Abstract
Silica immobilized sulfuric acid ([3-(propyl)sulfanyl]propyl]ester and N-propylsulfamic acid are employed as recyclable catalysts for the synthesis of 1,1-diacetates from the reaction between an aromatic aldehyde and acetic anhydride under mild- and solvent-free conditions at room temperature. These solid acids could be recycled several times without losing any catalytic activity.
Introduction
Selective protection and deprotection of carbonyl groups plays an important role in many multi-step organic syntheses Citation1. Ease of preparation and stability of 1,1-diacetates toward basic and neutral conditions make them as a preferred method of protection of aldehydes Citation1 Citation2. In addition, protection of aldehydes as the corresponding acylals can be achieved very easily in the presence of ketones. Moreover, acylals are used as good precursors for asymmetric allylic alkylation Citation3 and natural product synthesis Citation4 as well as the synthesis of 1-acetoxydienes and 2,2-dichlorovinylacetates for Diels–Alder reactions Citation5 Citation6. Also, they have been used as cross-linking agents for cellulose in cotton Citation7 and are useful intermediates in industry Citation5. Appropriate nucleophiles can convert the acylal functionality into other functional groups Citation8 Citation9. Generally, they are synthesized from acetic anhydride and aldehydes using strong protonic acids as catalysts, such as sulfuric Citation10, phosphoric Citation11, methanesulfuric Citation12, or perchloric acid Citation13, Lewis acids, such as FeCl3 Citation2, PCl3 Citation14, InCl3 Citation15, ZrCl4 Citation16, WCl6 Citation17, LiBF4 Citation18, Cu(BF4)2.xH2O Citation19, ceric ammonium nitrate (CAN) Citation20, Zn(BF4)2 Citation21, H6P2W18O62.24H2O Citation22, AlPW12O40 Citation23, Cu(OTf)2 Citation24, Sc(OTf)3 Citation25, LiOTf Citation26, and heterogeneous catalysts such as Nafion-H Citation27, expansive graphite Citation28, zeolites Citation29, montmorillonite clay Citation30 Citation31, and supported reagents Citation32–35. Other catalysts, such as iodine Citation36, N-bromosuccinimide Citation37, and trimethylchlorosilane/sodium iodide Citation38, have also been used for this transformation, but these procedures are often accompanied by longer reaction times, poor product yields, stringent conditions, good catalyst loading; require the use of toxic solvents; and so on. More recently, cobalt (II) bromide Citation39, SO3H-functionalized ionic liquids Citation40, ferrous methansulfonate Citation41, poly(N,N′-dibromo-N-ethylbenzene-1,3-disulfonamide) Citation42, NbCl5 Citation43, SbCl3 Citation44, silica-bonded S-sulfonic acid Citation45, and supported catalysts Citation46–50 were reported for the synthesis of 1,1-diacetates.
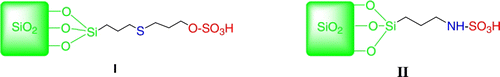
In recent years, heterogeneous catalysts have been prepared and applied as an alternative to traditional acids and homogeneous acids in catalyzing various organic transformations. The advantages of heterogeneous catalysts are environmentally friendly, easy catalyst separation, high-yielding, and recyclability. We recently applied several heterogeneous catalysts to discover different novel useful synthetic methodologies Citation51–55. In continuation of the work, we have observed that sulfuric acid ([3-(3-silicapropyl)sulfanyl]-propyl]ester (I), and silica-bonded N-propylsulfamic acid (II) () are efficient heterogeneous catalysts for the synthesis of aromatic 1,1-diacetates ().
Figure 1. The structure of catalysts I and II.
Results and discussion
Solid sulfuric acid I was prepared by the reaction of 3-(3-silicapropylthio)-1-propanol with chlorosulfonic acid in chloroform. Elemental analysis showed the S content to be 15.51%. Typically a loading at approximately 0.35 mmol/g is obtained. On the other hand, when the washed sulfonated product I was placed in an aqueous NaCl solution, the solution pH dropped virtually instantaneously to pH ≈ 1.93, as ion exchange occurred between protons and sodium ions proton exchange capacity: 0.33 mmol/g of sulfuric acid I which is in good agreement with the result obtained from TGA.
The thermo gravimetric curve (TGA) seems to indicate two-stage decomposition which is considered to be due to removal of physically absorbed water (100–130°C) or basic silica sulfonic acid (150–230°C), respectively, and is involved with a total overall weight loss of catalyst 43.1% (Figure 6 supporting information). The TGA of silica-bonded N-propylsulfamic acid (II) was shown a total overall weight loss of catalyst 50% (Figure 18 supporting information).
In the FT-IR spectrum of catalyst, the major peaks for silica (SiO2) are broad antisymmetric Si–O–Si stretching from 1300 to 1010.6 cm−1 and symmetric Si–O–Si stretching near 880–852.5 cm−1. For sulfuric acid functional group, the FT-IR absorption range of the O=S=O asymmetric and symmetric stretching modes lie in 1170 and 1060 cm−1, respectively, the S–O stretching mode lies around 574.7 cm−1. FT-IR spectrum shows the overlap asymmetric and symmetric stretching bands of SO2 with Si–O–Si stretching bands in the silica functionalized alkyl-sulfuric acid. The spectrum also shows a broad OH stretching absorption around 3600 to 2520 cm−1 ( supporting information).
The BET surface area of sulfuric acid ([3-(3-silicapropyl)sulfanyl]propyl)ester 4.35 m2g−1 and a total pore volume 0.63 cm3g−1 were measured (figures 3–5 supporting information) and also, for silica-bonded N-propylsulfamic acid the BET 2.91 m2g−1 and a total pore volume 0.456 cm3g−1 were obtained (figures 15–17 supporting information).
To study the effect of catalyst loading on the protection of aromatic aldehydes as the corresponding 1,1-diacetates the reaction of 4-methylbenzaldehyde with acetic anhydride was chosen as a model reaction (). The results show clearly I and II are effective catalysts for this transformation and in the absence of I or II the reaction did not take place, even after 12 h. Although lower catalyst loading 0.01 g of I or II accomplished this protection, however, 0.03 g of I or II per 1 mmol of aldehyde was optimum in terms of reaction time and isolated yield. Also, as shown in , the best result in the case of time and isolated yield was obtained by using 15 mmol of acetic anhydride.
Table 1. Conversion of p-methylbenzaldehyde to the corresponding diacetate with acetic anhydride in the presence of different amounts of catalysts I and II under solvent-free conditions and at room temperature.a
The model reaction was also examined in various solvents as well as under solvent-free conditions in the presence of 0.03 g mmol−1 of I or II ().
Table 2. Conversion of p-methylbenzaldehyde to the corresponding diacetate in different solvents and under solvent-free conditions in the presence of I and II (0.03 g).
The yield of the reaction under solvent-free conditions was the highest and the reaction time was shorter. In protic solvents such as water and ethanol this protection reaction proceeded in longer reaction times and with very poor yields, which maybe related to the instability of acetic anhydride in protic solvents. Therefore, we employed the optimized conditions (0.03 g mmol−1 of I or II and solvent-free conditions) for the conversion of various aryl aldehydes into the corresponding acylals ().
The results of the solvent-free preparation of acylals from aromatic aldehydes 1 in the presence of I and II at room temperature are shown in . 1,1-Diacetate 2a was obtained in high yield in 5 minutes by the reaction of benzaldehyde with acetic anhydride. Benzaldehydes with electron-donating or electron-withdrawing groups, i.e. 4-methylbenzaldehyde 1b and 2-methoxybenzaldehyde 1e, or 4-nitrobenzaldehyde 1c and 2-nitrobenzaldehyde 1l, were converted into the corresponding acylals 2b, 2e, 2c, and 2l in high yields after very short reaction times. The acid sensitive substrate thiophene-2-carbaldehyde 1o gave the expected acylal 2o in 75–76% yield without any by-products (, entry o). Aliphatic aldehyde such as hexanal was treated with acetic anhydride and gave the corresponding product 1p in 69–70% yield (, entry p).
Table 3. Preparation of various acylals in the presence of I and II under solvent-free conditions at room temperature.a,b
We also investigated the reactions of 2-hydroxybenzaldehyde 1k under the above conditions and observed that both the carbonyl and phenolic groups were acylated (, entry k). Several aliphatic and aromatic ketones including cyclohexanone, acetophenone, and 4-chloroacetophenone were not reactive under the described experimental conditions, even after 2 h.
Next, we studied the competitive acylation reactions of aromatic aldehydes in the presence of ketones using I or II. In the presence of ketones, exclusive acylation of the aldehyde functions was observed. The chemoselective acylations of benzaldehyde and 4-chlorobenzaldehyde in the presence of acetophenone and 4-chloroacetophenone are shown in .
Table 4. Competitive acylal formation from aldehydes in the presence of ketones using I or II under solvent-free conditions.a
Finally, a comparative study of I and II with other recently reported catalysts for acylation of 4-chlorobenzaldehyde, as a model compound was made which revealed that I or II is an equally efficient but much cheaper and reusable catalyst ().
Table 5. Comparison of the efficiency of I and II with some of the reported procedure on the reaction of 4-chlorobenzaldelyde with acetic anhydride.
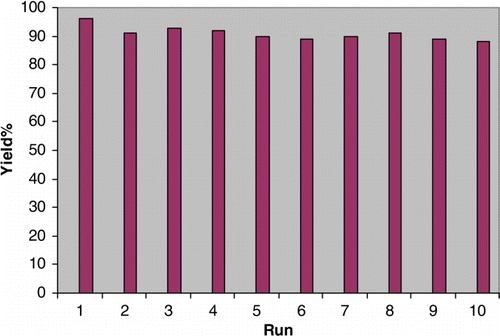
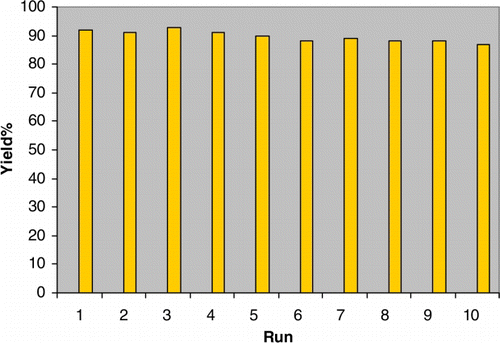
The possibility of recycling the catalyst was examined using the reaction of p-methylbenzaldehyde and acetic anhydride under the optimized conditions. Upon completion, the reaction mixture was filtered and the remaining solid was washed with dichloromethane, dried in air, and the catalyst reused in the next reaction. The recycled catalyst could be reused 10 times in the presence of I and II without any additional treatment. No observation of any appreciable loss in the catalytic activity of I and II was observed ( and ). Also, the characterization (TGA, IR, XRD, and BET) of after recovered catalysts I and II was run and confirmed the same as before the reaction (see supporting information). TGA of recycled catalysts confirmed the same thermal stability until 250°C. The XRD confirmed that there is no change in the pattern of catalysts. The IR of recycled catalysts slightly changed and in most cases shown broader peaks. In addition, the BET of recycled catalysts slightly changed as for catalyst I the BET before the reaction was 4.35 m2g−1 while after recycling 5.67 m2g−1 and for catalyst II the BET was 2.91 m2g−1 and after recycling 1.89 m2g−1.
Figure 2. Recyclability of I (0.03 g) in the reaction of p-methylbenzaldehyde (1 mmol) and acetic anhydride (15 mmol) at room temperature. Reaction time=7 min.
In conclusion, we have shown that silica-bonded functionalized sulfonic acids I and II, which can be prepared from commercially available and cheap starting materials, catalyzed efficiently the synthesis of aromatic 1,1-diacetates from aryl aldehydes. The mild reaction conditions and simplicity of the procedure and reusability of catalysts offer improvements over many existing methods.
Figure 3. Recyclability of II (0.03 g) in the reaction of p-methylbenzaldehyde (1 mmol) and acetic anhydride (15 mmol) at room temperature. Reaction time=7 min.
Experimental section
General
Chemicals were purchased from Fluka, Merck, and Aldrich chemical companies. All the products are known compounds and were characterized by comparison of their IR, 1H NMR, and 13C NMR spectroscopic data and their melting points with reported values Citation10–25. Silica immobilized sulfuric acid ([3-(propyl)sulfanyl]propyl]ester (I)) and silica-bonded N-propylsulfamic acid (II) were prepared according to our previously reported procedures Citation51 Citation52.
Catalyst preparation
3-(3-Silicapropylthio)-1-propanol
3-Chloro-1-propanol (5 mmol, 0.473 g) was added to a magnetically stirred mixture of 3-mercaptopropylsilica (10 g) in toluene (30 mL), then some drops of triethyl amine was added and the resulting mixture refluxed for 24 h. The mixture was filtered and the solid was washed with ethanol (3×20 mL), and then dried in an oven. 3-(3-Silicapropylthio)-1-propanol was obtained as a cream powder (10.3 g).
Sulfuric acid ([3-(3-silicapropyl)sulfanyl]propyl)ester (I)
To a mixture of 3-(3-silicapropylthio)-1-propanol (5 g) in chloroform (20 mL), chlorosulfonic acid (0.19 g, 1.65 mL) was added dropwise at 0°C during 2 h. After addition was complete, the mixture was stirred for 2 h until HCl gas evolution was stopped. Then, the mixture was filtered and washed with ethanol (30 mL) and dried at room temperature to obtain sulfuric acid ([3-(3-silicapropyl)sulfanyl]propyl)ester as a cream powder (5.13 g). Sulfur content of the samples by conventional elemental analysis was 15.51% Citation51.
Silica-bonded N-propylsulfamic acid (II)
To a mixture of 3-aminopropylsilica (5 g) in chloroform (20 mL) chlorosulfonic acid (0.2 g, 1.75 mL) was added dropwise at 0°C during 2 h. After addition was complete, the mixture was stirred for 2 h until HCl gas evolution was stopped. Then, the mixture was filtered and washed with ethanol (30 mL) and dried at room temperature to obtain silica-bonded N-propylsulfamic acid (2) as cream powder (5.13 g). Sulfur content of the samples by conventional elemental analysis was 9.29% Citation52.
General procedure for acylation
To a mixture of aldehyde (1 mmol) and acetic anhydride (15 mmol) was added catalyst (I or II) 0.03 g and the mixture was stirred at room temperature. When the reaction was complete as judged by TLC, CH2Cl2 (5 mL) was added and the reaction mixture was filtered and the remaining solid was washed with CH2Cl2 (3×5 mL) in order to separate the catalyst. The CH2Cl2 layer was washed with water (2×10 mL) and dried over anhydrous MgSO4. After removal of the solvent in vacuo, the obtained residue was recrystallized from ethanol.
Spectral data
2m: IR(KBr): 3005, 2820, 1756, 1580, 1430, 1367, 1240, 1200, 1158, 1060, 998, 942, 903, 802, 705 cm−1. 1H NMR (500 MHz, CDCl3): δ (ppm) 2.12 (s, 12H), 7.44 (t, 1H, J=7.5 Hz), 7.55 (d, 2H, J=7.5 Hz), 7.65 (s, 1H), 7.68 (s, 2H). 13C NMR (125 MHz, CDCl3): δ (ppm) 21.27, 89.66, 125.44, 128.59, 129.42, 136.49, 169.13. Analysis Calculated for C16H18O8: C, 56.80; H, 5.36; found: C, 56.51; H, 5.14.
Acknowledgements
We are thankful to Islamic Azad University Research Council for partial support of this work, Dr Mohammad Mahdi Doroodmand for helpful comments, and University of Manchester for running BET and XRD.
References
- Greene , T.W. ; Wuts , P.G.M. Greene's Protective Groups in Organic Synthesis , 4th ed. ; New York : Wiley , 2007 .
- Kochhar , K.S. ; Bal , B.S. ; Deshpande , R.P. ; Rajadhyaksha , S.N. ; Pinnick , H.W. J. Org. Chem . 1983 , 48 , 1765 – 1767 . doi: 10.1021/jo00158a036
- Trost , B.M. ; Lee , C. J. Am. Chem. Soc . 2001 , 123 , 12191 – 12201 . doi: 10.1021/ja0118338
- Sandberg , M. ; Sydnes , L.K. Org. Lett . 2000 , 2 , 687 – 689 . doi: 10.1021/ol005535b
- Held , H. ; Rengstle , A. ; Mayer , A. Ullman's Encyclopedia of Industrial Chemistry , 5th ed. ; Gerhartz W. ; A1 , Wiley-VCH : New York , 1985 ; 68 .
- Freeman , F. ; Karcherski , E.M. J. Chem. Eng. Data 1977 , 22 , 355 – 357 . doi: 10.1021/je60074a038
- Frick ., J.G. Jr ; Harper ., R.J. Jr J. Appl. Polym. Sci . 1984 , 29 , 1433 – 1447 . doi: 10.1002/app.1984.070290436
- Van Heerden , F.R. ; Huyser , J.J. ; Bradley , D. ; Williams , G. ; Holzapfel , C.W. Tetrahedron Lett . 1998 , 39 , 5281 – 5284 . doi: 10.1016/S0040-4039(98)01000-4
- Sandberg , M. ; Sydnes , L.K. Tetrahedron Lett . 1998 , 39 , 6361 . doi: 10.1016/S0040-4039(98)01309-4
- Tomita , M. ; Kikuchi , T. ; Bessho , K. ; Hori , T. ; Inubushi , Y. Chem. Pharm. Bull . 1963 , 11 , 1484 – 1490 . doi: 10.1248/cpb.11.1484
- Davey , W. ; Gwilt , J.R. J. Chem. Soc . 1957 , 1008 – 1014 . doi: 10.1039/jr9570001008
- Freeman , F. ; Karchefski , E.M. J. Chem. Eng. Data 1977 , 22 , 355 – 357 . doi: 10.1021/je60074a038
- Marshall , J.A. ; Wuts , P.G.M. J. Org. Chem . 1977 , 42 , 1794 – 1798 . doi: 10.1021/jo00430a027
- Michie , J.K. ; Miller , J.A. Synthesis 1981 , 10 , 824 . doi: 10.1055/s-1981-29613
- Yadav , J.S. ; Reddy , B.V.S. ; Srinivas , C. Synth. Commun . 2002 , 32 , 2169 – 2174 .
- Smitha , G. ; Reddy , C.S. Tetrahedron 2003 , 59 , 9571 – 9576 . doi: 10.1016/j.tet.2003.10.002
- Karimi , B. ; Ebrahiminan , G.R. ; Seradj , H. Synth. Commun . 2002 , 32 , 669 – 673 . doi: 10.1081/SCC-120002503
- Yadav , J.S. ; Reddy , B.V.S. ; Venugopal , C. ; Ramalingam , T. Synlett 2002 , 4 , 604 – 606 . doi: 10.1055/s-2002-22705
- Chakraborti , A.K. ; Thilagavathi , R. ; Kumar , R. Synthesis 2004 , 6 , 831 – 833 . doi: 10.1055/s-2004-816000
- Roy , S.C. ; Banerjee , B. Synlett 2002 , 10 , 1677 – 1678 . doi: 10.1055/s-2002-34243
- Ranu , B.C. ; Dutta , J. Chem. Lett . 2003 , 32 , 366 – 367 . doi: 10.1246/cl.2003.366
- Romanelli , G.P. ; Thomas , H.J. ; Baronetti , G.T. ; Autino , J.C. Tetrahedron Lett . 2003 , 44 , 1301 – 1303 . doi: 10.1016/S0040-4039(02)02817-4
- Firouzabadi , H. ; Iranpoor , N. ; Nowrouzi , F. ; Amani , K. Tetrahedron Lett . 2003 , 44 , 3951 – 3954 . doi: 10.1016/S0040-4039(03)00811-6
- Chandra , K.L. ; Saravanan , P. ; Singh , V.K. Synlett 2000 , 3 , 359 – 360 .
- Aggarwal , V.K. ; Fonquerna , S. ; Vennall , G.P. Synlett 1998 , 8 , 849 – 850 . doi: 10.1055/s-1998-1799
- Karimi , B. ; Maleki , J. J. Org. Chem . 2003 , 68 , 4951 – 4954 . doi: 10.1021/jo026678+
- Olah , G.A. ; Mehrotra , A.K. Synthesis 1982 , 11 , 962 – 963 . doi: 10.1055/s-1982-30021
- Jin , T.-S. ; Du , G.-Y. ; Zhang , Z.-H. ; Li , T.-S. Synth. Commun . 1997 , 27 , 2261 – 2266 . doi: 10.1080/00397919708003380
- Ballini , R. ; Bordoni , M. ; Bosica , G. ; Maggi , R. ; Sartori , G. Tetrahedron Lett . 1998 , 39 , 7587 – 7590 . doi: 10.1016/S0040-4039(98)01649-9
- Li , T.-S. ; Zhang , Z.-H. ; Gao , Y.-J. Synth. Commun . 1998 , 28 , 4665 – 4671 . doi: 10.1080/00397919808004531
- Nagy , N.M. ; Jakab , M.A. ; Konya , J. ; Antus , S. Appl. Clay Sci . 2002 , 21 , 213 – 216 . doi: 10.1016/S0169-1317(02)00066-2
- Li , Y.-Q. ; Cheng , L.-H. Chin. Chem. Lett . 2001 , 12 , 565 – 568 .
- Li , Y.-Q. Synth. Commun . 2000 , 30 , 3913 – 3917 . doi: 10.1080/00397910008086948
- Jin , T.-S. ; Ma , Y.-R. ; Sun , X. ; Liang , D. ; Li , T.-S. J. Chem. Res., Synop . 2000 , 2 , 96 – 97 . doi: 10.3184/030823400103166526
- Jin , T.-S. ; Feng , G.-L. ; Yang , M.-N. ; Li , T.-S. Synth. Commun . 2004 , 34 , 1645 – 1651 . doi: 10.1081/SCC-120030752
- Deka , N. ; Kalita , D.J. ; Borah , R. ; Sarma , J.C. J. Org. Chem . 1997 , 62 , 1563 – 1564 . doi: 10.1021/jo961741e
- Karimi , B. ; Seradj , H. ; Ebrahimian , G.R. Synlett 2000 , 5 , 623 – 624 .
- Deka , N. ; Borah , R. ; Kalita , D.J. ; Sarma , J.C. J. Chem. Res. Synop . 1998 , 2 , 94 – 95 . doi: 10.1039/a703477f
- Meshram , G.A. ; Patil , V.D. Synth. Commun . 2010 , 40 , 442 – 449 . doi: 10.1080/00397910902985499
- Wang , Y.Y. ; Xu , Y.N. ; Wang , Z.Z. ; Dai , L.Y. Chin. Chem. Lett . 2010 , 21 , 524 – 528 . doi: 10.1016/j.cclet.2009.12.014
- Wang , M. ; Tian , G.F. ; Song , Z.G. ; Jiang , H. Chin. Chem. Lett . 2009 , 20 , 1034 – 1038 . doi: 10.1016/j.cclet.2009.05.008
- Ghorbani-Vaghei , R. ; Amiri , M. ; Moshfeghifar , N. ; Veisi , H. ; Dadamahaleh , S.A. J. Iran. Chem. Soc . 2009 , 6 , 754 – 760 . doi: 10.1007/BF03246166
- Gao , S.T. ; Zhao , Y. ; Li , C. ; Ma , J.J. ; Wang , C. Synth. Commun . 2009 , 39 , 2221 – 2229 . doi: 10.1080/00397910802654674
- Xu , R. ; Zhang , J. ; Tian , Y. ; Zhou , J. J. Iran Chem. Soc . 2009 , 6 , 443 – 447 . doi: 10.1007/BF03245855
- Niknam , K. ; Saberi , D. ; Nouri Sefat ., M. Tetrahedron Lett . 2009 , 50 , 4058 – 4062 . doi: 10.1016/j.tetlet.2009.04.096
- Khan , A.T. ; Choudhury , L.H. ; Ghosh , S. J. Mol. Catal. A, Chem . 2006 , 255 , 230 – 235 . doi: 10.1016/j.molcata.2006.04.008
- Wang , Q.-Y. ; Sheng , S.-R. ; Wei , M.-H. ; Xie , Z.-L. ; Liu , X.-L. Synth. Commun . 2007 , 37 , 1019 – 1026 . doi: 10.1080/00397910601168264
- Wang , M. ; Song , Z. ; Gong , H. ; Jiang , H. Synth. Commun . 2008 , 38 , 961 – 966 . doi: 10.1080/00397910701845720
- Saini , A. ; Kumar , S. ; Sandhu , J.S. Synth. Commun . 2008 , 38 , 106 – 113 . doi: 10.1080/00397910701650831
- Jermy , B.R. ; Pandurangan , A. Catal. Commun . 2008 , 9 , 577 – 583 . doi: 10.1016/j.catcom.2007.02.016
- Niknam , K. ; Saberi , D. ; Appl. Catal. A: Gen . 2009 , 366 , 220 – 225 . doi: 10.1016/j.apcata.2009.07.014
- Niknam , K. ; Saberi , D. Tetrahedron Lett . 2009 , 50 , 5210 – 5214 . doi: 10.1016/j.tetlet.2009.06.140
- Niknam , K. ; Saberi , D. ; Sadegheyan , M. ; Deris , A. Tetrahedron Lett . 2010 , 51 , 962 – 964 .
- Niknam , K. ; Saberi , D. ; Baghernejad , M. Phosphorus Sulfur Silicon 2010 , 185 , 875 – 882 . doi: 10.1080/10426500903019127
- Niknam , K. ; Panahi , F. ; Saberi , D. ; Mohagheghnejad , M. J. Heterocycl. Chem . 2010 , 47 , 292 – 300 .