–32. For ester, there is resonance-associated stability as a lone pair of electrons interacts with the p orbital of the carbonyl carbon, resulting in increased delocalization in the molecule. This stability would be lost with the addition of a reagent to the carbonyl group. Hence these compounds are more resistant to addition.
In order to select a more appropriate power level, the reaction has been carried out at different power levels ranging from 80 to 720 W. The results are shown graphically in . It is clear that the power level of 320 W gave the maximum yields of 95%, 92%, and 95% for compounds 9, 17, and 25, respectively. In order to evaluate the synergy between fly ash and microwave activation in this type of reaction, several experiments were tried (). Only in the case of dry media coupled with MW irradiation taking advantage of the synergy between both methodologies is the product formed in excellent yields of 95%, 92%, and 95% for compounds 9, 17, and 25, respectively. In order to find out the effect of microwave radiation on fly ash, the same reaction was tested under thermo-stated heating mantle at 60°C in the absence of microwave irradiation. The yields were 82%, 78%, and 85% for compounds 9, 17, and 25, respectively. Significant decreases in yields were obtained under conventional heating than obtained using the MW-assisted method. Even by extending reaction times, yields remain lower under thermal conditions when compared to MW activation.
Figure 2. Effect of microwave power level (in watts) for the formation of naphthyl cyclohexenones, indazolonols, and nitrohydrazones (compounds 9, 17, and 25).
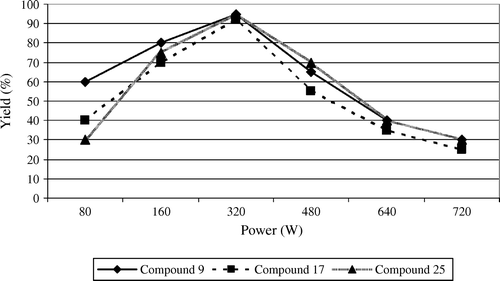
Table 2. Comparison of results under both MW and thermal conditions for compounds 9, 17, and 25.
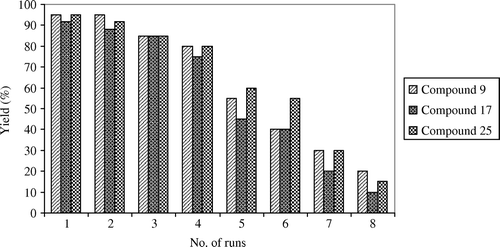
In addition, the recovery and reuse of activated fly ash catalyst is studied and the results are given in . Regeneration of fly ash catalyst shows that the gradual deactivation of fly ash in four runs () makes the process greener and environmentally benign. The catalyst is easily regenerated by washing with ethyl acetate followed by drying under microwave irradiation at a power level of 160 W for 120 s. It is shown that the catalyst is active up to four runs. However, there is a significant decrease in the activity after four runs since recycling leads to loss of efficiency of the catalyst owing to the absorption of organics. Organics on the solid surface result in the reduction of number of active centers. Organic species could be removed at higher temperature reactivating the catalyst.
Figure 3. Recovery and reuse of activated fly ash in catalyzing the synthesis of naphthyl cyclohexenones, indazolonols, and nitrohydrazones (compounds 9, 17, and 25).
Experimental
Chemistry
The progress of the reaction was monitored by thin layer chromatography (TLC) analysis. All the reported melting points were taken in open capillaries and were uncorrected. A Biotage Initiator microwave synthesizer (a scientific microwave oven) was used for irradiation. Infrared (IR) spectra were recorded in KBr (pellet forms) on a Thermo Nicolet-Avatar-330 Fourier transform infrared (FT-IR) spectrophotometer and only noteworthy absorption values (cm−1) were listed. 1H and 13C NMR (nuclear magnetic resonance) spectra were recorded at 400 and 100 MHz, respectively, on a Bruker Avance II 400 NMR spectrometer using DMSO-d 6 as solvent. Two-dimensional HOMOCOR and HSQC spectra were recorded on a Bruker DRX 500 NMR spectrometer. The ESI +ve MS (mass spectrometry) spectra were recorded on a Bruker Daltonics liquid chromatography–mass spectrometry spectrometer. Satisfactory microanalyses were obtained on a Carlo Erba 1106 CHN analyzer.
By adopting the literature precedent Citation41, (E)-1-(naphthalen-2-yl)-3-arylprop-2-en-1-ones 1–7 were prepared.
General procedure for the synthesis of ethyl 4-(naphthalen-2-yl)-2-oxo-6-arylcyclohex-3-enecarboxylates 9–16
Classical method
To a solution of sodium ethoxide (0.005 mol) in 30 mL of absolute ethanol, freshly distilled ethyl acetoacetate (0.005 mol) and respective (E)-1-(naphthalen-2-yl)-3-arylprop-2-en-1-ones (0.005 mol) in absolute ethanol (40 mL) was mixed. This mixture was refluxed in a water bath for 1–3 h by maintaining the temperature around 70–80°C. The reaction mixture was allowed to cool and filtered. Then the crude product was recrystallized from absolute ethanol to afford ethyl 4-(naphthalen-2-yl)-2-oxo-6-arylcyclohex-3-enecarboxylates 9–16.
Microwave method
A mixture of (E)-1-(naphthalen-2-yl)-3-arylprop-2-en-1-ones 1–8 (0.005 mol), ethyl acetoacetate (0.005 mol) and activated fly ash (20 mg) was added in an alumina bath and mixed properly with the aid of glass rod (10 s) and then irradiated in a microwave oven for 120–180 s at 320 W (monitored by TLC). After completion of the reaction, the reaction mixture was extracted with ethyl acetate (3×5 mL). The catalyst and other solid wastes were removed by filtration. The combined organic layer was washed with water three times and then dried over anhydrous MgSO4. The organic layer was concentrated in vacuo to furnish the products 9–16, which were purified by recrystallization in ethanol.
Ethyl 4-(naphthalen-2-yl)-2-oxo-6-phenylcyclohex-3-enecarboxylate 9
IR (KBr) ν (cm−1): 3057, 2987, 2929, 1738, 1659, 1606, 1292, 1155, 847, 816, 766, 704; 1H NMR (δ ppm): 0.92 (3H, t, CH2CH 3 at C-1, J=7.1 Hz), 3.22–3.28 (2H, H5, m), 3.65–3.73 (1H, H6, m), 3.91 (2H, q, CH 2 CH3 at C-1, J=7.0 Hz), 4.17 (1H, H1, d, J=13.4 Hz), 6.73 (1H, s, H3), 7.24–8.33 (12H, m, Harom); 13C NMR (δ ppm): 13.7 CH2 CH3 at C-1, 35.1 C-6, 43.8 C-5, 58.7 CH2CH3 at C-1, 59.9 C-1, 123.1 C-3, 169.2 C = O at C-1, 194.2 C-2, 123.4–128.8 –Carom, C-4 carbon may be merged with –Carom, 132.7, 133.6, 134.3, 141.5 ipso carbons.
Ethyl 4-(naphthalen-2-yl)-2-oxo-6-p-tolylcyclohex-3-enecarboxylate 10
IR (KBr) ν (cm−1): 3065, 3027, 2979, 2923, 2863, 1740, 1660, 1604, 1148, 897, 849, 816, 753; 1H NMR (δ ppm): 0.96 (3H, t, CH2CH 3 at C-1, J=7.1 Hz), 2.28 (3H, s, CH3 at phenyl ring), 3.15–3.17 (2H, H5, m), 3.62–3.66 (1H, H6, m), 3.92 (2H, q, CH 2 CH3 at C-1, J=6.9 Hz), 4.11 (1H, H1, d, J=13.3 Hz), 6.71 (1H, s, H3), 7.13–8.30 (11H, m, Harom); 13C NMR (δ ppm): 14.2 CH2 CH3 at C-1, 21.0 CH3 at phenyl ring, 35.7 C-6, 43.8 C-5, 59.2 CH2CH3 at C-1, 60.3 C-1, 123.5 C-3, 169.6 C = O at C-1, 194.7 C-2, 123.7–129.2 –Carom, C-4 carbon may be merged with –Carom, 133.1, 134.0, 134.7, 136.5, 138.9 ipso carbons.
Ethyl 6-(4-fluorophenyl)-4-(naphthalen-2-yl)-2-oxocyclohex-3-enecarboxylate 11
IR (KBr) ν (cm−1): 3054, 2989, 2925, 1739, 1656, 1603, 1225, 1153, 888, 841, 815, 752; 1H NMR (δ ppm): 0.94 (3H, t, CH2CH 3 at C-1, J=7.0 Hz), 3.19–3.21 (2H, H5, m), 3.68–3.75 (1H, H6, m), 3.93 (2H, q, CH 2 CH3 at C-1, J=7.0 Hz), 4.16 (1H, H1, d, J = 13.4 Hz), 6.73 (1H, s, H3), 7.16–8.33 (11H, m, Harom); 13C NMR (δ ppm): 13.8 CH2 CH3 at C-1, 35.0 C-6, 43.0 C-5, 58.8 CH2CH3 at C-1, 59.9 C-1, 123.0 C-3, 169.1 C = O at C-1, 194.1 C-2, 114.9–129.6 –Carom, C-4 carbon may be merged with –Carom, 132.7, 1333.6, 134.2, 137.7, 160.0 ipso carbons.
Ethyl 6-(4-methoxyphenyl)-4-(naphthalen-2-yl)-2-oxocyclohex-3-enecarboxylate 12
IR (KBr) ν (cm−1): 3056, 2987, 2932, 2838, 1738, 1657, 1606, 1252, 888, 831, 815, 749; 1H NMR (δ ppm): 0.96 (3H, t, CH2CH 3 at C-1, J = 7.0 Hz), 3.16–3.29 (2H, H5, m), 3.59–3.67 (1H, H6, m), 3.73 (3H, s, OCH3 at phenyl ring), 3.92 (2H, q, CH 2 CH3 at C-1, J=7.0 Hz), 4.10 (1H, H1, d, J=14.3 Hz), 6.71 (1H, s, H3), 6.89–8.32 (11H, m, Harom); 13C NMR (δ ppm): 14.2 CH2 CH3 at C-1, 35.7 C-6, 43.5 C-5, 55.4 OCH3 at phenyl ring, 59.5 CH2CH3 at C-1, 60.2 C-1, 123.5 C-3, 169.7 C = O at C-1, 194.7 C-2, 114.1–129.0 –Carom, C-4 carbon may be merged with –Carom, 129.2, 133.1, 134.0, 134.7, 159.1 ipso carbons.
Ethyl 6-(4-chlorophenyl)-4-(naphthalen-2-yl)-2-oxocyclohex-3-enecarboxylate 13
IR (KBr) ν (cm−1): 3056, 3022, 2978, 2928, 1738, 1658, 1257, 1147, 890, 850, 818, 749; 1H NMR (δ ppm): 0.95 (3H, t, CH2CH 3 at C-1, J=7.0 Hz), 3.19–3.29 (2H, H5, m), 3.68–3.75 (1H, H6, m), 3.93 (2H, q, CH 2 CH3 at C-1, J=7.1 Hz), 4.18 (1H, H1, d, J=13.4 Hz), 6.73 (1H, s, H3), 7.40–8.32 (11H, m, Harom); 13C NMR (δ ppm): 13.8 CH2 CH3 at C-1, 34.8 C-6, 43.1 C-5, 58.5 CH2CH3 at C-1, 60.0 C-1, 123.0 C-3, 169.1 C = O at C-1, 193.9 C-2, 123.3–129.5 –Carom, C-4 carbon may be merged with –Carom, 132.7, 133.6, 134.2, 134.9, 140.5 ipso carbons.
Ethyl 6-(4-bromophenyl)-4-(naphthalen-2-yl)-2-oxocyclohex-3-enecarboxylate 14
IR (KBr) ν (cm−1): 3058, 3027, 2977, 2929, 2907, 2863, 1732, 1657, 1598, 1170, 1145, 892, 817, 748, 711; 1H NMR (δ ppm): 0.96 (3H, t, CH2CH 3 at C-1, J=7.0 Hz), 3.19–3.21 (2H, H5, m), 3.67–3.74 (1H, H6, m), 3.94 (2H, q, CH 2 CH3 at C-1, J=7.0 Hz), 4.18 (1H, H1, d, J=13.4 Hz), 6.73 (1H, s, H3), 7.42–8.32 (11H, m, Harom); 13C NMR (δ ppm): 13.8 CH2 CH3 at C-1, 34.8 C-6, 43.2 C-5, 58.5 CH2CH3 at C-1, 60.0 C-1, 123.1 C-3, 169.1 C = O at C-1, 194.0 C-2, 120.1–129.9 –Carom, C-4 carbon may be merged with –Carom, 131.3, 132.7, 133.6, 134.2, 141.0 ipso carbons.
Ethyl 4-(naphthalen-2-yl)-6-(4-nitrophenyl)-2-oxo-cyclohex-3-enecarboxylate 15
IR (KBr) ν (cm−1): 3057, 2978, 2925, 2852, 1734, 1658, 1599, 1278, 1149, 893, 856, 815, 743; 1H NMR (δ ppm): 0.94 (3H, t, CH2CH 3 at C-1, J=6.9 Hz), 3.17–3.26 (2H, H5, m), 3.68–3.73 (1H, H6, m), 3.92 (2H, q, CH 2 CH3 at C-1, J=6.8 Hz), 4.17 (1H, H1, d, J=13.7 Hz), 6.73 (1H, s, H3), 6.98–8.22 (11H, m, Harom); 13C NMR (δ ppm): 13.5 CH2 CH3 at C-1, 34.8 C-6, 43.3 C-5, 58.7 CH2CH3 at C-1, 60.3 C-1, 123.2 C-3, 169.8 C = O at C-1, 193.2 C-2, 123.8–129.1 –Carom, C-4 carbon may be merged with –Carom, 132.6, 133.6, 134.3, 134.8, 140.1 ipso carbons.
Ethyl 6-(3-chlorophenyl)-4-(naphthalen-2-yl)-2-oxocyclohex-3-enecarboxylate 16
IR (KBr) ν (cm−1): 3058, 2977, 2927, 2869, 1658, 1733, 1597, 1170, 1147, 888, 851, 815, 786; 1H NMR (δ ppm): 0.94 (3H, t, CH2CH 3 at C-1, J=7.1 Hz), 3.19–3.28 (2H, H5, m), 3.66–3.73 (1H, H6, m), 3.91 (2H, q, CH 2 CH3 at C-1, J=7.1 Hz), 4.16 (1H, H1, d, J=13.7 Hz), 6.74 (1H, s, H3), 7.08–8.29 (11H, m, Harom); 13C NMR (δ ppm): 13.7 CH2 CH3 at C-1, 34.8 C-6, 43.2 C-5, 58.6 CH2CH3 at C-1, 60.1 C-1, 123.2 C-3, 169.0 C = O at C-1, 193.7 C-2, 123.1–129.8 –Carom, C-4 carbon may be merged with –Carom, 132.5, 133.6, 134.0, 134.7, 140.3 ipso carbons.
General method for the synthesis of 4,5-dihydro-6-(naphthalen-2-yl)-4-aryl-2H-indazol-3-ols 17–24
Classical method
A solution of ethyl 4-(naphthalen-2-yl)-2-oxo-6-arylcyclohex-3-enecarboxylates 9–16 (0.001 mol) in ethanol (40 mL) was treated with hydrazine hydrate (0.001 mol) and anhydrous sodium acetate (0.001 mol) and refluxed for 6–8 h. The reaction mixture was cooled and then poured over crushed ice. The crude products were recrystallized twice using ethanol as solvent to afford the product.
Microwave method
A mixture of 4-(naphthalen-2-yl)-2-oxo-6-arylcyclohex-3-enecarboxylates 9–16 (0.005 mol), hydrazine hydrate (0.005 mol), and activated fly ash (20 mg) was added in an alumina bath and mixed properly with the aid of glass rod (10 s) and then irradiated in a microwave oven for 120–240 s at 320 W (monitored by TLC). After completion of the reaction, the reaction mixture was extracted with ethyl acetate (3×5 mL). The catalyst and other solid wastes were removed by filtration. The combined organic layer was dried over anhydrous MgSO4 and concentrated in vacuo to furnish the products 17–24, which were purified by recrystallization in ethanol.
4,5-Dihydro-6-naphthalen-2-yl-4-phenyl-2H-indazol-3-ol 17
IR (KBr) ν (cm−1): 3399, 3213, 3055, 3016, 2929, 2863, 2728, 1598, 1500, 848, 815, 747, 699; 1H NMR (δ ppm): 3.01–3.09 (1H, m, H5a), 3.25–3.29 (1H, m, H5e), 4.22–4.24 (1H, dd, J 4a,5a=8.1 Hz, J 4a,5e=3.2 Hz), 6.94 (1H, s), 7.08–8.26 (12H, m, Harom), 9.59 (1H, bs, H2), 11.61 (1H, bs, H3); in the D2O-exchanged 1H NMR spectrum, two broad singlets, one at 9.59 ppm due to labile NH proton and another at 11.61 ppm due to OH proton disappeared; 13C NMR (δ ppm): 33.9 C-4, 36.1 C-5, 98.6 C-9, 113.8 C-7, 142.2 C-8, 158.2 C-3, 123.3–128.5 Carom, C-6 carbon may be merged with Carom, 132.2, 133.0, 134.7, 135.8, 137.2, 141.2 ipso carbons.
4,5-Dihydro-6-naphthalen-2-yl-4-p-tolyl-2H-indazol-3-ol 18
IR (KBr) ν (cm−1): 3388, 3196, 3050, 3022, 2919, 2858, 2721, 1596, 1509, 848, 813, 772, 745, 670; 1H NMR (δ ppm): 2.18 (3H, s, CH3 at phenyl ring), 3.01–3.06 (1H, m, H5a), 3.21–3.28 (1H, m, H5e), 4.17–4.20 (1H, dd, J 4a,5a=8.3 Hz, J 4a,5e=3.6 Hz), 6.98 (1H, s), 7.00–7.96 (11H, m, Harom), 9.65 (1H, bs, H2), 11.70 (1H, bs, H3); in the D2O-exchanged 1H NMR spectrum, two broad singlets, one at 9.65 ppm due to labile NH proton and another at 11.70 ppm due to OH proton disappeared; 13C NMR (δ ppm): 33.9 C-4, 36.1 C-5, 98.6 C-9, 113.8 C-7, 142.2 C-8, 158.3 C-3, 123.3–128.5 Carom, C-6 carbon may be merged with Carom, 132.2, 133.0, 134.7, 135.8, 137.2, 141.2 ipso carbons.
4-(4-Fluorophenyl)-4,5-dihydro-6-(naphthalen-2-yl)-2H-indazol-3-ol 19
IR (KBr) ν (cm−1): 3382, 3213, 3051, 3022, 2919, 2858, 2721, 1596, 1510, 848, 814, 773, 745, 671; 1H NMR (δ ppm): 3.01–3.09 (1H, m, H5a), 3.21–3.28 (1H, m, H5e), 4.17–4.20 (1H, dd, J 4a,5a=8.3 Hz, J 4a,5e=3.6 Hz), 6.98 (1H, s), 7.00–7.96 (11H, m, Harom), 9.85 (1H, bs, H2), 11.48 (1H, bs, H3); in the D2O-exchanged 1H NMR spectrum, two broad singlets, one at 9.85 ppm due to labile NH proton and another at 11.48 ppm due to OH proton disappeared; 13C NMR (δ ppm): 33.9 C-4, 36.1 C-5, 98.6 C-9, 113.8 C-7, 142.2 C-8, 158.3 C-3, 123.3–128.5 Carom, C-6 carbon may be merged with Carom, 133.0, 135.0, 135.8, 137.2, 141.0, 156.4 ipso carbons.
4,5-Dihydro-4-(4-methoxyphenyl)-6-(naphthalen-2-yl)-2H-indazol-3-ol 20
IR (KBr) ν (cm−1): 3380, 3172, 3053, 2994, 2931, 2891, 2832, 1604, 1508, 817, 746, 774, 705, 673; 1H NMR (δ ppm): 3.01–3.06 (1H, m, H5a), 3.20–3.27 (1H, m, H5e), 3.64 (3H, s, OCH3 at phenyl ring), 4.16–4.19 (1H, dd, J 4a,5a=8.2 Hz, J 4a,5e=3.6 Hz), 6.74 (1H, s), 6.76–7.97 (11H, m, Harom), 9.86 (1H, bs, H2), 11.34 (1H, bs, H3); in the D2O-exchanged 1H NMR spectrum, two broad singlets, one at 9.86 ppm due to labile NH proton and another at 11.34 ppm due to OH proton disappeared; 13C NMR (δ ppm): 33.5 C-4, 36.2 C-5, 55.0 OCH3 at phenyl ring, 98.8 C-9, 113.8 C-7, 141.0 C-8, 157.5 C-3, 113.4–129.2 Carom, C-6 carbon may be merged with Carom, 132.2, 133.0, 135.8, 137.2, 160.1 ipso carbons.
4-(4-Chlorophenyl)-4,5-dihydro-6-(naphthalen-2-yl)-2H-indazol-3-ol 21
IR (KBr) ν (cm−1): 3415, 3180, 3053, 2967, 2925, 2874, 2716, 2574, 1597, 1489, 815, 848, 744, 670; 1H NMR (δ ppm): 3.01–3.06 (1H, m, H5a), 3.23–3.28 (1H, m, H5e), 4.22–4.26 (1H, dd, J 4a,5a=8.3 Hz, J 4a,5e=3.9 Hz), 6.94 (1H, s), 7.18–7.90 (11H, m, Harom), 9.90 (1H, bs, H2), 11.49 (1H, bs, H3); in the D2O-exchanged 1H NMR spectrum, two broad singlets, one at 9.90 ppm due to labile NH proton and another at 11.49 ppm due to OH proton disappeared; 13C NMR (δ ppm): 33.8 C-4, 35.8 C-5, 97.9 C-9, 113.8 C-7, 144.2 C-8, 156.3 C-3, 123.3–129.1 Carom, C-6 carbon may be merged with Carom, 130.4, 132.3, 133.0, 135.7, 137.0 ipso carbons.
4-(4-Bromophenyl)-4,5-dihydro-6-(naphthalen-2-yl)-2H-indazol-3-ol 22
IR (KBr) ν (cm−1): 3355, 3180, 3052, 2924, 2869, 2732, 1596, 1536, 849, 815, 745, 667; 1H NMR (δ ppm): 3.00–3.05 (1H, m, H5a), 3.23–3.27 (1H, m, H5e), 4.21–4.24 (1H, dd, J 4a,5a=8.2 Hz, J 4a,5e=4.0 Hz), 6.94 (1H, s), 7.12–7.96 (11H, m, Harom), 9.84 (1H, bs, H2), 11.57 (1H, bs, H3); in the D2O-exchanged 1H NMR spectrum, two broad singlets, one at 9.84 ppm due to labile NH proton and another at 11.57 ppm due to OH proton disappeared; 13C NMR (δ ppm): 33.9 C-4, 35.8 C-5, 97.8 C-9, 113.9 C-7, 144.6 C-8, 158.1 C-3, 118.9–129.5 Carom, C-6 carbon may be merged with Carom, 130.3, 130.9, 131.1, 132.3, 133.0, 135.7, 137.0 ipso carbons.
4,5-Dihydro-6-naphthalen-6-yl-4-(4-nitrophenyl)-2H-indazol-3-ol 23
IR (KBr) ν (cm−1): 3371, 3191, 3054, 2922, 2847, 1598, 1522, 851, 813, 740, 684; 1H NMR (δ ppm): 3.01–3.06 (1H, m, H5a), 3.23–3.28 (1H, m, H5e), 4.21–4.24 (1H, dd, J 4a,5a=8.1 Hz, J 4a,5e=3.8 Hz), 6.93 (1H, s), 6.96–7.88 (11H, m, Harom), 9.72 (1H, bs, H2), 11.52 (1H, bs, H3); in the D2O-exchanged 1H NMR spectrum, two broad singlets, one at 9.72 ppm due to labile NH proton and another at 11.52 ppm due to OH proton disappeared; 13C NMR (δ ppm): 33.7 C-4, 35.6 C-5, 97.6 C-9, 113.8 C-7, 144.5 C-8, 158.4 C-3, 124.9–128.9 Carom, C-6 carbon may be merged with Carom, 130.6, 130.9, 131.1, 132.6, 133.2, 135.6, 137.2 ipso carbons.
4-(3-Chlorophenyl)-4,5-dihydro-6-(naphthalen-2-yl)-2H-indazol-3-ol 24
IR (KBr) ν (cm−1): 3410, 3360, 3196, 3054, 3011, 2929, 2808, 2765, 2716, 1595, 1563, 1083, 1188, 848, 814, 782, 746; 1H NMR (δ ppm): 3.03–3.07 (1H, m, H5a), 3.24–3.28 (1H, m, H5e), 4.23–4.26 (1H, dd, J 4a,5a=8.2 Hz, J 4a,5e=3.8 Hz), 6.91 (1H, s), 7.13–7.69 (11H, m, Harom), 9.92 (1H, bs, H2), 11.51 (1H, bs, H3); in the D2O-exchanged 1H NMR spectrum, two broad singlets, one at 9.92 ppm due to labile NH proton and another at 11.51 ppm due to OH proton disappeared; 13C NMR (δ ppm): 33.7 C-4, 35.8 C-5, 97.8 C-9, 113.5 C-7, 144.3 C-8, 156.7 C-3, 123.2–128.9 Carom, C-6 carbon may be merged with Carom, 130.2, 132.4, 133.2, 135.7, 137.3 ipso carbons.
General method for the synthesis of (2E)-ethyl-2-(2-(2,4-dinitrophenyl)hydrazono)-4-(naphthalen-2-yl)-6-arylcyclohex-3-ene carboxylates 25–32
Classical method
A solution of ethyl 4-(naphthalen-2-yl)-2-oxo-6-arylcyclohex-3-enecarboxylates 9–16 (0.001 mol) in ethanol (40 mL) was treated with 2,4-dintrophenylhydrazine (0.001 mol) and two to four drops of sulfuric acid and refluxed for 2–3 h. The reaction mixture was cooled and then poured over crushed ice. The crude products were recrystallized twice using ethanol as solvent to afford the product.
Microwave method
A mixture of 4-(naphthalen-2-yl)-2-oxo-6-arylcyclohex-3-enecarboxylates 9–16 (0.005 mol), 2,4-dinitrophenylhydrazine (0.005 mol) and activated fly ash (20 mg) was added in an alumina bath and mixed properly with the aid of glass rod (10 s) and then irradiated in a microwave oven for 120–180 s at 320 W (monitored by TLC). After completion of the reaction, the reaction mixture was extracted with ethyl acetate (3×5 mL). The catalyst and other solid wastes were removed by filtration. The combined organic layer was dried over anhydrous MgSO4 and concentrated in vacuo to furnish the products 25–32, which were purified by recrystallization in ethanol.
(2E)-Ethyl-2-(2-(2,4-dinitrophenyl)hydrazono)-4-(naphthalen-2-yl)-6-phenylcyclohex-3-enecarboxylate 25
IR (KBr) ν (cm−1): 3323, 3099, 3060, 2989, 2924, 2847, 1737, 1656, 1512, 1330, 818, 763, 704, 623; 1H NMR (δ ppm): 0.92 (3H, t, CH2CH 3 at C-1, J=7.0 Hz), 3.20–3.21 (2H, H5, m), 3.66–3.72 (1H, H6, m), 3.91 (2H, q, CH 2 CH3 at C-1, J=7.0 Hz), 4.16 (1H, H1, d, J=13.4 Hz), 6.73 (1H, s, H3), 7.25–8.80 (15H, m, Harom), 9.97 (1H, bs, NH); in the D2O-exchanged 1H NMR spectrum, a broad singlet at 9.97 ppm due to labile NH proton disappeared; 13C NMR (δ ppm): 13.8 CH2 CH3 at C-1, 35.1 C-5, 43.8 C-6, 59.9 CH2CH3 at C-1, 58.7 C-1, 115.5 C-3, 169.2 C = O at C-1, 158.7 C-2, 123.1–128.8 –Carom, C-4 carbon may be merged with –Carom, 129.5, 132.7, 133.6, 134.3, 141.5, 149.5 ipso carbons.
(2E)-Ethyl-2-(2-(2,4-dinitrophenyl)hydrazono)-4-(naphthalen-2-yl)-6-p-tolylcyclohex-3-enecarboxylate 26
IR (KBr) ν (cm−1): 3323, 3096, 3049, 3022, 2972, 2923, 2858, 1739, 1657, 1512, 1330, 817, 747, 705, 629; 1H NMR (δ ppm): 0.95 (3H, t, CH2CH 3 at C-1, J=7.0 Hz), 2.28 (3H, s, CH3 at phenyl ring), 3.15–3.17 (2H, H5, m), 3.60–3.68 (1H, H6, m), 3.92 (2H, q, CH 2 CH3 at C-1, J=7.0 Hz), 4.12 (1H, H1, d, J=13.4 Hz), 6.71 (1H, s, H3), 7.14–8.80 (14H, m, Harom), 9.97 (1H, bs, NH); in the D2O-exchanged 1H NMR spectrum, a broad singlet at 9.97 ppm due to labile NH proton disappeared; 13C NMR (δ ppm): 13.8 CH2 CH3 at C-1, 20.6 CH3 at phenyl ring, 35.3 C-5, 43.4 C-6, 59.9 CH2CH3 at C-1, 58.9 C-1, 115.5 C-3, 169.3 C = O at C-1, 158.7 C-2, 123.1–128.9 –Carom, C-4 carbon may be merged with –Carom, 129.5, 132.7, 133.6, 134.3, 136.1, 138.6, 149.1 ipso carbons.
(2E)-Ethyl-2-(2-(2,4-dinitrophenyl)hydrazono)-6-(4-fluorophenyl)-4-(naphthalen-2-yl)-cyclohex-3-enecarboxylate 27
IR (KBr) ν (cm−1): 3319, 3100, 3071, 2923, 2852, 1739, 1654, 1510, 1332, 839, 749, 704, 627; 1H NMR (δ ppm): 0.93 (3H, t, CH2CH 3 at C-1, J=7.0 Hz), 3.18–3.20 (2H, H5, m), 3.46–3.74 (1H, H6, m), 3.92 (2H, q, CH 2 CH3 at C-1, J=7.0 Hz), 4.15 (1H, H1, d, J=14.4 Hz), 6.72 (1H, s, H3), 7.15–8.80 (14H, m, Harom), 9.96 (1H, bs, NH); in the D2O-exchanged 1H NMR spectrum, a broad singlet at 9.96 ppm due to labile NH proton disappeared; 13C NMR (δ ppm): 13.8 CH2 CH3 at C-1, 35.1 C-5, 43.1 C-6, 60.1 CH2CH3 at C-1, 59.0 C-1, 115.2 C-3, 169.5 C = O at C-1, 158.8 C-2, 115.0–129.6 –Carom, C-4 carbon may be merged with –Carom, 129.7, 132.7, 133.7, 134.3, 137.8, 149.2, 162.5 ipso carbons.
(2E)-Ethyl-2-(2-(2,4-dinitrophenyl)hydrazono)-6-(4-methoxyphenyl)-4-(naphthalen-2-yl)-cyclohex-3-enecarboxylate 28
IR (KBr) ν (cm−1): 3319, 3054, 2994, 2928, 2841, 1737, 1655, 1512, 1332, 828, 747, 704, 628; 1H NMR (δ ppm): 0.95 (3H, t, CH2CH 3 at C-1, J=7.1 Hz), 3.14–3.16 (2H, H5, m), 3.54–3.62 (1H, H6, m), 3.73 (3H, s, OCH3 at phenyl ring), 3.92 (2H, q, CH 2 CH3 at C-1, J=7.0 Hz), 4.08 (1H, H1, d, J = 13.4 Hz), 6.88 (1H, s, H3), 6.89–8.89 (14H, m, Harom), 9.97 (1H, bs, NH); in the D2O-exchanged 1H NMR spectrum, a broad singlet at 9.97 ppm due to labile NH proton disappeared; 13C NMR (δ ppm): 13.8 CH2 CH3 at C-1, 35.3 C-5, 43.1 C-6, 55.0 OCH3 at phenyl ring, 59.9 CH2CH3 at C-1, 59.2 C-1, 115.4 C-3, 169.4 C = O at C-1, 158.7 C-2, 113.7–129.4 –Carom, C-4 carbon may be merged with –Carom, 132.7, 133.0, 133.5, 133.6, 134.38, 134.30, 149.1, 158.3 ipso carbons.
(2E)-Ethyl-2-(2-(2,4-dinitrophenyl)hydrazono)-6-(4-chlorophenyl)-4-(naphthalen-2-yl)-cyclohex-3-enecarboxylate 29
IR (KBr) ν (cm−1): 3323, 3088, 2972, 2924, 2847, 1736, 1645, 1514, 1326, 823, 744, 629; 1H NMR (δ ppm): 0.92 (3H, t, CH2CH 3 at C-1, J=7.1 Hz), 3.15–3.20 (2H, H5, m), 3.69–3.75 (1H, H6, m), 3.95 (2H, q, CH 2 CH3 at C-1, J=7.1 Hz), 4.16 (1H, H1, d, J=13.4 Hz), 6.74 (1H, s, H3), 7.27–8.90 (14H, m, Harom), 9.99 (1H, bs, NH); in the D2O-exchanged 1H NMR spectrum, a broad singlet at 9.99 ppm due to labile NH proton disappeared; 13C NMR (δ ppm): 13.8 CH2 CH3 at C-1, 34.9 C-5, 43.1 C-6, 60.0 CH2CH3 at C-1, 58.6 C-1, 115.4 C-3, 169.1 C = O at C-1, 158.6 C-2, 120.9–129.4 –Carom, C-4 carbon may be merged with –Carom, 129.5, 131.6, 132.7, 133.0, 133.6, 134.2, 140.5, 149.1 ipso carbons.
(2E)-Ethyl-2-(2-(2,4-dinitrophenyl)hydrazono)-6-(4-bromophenyl)-4-(naphthalen-2-yl)-cyclohex-3-enecarboxylate 30
IR (KBr) ν (cm−1): 3324, 3097, 2978, 2923, 2847, 1735, 1643, 1514, 1330, 826, 744, 705, 629; 1H NMR (δ ppm): 0.95 (3H, t, CH2CH 3 at C-1, J=7.0 Hz), 3.15–3.20 (2H, H5, m), 3.57–3.73 (1H, H6, m), 3.93 (2H, q, CH 2 CH3 at C-1, J=7.1 Hz), 4.17 (1H, H1, d, J=14.4 Hz), 6.73 (1H, s, H3), 7.42–8.80 (14H, m, Harom), 9.97 (1H, bs, NH); in the D2O-exchanged 1H NMR spectrum, a broad singlet at 9.97 ppm due to labile NH proton disappeared; 13C NMR (δ ppm): 13.8 CH2 CH3 at C-1, 34.8 C-5, 43.2 C-6, 60.1 CH2CH3 at C-1, 58.5 C-1, 115.5 C-3, 169.1 C = O at C-1, 158.6 C-2, 120.2–129.5 –Carom, C-4 carbon may be merged with –Carom, 129.9, 131.3, 132.7, 133.1, 133.6, 134.2, 141.0, 149.1 ipso carbons.
(2E)-Ethyl-2-(2-(2,4-dinitrophenyl)hydrazono)-4-(naphthalen-2-yl)-6-(4-nitrophenyl) cyclohex-3-enecarboxylate 31
IR (KBr) ν (cm−1): 3324, 3088, 2922, 2858, 1735, 1640, 1518, 1324, 828, 742, 705, 630; 1H NMR (δ ppm): 0.91 (3H, t, CH2CH 3 at C-1, J=6.9 Hz), 3.14–-3.19 (2H, H5, m), 3.67–3.73 (1H, H6, m), 3.94 (2H, q, CH 2CH3 at C-1, J=7.0 Hz), 4.17 (1H, H1, d, J=13.2 Hz), 6.76 (1H, s, H3), 7.21–8.78 (14H, m, Harom), 9.96 (1H, bs, NH); in the D2O-exchanged 1H NMR spectrum, a broad singlet at 9.96 ppm due to labile NH proton disappeared; 13C NMR (δ ppm): 13.6 CH2 CH3 at C-1, 35.0 C-5, 43.3 C-6, 60.3 CH2CH3 at C-1, 59.2 C-1, 115.7 C-3, 169.8 C = O at C-1, 158.3 C-2, 121.3–129.7 –Carom, C-4 carbon may be merged with –Carom, 129.4, 132.3, 133.6, 134.2, 137.7, 145.4, 149.2 ipso carbons.
(2E)-Ethyl-2-(2-(2,4-dinitrophenyl)hydrazono)-6-(3-chlorophenyl)-4-(naphthalen-2-yl)-cyclohex-3-enecarboxylate 32
IR (KBr) ν (cm−1): 3324, 3098, 3060, 2972, 2924, 2852, 1734, 1657, 1512, 1330, 820, 787, 747, 698; 1H NMR (δ ppm): 0.94 (3H, t, CH2CH 3 at C-1, J=7.1 Hz), 3.19–3.21 (2H, H5, m), 3.45–3.74 (1H, H6, m), 3.93 (2H, q, CH 2 CH3 at C-1, J=7.2 Hz), 4.13 (1H, H1, d, J=14.1 Hz), 6.77 (1H, s, H3), 7.21–8.77 (14H, m, Harom), 9.94 (1H, bs, NH); in the D2O-exchanged 1H NMR spectrum, a broad singlet at 9.94 ppm due to labile NH proton disappeared; 13C NMR (δ ppm): 13.7 CH2 CH3 at C-1, 34.7 C-5, 43.3 C-6, 60.2 CH2CH3 at C-1, 58.7 C-1, 115.4 C-3, 169.3 C = O at C-1, 158.3 C-2, 124.8–129.1 –Carom, C-4 carbon may be merged with –Carom, 129.2, 131.4, 132.6, 133.2, 133.7, 134.2, 140.2, 149.3 ipso carbons.
Conclusion
It has been demonstrated that activated fly ash is a highly effective catalyst for an efficient and rapid synthesis of an array of naphthyl-substituted cyclohexenone carboxylates, indazolonols, and nitrohydrazones by using activated fly ash as catalyst in “dry media.” All the synthesized compounds are characterized by their physical and analytical characterization, including melting point, elemental analysis, MS, FT-IR, one-dimensional NMR (1H and D2O-exchanged 1H and 13C), and two-dimensional HOMOCOR and HSQC spectroscopic data (see Supplementary Content). This novel catalyst provides a clean and convenient alternative methodology for the synthesis of title compounds. The method not only offers a substantial increase in the yield over the conventional method but also eliminates the usage of solvents such as ethanol, corrosive bases such as piperidine, and sodium hydroxide and shortens the reaction time from several hours of reflux to few seconds Citation42 Citation43. This reaction may have wide applicability in building a variety of heterocycles by choosing 4-(naphthalen-2-yl)-2-oxo-6-arylcyclohex-3-enecarboxylates as synthon, which has three versatile functional groups, i.e., ketone, olefin, and ester, for the synthesis of structurally diverse organic compounds. Hence, it may attract many synthetic organic chemists for utility of these compounds in organic synthesis.
Acknowledgements
Authors are thankful to NMR Research Centre, Indian Institute of Science, Bangalore, India, for recording spectra. One of the authors, V. Kanagarajan, is grateful to Council of Scientific and Industrial Research (CSIR), New Delhi, India, for providing financial support in the form of CSIR-Senior Research Fellowship (SRF) in Organic Chemistry.
References
- Cecchi , L. , Melani , F. , Filacchioni , G. and Tredici , M. 1984 . Farmaco , 39 : 945 – 952 .
- Ko , J.H. , Yeon , S.W. , Ryu , J.S. , Yong , K.T. , Ha , S.E. , Jung , Y.H. , Eun , P.R. and Kyu , R.C. 2006 . Bioorg. Med. Chem. Lett. , 16 : 6001 – 6005 .
- Gerpe , A. , Aguirre , G. , Boiani , L. , Cerecetto , H. , Gonzalez , M. , Olea-Azar , C. , Rigol , C. , Maya , J.D. , Morello , A. , Piro , O.E. , Aran , V.J. , Azqueta , A. , de Cerain , A.L. , Monge , A. , Rojas , M.A. and Yaluff , G. 2006 . Bioorg. Med. Chem. , 14 : 3467 – 3480 .
- Li , X. , Chu , S. , Feher , V.A. , Khalili , M. , Nie , Z. , Margosiak , S. , Nikulin , V. , Levin , J. , Sprankle , K.G. , Tedder , M.E. , Almassy , R. , Appelt , K. and Yager , K.M. 2003 . J. Med. Chem. , 46 : 5663 – 5673 .
- Selwood , D.L. , Brummell , D.G. , Budworth , J. , Burtin , G.E. , Campbell , R.O. , Chana , S. , Charles , I.G. , Fernandez , P.A. , Glen , R.C. , Goggin , M.C. , Hobbs , A.J. , Kling , M.R. , Liu , Q. , Madge , D.J. , Meillerais , S. , Powell , K.L. , Reynolds , K. , Spacey , G.D. , Stables , J.N. , Tatlock , M.A. , Wheelers , K.A. , Wishart , G. and Woo , C. 2001 . J. Med. Chem. , 44 : 78 – 93 .
- Finn , R. 2007 . Ob. Gyn. News , 42 : 28
- Pelicano , H. , Martin , D.S. , Xu , R.H. and Huang , P. 2006 . Oncogene , 25 : 4633 – 4646 .
- Southan , G.J. and Szabo , C. 1996 . Biochem. Pharmacol. , 51 : 383 – 394 .
- Kimura , Y. , Hatanaka , K. , Naitou , Y. , Maeno , K. , Shimada , I. , Koakutsu , A. , Wanibuchi , F. and Yamaguchi , T. 2004 . Eur. J. Pharmacol. , 483 : 37 – 43 .
- May , J.A. , Sharif , N.A. , Chen , H.H. , Liao , J.C. , Kelly , C.R. , Glennon , R.A. , Young , R. , Li , J.X. , Rice , K.C. and France , C.P. 2009 . Pharmacol. Biochem. Behav. , 91 : 307 – 314 .
- Arctander , S. 1969 . Perfume and Flavour Chemicals (Aroma Chemicals) , Monclair, NJ : Author .
- Dimmock , J.R. , Vashishtha , S.C. and Stables , J.P. 2000 . Eur. J. Med. Chem. , 35 : 241 – 248 .
- Ergenc , N. and Gunay , N.S. 1998 . Eur. J. Med. Chem. , 33 : 143 – 148 .
- Todeschini , A.R. , Miranda , A.L. , Silva , C.M. , Parrini , S.C. and Barreiro , E.J. 1998 . Eur. J. Med. Chem. , 33 : 189 – 199 .
- Lima , P.C. , Lima , L.M. , Silva , K.C. , Leda , P.H. , Miranda , A.L.P. , Fraga , C.A.M. and Barreiro , E. 2000 . Eur. J. Med. Chem. , 35 : 187 – 203 .
- Fraga , A.G.M. , Rodrigues , C.R. , Miranda , A.L.P. , Barreiro , E.J. and Fraga , C.A.M. 2000 . Eur. J. Pharm. Sci. , 11 : 285 – 290 .
- Walcourt , A. , Loyevsky , M. , Lovejoy , D.B , Gordeuk , V.R. and Richardson , D.R. 2004 . Int. J. Biochem. Cell Biol. , 36 : 401 – 407 .
- Kucukguzel , S.G. , Rollas , S. , Erdeniz , H. and Kiraz , M. 1999 . Eur. J. Med. Chem. , 34 : 153 – 160 .
- Silva , A.G. , Zapata-Suto , G. , Kummerle , A.E. , Fraga , C.A.M. , Barreiro , E.J. and Sudo , R.T. 2005 . Bioorg. Med. Chem. , 13 : 3431 – 3437 .
- Pandey , J. , Pal , R. , Dwivedi , A. and Hajela , K. 2002 . Arzneimittelforschung , 52 : 39 – 44 .
- Watt , J.D. and Throne , D.J. 1965 . J. Appl. Chem. , 15 : 595 – 604 .
- Throne , D.J. and Watt , J.D. 1966 . J. Appl. Chem. , 16 : 33 – 39 .
- Saraswathy , V. , Muralidharan , S. , Thangavel , K. and Srinivasan , S. 2003 . Cement Concr. Res. , 25 : 673 – 680 .
- Gopalakrishnan , M. , Sureshkumar , P. , Kanagarajan , V. , Thanusu , J. and Govindaraju , R. 2006 . Arkivoc , 13 : 130 – 141 .
- Gopalakrishnan , M. , Thanusu , J. and Kanagarajan , V. 2007 . J. Korean Chem. Soc. , 51 : 520 – 525 .
- Khatri , C. and Rani , A. 2008 . Fuel , 87 : 2886 – 2892 .
- Narkhede , H.P. , More , U.B. , Dalal , D.S. , Pawar , N.S. , More , D.H. and Mahulikar , P.P. 2007 . Synth. Commun. , 37 : 573 – 577 .
- Narkhede , H.P. , More , U.B. , Dalal , D.S. and Mahulikar , P.P. 2008 . Synth. Commun. , 38 : 2413 – 2418 .
- Gopalakrishnan , M. , Thanusu , J. and Kanagarajan , V. 2008 . J. Korean Chem. Soc. , 52 : 523 – 529 .
- Kanagarajan , V. , Thanusu , J. and Gopalakrishnan , M. 2009 . Green Chem. Lett. Rev. , 2 : 161 – 167 .
- Wang , S. and Wu , H.W. 2006 . J. Hazard. Mater. , 136 : 482 – 501 .
- Ng , P.F. , Li , L. , Wang , S. , Zhu , Z.H. , Lu , G.Q. and Yan , Z.F. 2007 . Environ. Sci. Technol. , 41 : 3758 – 3762 .
- Wang , S. , Ma , Q. and Zhu , Z.H. 2008 . Fuel , 87 : 3469 – 3473 .
- Wang , S. 2008 . Environ. Sci. Technol. , 42 : 7055 – 7063 .
- Gopalakrishnan , M. , Thanusu , J. and Kanagarajan , V. 2008 . J. Korean Chem. Soc. , 52 : 47 – 51 .
- Gopalakrishnan , M. , Sureshkumar , P. , Thanusu , J. , Kanagarajan , V. and Ezhilarasi , M.R. 2008 . Lett. Org. Chem. , 5 : 142 – 147 .
- Gopalakrishnan , M. , Thanusu , J. and Kanagarajan , V. 2008 . J. Sulfur Chem. , 29 : 179 – 185 .
- Lidstrom , P. ; Tierney , J.P. ; Wathey , B. ; Westman , J. ; Microwave-Assisted Organic Synthesis ; Oxford : Blackwell , 2005 .
- Loupy , A. ; Microwaves in Organic Synthesis ; Weinheim : Wiley-VCH , 2006 .
- Leadbeater , N.E. and Schmink. , J.R. 2007 . Tetrahedron , 63 : 6764 – 6773 .
- Rajanarendar , E. , Kalyan Rao , E. and Raju , S. 2009 . Indian J. Chem. , 48B : 749 – 753 .
- Padmavathi , V. , Sharmila , K. , Balaiah , A. , Somasekhar Reddy , A. and Bhaskar Reddy , D. 2001 . Synth. Commun. , 31 : 2119 – 2126 .
- Guthrie , W. and Wang , X.P. 1991 . Can. J. Chem. , 69 : 339
Supplementary Content
Spectral analysis of 9, 17, and 25
In order to discuss the spectral data of ethyl 4-(naphthalen-2-yl)-2-oxo-6-arylcyclohex-3-enecarboxylates 9–16, ethyl 4-(naphthalen-2-yl)-2-oxo-6-phenyl cyclohex-3-enecarboxylate 9 is chosen as the representative compound. The FT-IR spectrum of ethyl 4-(naphthalen-2-yl)-2-oxo-6-phenylcyclohex-3-enecarboxylate 9 shows two strong characteristic absorptions at 1738 and 1659 cm−1 due to ester carbonyl and ketone functional groups, respectively. The band at 1606 cm−1 is due to the presence of C = C stretching frequency. The absorption frequency at 3057 cm−1 is assigned to aromatic C–H stretching vibration, and the absorption frequencies at 2987 and 2929 cm−1 are assigned to aliphatic C–H stretching vibration. The observed ester carbonyl, ketone, and C = C stretching vibrational bands are supporting evidence for the formation of synthesized compound 9. In the 1H NMR spectrum of 9, a triplet is observed at 0.92 ppm (J=7.1 Hz) corresponding to three protons and this signal is due to ester methyl protons at C-1. A quartet observed at 3.91 ppm (J=7.0 Hz) corresponding to two protons and this signal is due to ester methylene protons at C-1. Two multiplets are obtained in the range 3.22–3.28 and 3.65–3.73 ppm, and they are due to H-5 and H-6 protons. The doublet at 4.17 ppm (J=13.4 Hz) has been assigned to H-1 proton. The singlet observed in downfield region at 6.73 ppm is due to H-3 proton. The aromatic protons appeared as a multiplet in the range 7.24–8.33 ppm. Two 13C resonances at 194.2 and 169.2 6 ppm are assigned to C-2 carbonyl carbon and ester carbonyl carbon, respectively. The 13C resonances at 35.1 and 43.8 ppm are due to the C-6 and C-5 carbons, respectively. The 13C resonances observed at 58.7 and 13.7 ppm are assigned to ester methylene and methyl carbons at C-1, respectively. The signal observed at 59.9 ppm is assigned to C-1 carbon, whereas the signal at 123.1 ppm is assigned to C-3 carbon. The aromatic carbons are observed in the range of 123.4–128.8 ppm. The C-4 carbon may be merged with the aromatic carbons. The remaining 13C signals at 132.7, 133.6, 134.3, and 141.5 ppm are due to ipso carbons.In order to discuss the spectral data of 4,5-dihydro-6-(naphthalen-2-yl)-4-aryl-2H-indazol-3-ols 17–24, 4,5-dihydro-6-naphthalen-2-yl-4-phenyl-2H-indazol-3-ol 17 is chosen as the representative compound. The FT-IR spectrum of the 4,5-dihydro-6-naphthalen-6-yl-4-phenyl-2H-indazol-3-ol 17 shows that two characteristic absorption frequencies at 3399 and 3213 cm−1 suggest the presence of –OH and –NH functional groups. The absorption frequency at 1598 cm−1 is assigned to C = N stretching vibration. The band at 1500 cm−1 is due to the presence of C = C stretching frequency. Besides these, aromatic CH stretching frequencies are observed at 3055 and 3016 cm−1 and the aliphatic CH stretching frequencies are observed at 2929, 2863, and 2728 cm−1. The observed –OH, –NH, C = N, and C = C stretching vibrational bands are supporting evidence for the formation of synthesized compound 17. In the 1H NMR spectrum of compound 17, a doublet of doublet is observed for H-4 proton and two coupling constants are extracted from it, namely, J 4a,5a=8.1 Hz and J 4a,5e=3.2 Hz. Two multiplets in the region 3.01–3.09 and 3.25–3.29 ppm are assigned to H5a and H5e, respectively. A singlet at 6.94 ppm is conveniently assigned to H-7 proton. The aromatic protons appear as a multiplet around 7.08–8.26 ppm. The labile OH and NH protons (exchangeable with D2O) appear as a broad singlet at 11.61 and 9.59 ppm, respectively. In the HOMOCOR spectrum of 17, the signal at 4.22–4.24 ppm shows cross-peaks with the signals at 3.01–3.09 and 3.25–3.29 ppm. The signal at 3.25–3.29 ppm shows cross-peaks at 3.01–3.09 and 4.22–4.24 ppm. Similarly, the signal at 3.01–3.09 ppm shows cross-peaks at 3.25–3.29 and 4.22–4.24 ppm. The signal at 4.22–4.24 ppm must be due to H-4 proton, since this can have coupling with H5a and H5e protons. Consequently, two signals at 3.01–3.09 and 3.25–3.29 ppm must be due to the H5a and H5e protons. The signal at 6.94 ppm shows a cross-peak with 3.25–3.29 ppm and vice versa. Hence the signal at 6.94 ppm is conveniently assigned to H-7 proton and the signal at 3.25–3.29 ppm is assigned to H5e. In the 13C NMR spectrum of 17, two resonances in the aliphatic region at 34.3 and 35.9 ppm are due to C-4 and C-5 carbons, respectively. The remaining 13C resonances in quaternary carbon signals at 158.2, 113.8, 145.3, and 98.4 are due to C-3, C-7, C-8, and C-9 carbons, respectively. The aromatic carbons are observed in the range of 123.3–129.6 ppm. The 13C resonances at 132.2, 133.0, 135.8, 137.2, and 138.1 are due to ipso carbons. In the HSQC spectrum of 17, one bond correlation (34.3/4.22–4.24 ppm) is observed between C-4 and H-4. The 13C resonance at 35.9 has correlations with methylene protons H5a and H5e (35.9/3.01–3.09; 35.9/3.25–3.29) and hence C-5 resonates at 35.9 ppm. The 13C resonance at 113.8 ppm has correlations with a singlet at 6.94 ppm. So the signal at 6.94 ppm is conveniently assigned to H-7 proton, and the carbon signal at 113.8 ppm is assigned to C-7. In the HSQC, the 13C resonances at 98.4, 145.3, and 158.2 ppm have no correlations with protons and hence these resonances are due to quaternary carbons. The 13C resonances at 98.4, 145.3, and 158.2 ppm are due to C-9, C-8, and C-3 carbons, respectively. The C-6 carbon may be merged with the aromatic carbons. Among the quaternary carbons, the 13C resonances at 132.2, 133.0, 135.8, 137.2, and 138.1 are due to ipso carbons.
In order to discuss the spectral data of (2E)-ethyl-2-(2-(2,4-dinitrophenyl)hydrazono)-4-(naphthalen-2-yl)-6-arylcyclohex-3-ene carboxylates 25–32, (2E)-ethyl-2-(2-(2,4-dinitrophenylhydrazono)-4-(naphthalen-2-yl)-6-phenylcyclohex-3-enecarboxylate 25 is chosen as the representative compound. The FT-IR spectrum of (2E)-ethyl-2-(2-(2,4-dinitrophenylhydrazono)-4-(naphthalen-2-yl)-6-phenylcyclohex-3-ene carboxylate 25 shows that a characteristic absorption frequency at 3323 cm−1 suggests the presence of the –NH functional group. The absorption frequency at 1656 cm−1 is assigned to C = N stretching vibration. The band at 1512 cm−1 is due to the presence of C = C stretching frequency. Nitro functional groups show a characteristic stretching frequency around 1330 cm−1. Besides these, aromatic CH stretching frequencies are observed at 3099 and 3060 cm−1 and the aliphatic CH stretching frequencies are observed at 2989, 2924, and 2847 cm−1. The observed –NH, C = N, C = C, and NO2 stretching vibrational bands are supporting evidence for the formation of synthesized compound 25. In the 1H NMR spectrum of 25, a triplet is observed at 0.92 ppm (J=7.0 Hz) corresponding to three protons and this signal is due to ester methyl protons at C-1. A quartet is observed at 3.91 ppm (J=7.0 Hz) corresponding to two protons and this signal is due to ester methylene protons at C-1. Two multiplets are obtained in the range 3.20–3.21 and 3.66–3.72 ppm and they are due to H-5 and H-6 protons. The doublet at 4.16 ppm (J=13.4 Hz) has been assigned to H-1 proton. The singlet observed in downfield region at 6.73 ppm is due to H-3 proton. The aromatic protons appeared as a multiplet in the range 7.25–8.80 ppm. The labile NH proton (exchangeable with D2O) appears as a broad singlet at 9.97 ppm. In the HOMOCOSY spectrum of 25, the signal at 0.92 ppm shows a cross-peak with the signal at 3.91 ppm and vice versa. From the above cross-peaks it is confirmed that the triplet observed at 0.92 ppm corresponds to ester methyl protons at C-1, whereas the quartet at 3.91 ppm corresponds to ester methylene protons at C-1. The signal at 4.16 ppm gives a cross-peak with the multiplet at 3.66–3.72 ppm. Similarly, the multiplet at 3.66–3.72 ppm gives cross-peaks with the signal at 4.16 ppm as well as the signal at 3.20–3.21 ppm. Also, the signal at 6.73 ppm gives correlations with the signal at 3.20–3.21 ppm and vice versa. From the above correlations, it reveals that the singlet at 6.73 ppm corresponds to H-3 proton and the multiplet at 3.66–3.72 ppm corresponds to H-6 proton. The doublet at 4.16 ppm is conveniently assigned to H-1 proton and the multiplet at 3.20–3.21 ppm is unambiguously assigned to H-5 proton. In the 13C NMR spectrum of 25, five resonances in the aliphatic region at 13.8, 35.1, 43.8, 58.7, and 59.9 ppm are observed. They are all due to CH2 CH3 at C-1, C-5, C-6, C-1 and CH2CH3 at C-1, respectively. C-3 carbon resonates at 115.5 ppm. The remaining 13C resonances in quaternary carbon signals at 169.2 and 158.7 ppm are due to C = O at C-1 and C-2 carbons, respectively. The aromatic carbons are observed in the range of 123.1–128.8 ppm. C-4 carbon may be merged with aromatic carbon signals. The 13C resonances at 129.5, 132.7, 133.6, 134.3, 141.5, and 149.5 are due to ipso carbons. In the HSQC spectrum of 25, one bond correlation (13.8/0.92 ppm) is observed between CH2 CH3 at C-1 and CH2CH 3 at C-1. The 13C resonance at 59.9 has correlations with ester methylene protons CH 2CH3 at C-1. Another one-bond correlation (35.1/3.20–3.21 ppm) is observed between C-5 and H-5. The 13C resonance at 43.8 ppm has one-bond correlation with a multiplet around 3.66–3.72 ppm. Hence, the signal at 43.8 ppm corresponds to C-6 carbon, whereas the multiplet at 3.66–3.72 ppm is assigned to H-6 proton. Another aliphatic carbon that resonances at 58.7 ppm shows one-bond correlation with a doublet at 4.16 ppm. From this correlation, it is revealed that the doublet at 4.16 ppm corresponds to H-1 proton of the cyclohexenone moiety and the 13C signal at 58.7 is assigned to C-1 carbon. The 13C resonance at 115.5 ppm has correlations with a singlet at 6.73 ppm. So the signal at 6.73 ppm is conveniently assigned to H-7 proton and the carbon signal at 115.5 ppm is assigned to C-7. In the HSQC, the 13C resonances at 169.2 and 158.7 ppm have no correlations with protons and hence it is due to quaternary carbons, ester C = O at C-1 and C = N, respectively. The C-6 carbon may be merged with the aromatic carbons. Among the quaternary carbons, the 13C resonances at 129.5, 132.7, 133.6, 134.3, 141.5, and 149.5 ppm are due to ipso carbons.