Abstract
An efficient, general, and atom-economic procedure for the synthesis of 2-amino-6 methyl-4-aryl-8-[(E)-arylmethylidene]-5, 6, 7, 8-4H-pyrano [3, 2-c]pyridine-3-carbonitriles has been developed by reaction of 3,5-bis[(E)-arylmethylidene]-tetrahydro-4(1H)-pyridinones with malononitrile. The reaction proceeds under green media (EtOH/H2O, 1:1) and in the presence of diammonium hydrogen phosphate (10%) or piperidine (10%) to afford pyranopyridines at room temperature with good high yields.
Introduction
The development of efficient and mild methods for the synthesis of heterocyclic compounds represents a broad area of organic synthesis Citation1. Fused heterocyclic compounds are an important class of compounds in medicinal chemistry Citation2. Recently pyrano [3, 2-c]pyridone derivatives have received considerable attention due to their biological activities. The pyrano [3,2-c]pyridone scaffold is broadly represented by pyranopyridone alkaloids manifesting diverse biological activities Citation3. Some of the compounds containing these structures exhibit cancer cell growth inhibition and have a high potential as anti-cancer drug candidates. It was shown that these compounds have also antiproliferative and antitubulin activities Citation4. Some of the tetrahydro-4H-pyrano [3,2-c]pyridine derivatives have shown the inhibitory effect on Mycobacterium tuberculosis H37Rv (MTB) Citation5 growth, in in-vitro culture and could be useful for the treatment of tuberculosis Citation5 Citation6. There has been considerable interest in the development of preparative methods for the synthesis of pyrano [3,2-c]pyridone derivatives. One of the starting materials for the synthesis of tetrahydro-4H-pyrano [3,2-c]pyridine derivatives is bis-(arylidene)-4-piperidones. It has been reported that these compounds show cytotoxic activities Citation7. They have also been used as starting materials for the synthesis of many biologically active and heterocyclic compounds Citation8–11. For example, N-alkyl-4-piperidinones are used as efficient starting material for the synthesis of fentanyl which is considered the safest opioid medication on the market Citation12.
There are some reports describing synthesis of these compounds which include using a strong base such as sodium ethoxide Citation5, refluxing in n-butanol Citation6, and use of MW irradiation in DMF at 140°C Citation13 Citation14. Some of the reported methods have side effects such as long reaction time, harsh reaction conditions, and inconsistent yields. Therefore, the development of new synthetic strategies and methodologies using mild reaction conditions is very attractive. In addition, reduction or elimination of organic solvents or using solvents that have little to no detrimental effects on the environment is highly desirable Citation15–20. Thus, organic reactions in water Citation21–31 have drawn the attention of researchers due to their unique characteristics like nonflammability, high dielectric constant, high boiling point, hydrogen bond donor/acceptor properties and above all economic benefits.
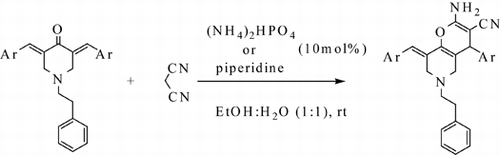
In continuation to our previous efforts in the synthesis of the pyran skeleton Citation32–36, herein we wish to report an efficient method for the synthesis of some tetrahydro-4H-pyrano [3,2-c]pyridine derivatives via reaction of (E)-3,5 bis (arylidene)-4-piperidinones with malononitrile in the presence of diammonium hydrogen phosphate (DAHP, 10%) or piperidine (10%) in aqueous media at room temperature ().
Scheme 1. Synthesis of 2-amino-4-aryl-8[(E)-arylmethylidene]-5,6,7,8,-4H pyrano[3,2-c] pyridine 3a-f in green media.
![Scheme 1. Synthesis of 2-amino-4-aryl-8[(E)-arylmethylidene]-5,6,7,8,-4H pyrano[3,2-c] pyridine 3a-f in green media.](/cms/asset/e1acfa01-311f-4ff0-802f-fec20637f1e6/tgcl_a_709280_o_sch0001g.gif)
Results and discussions
The designed target compounds depicted in were obtained by reaction of the starting material (1-phenylethyl)-4-piperidinone with variety of aromatic or heteroaromatic aldehydes under aldol condensation reaction in basic conditions to produce the (E)-3,5 bis (arylidene)-4-piperidinones. Reaction of desired (E)-3,5 bis (arylidene)-4-piperidinones with malononitrile in the presence of either DAHP (a neutral catalyst) Citation37 Citation38 in catalytic amount (10%) or also (10%) piperidine at room temperature afforded solely tetrahydro-4H-pyrano [3,2-c]pyridines in high yields. Neither crystallization nor column chromatographic purification was necessary. The results are summarized in . To test the generality and versatility of this procedure, we examined a number of different substituted arylaldehydes using the optimized experimental conditions. As shown in , the aromatic aldehydes, having different substituents such as halogen, nitro group and also a heteroaryl aldehyde such as thiophene carbaldehyde were converted to the corresponding pyrano [3,2-c]pyridine skeletons in high yields.
Table 1. Synthesis of tetrahydro-4H-pyrano [3,2-c]pyridines in aqueous media
Diammonium hydrogen phosphate as a neutral catalyst is inexpensive, water soluble, nontoxic, and commercially available and can be used in the laboratory without special precautions Citation37 Citation38, and it was used for the synthesis of different organic compounds Citation39–44. Reaction of (E)-3,5 bis (phenylmethylidene)-4-piperidinone(1a) with malononitrile was selected as a model reaction, control experiments without catalytic amount (10%) of DAHP or piperidine under the same reaction conditions showed that no reaction was occurred at all. The amount of these basic catalysts was checked using 5, 10, and 20% of DAHP or piperidine. The best yields were obtained in the presence of (10%) of piperidine or DAHP, 87, and 85%, respectively. A decrease in the amount of catalyst (5%) resulted in a significant yield reductions (54% and 52%) and also long reaction time (6 h), while an increased amount of catalyst revealed negligible effect on the efficiency of the reaction.
The proposed mechanism for the synthesis of pyrano [3,2-c]pyridine derivatives may tentatively be visualized to occur via a tandem sequence of reactions. First the deprotonated form of malononitrile could add to the α,β unsaturated carbonyl arylidene piperidinones. Finally cyclization and tautomerization led to the desired products. This reaction could be categorized as sequential Michael/cyclization reaction.
From an environmental point of view, it is desirable to minimize the amount of waste for each organic transformation. Reactions were done in aqueous media as a green solvent and also the obtained products were pure and did not require further purification.
The structure of the products was elucidated using NMR spectroscopic data and ESI Mass spectrometry data. The 1H-NMR spectrum for 4a has characteristic peaks for the pyran ring (H-4) at δ 4.04 ppm, and the benzylidene proton at δ 6.91. The four piperidine protons with geminal coupling (J=15 Hz) produced four different doublet peaks between 2.71 and 3.62 ppm. In the 13C-NMR spectra the carbon signals at 57.5, 120.5, and 121.6 were assigned to C-3, C-9, and the nitrile group, respectively. The ESI mass spectrometry data confirmed the molecular weight and also the high purity. The structures of all the synthetic starting materials are in accord with their analytical and NMR spectroscopic data.
In conclusion, we demonstrated an efficient, atom-economic synthesis of tetrahydro-4H-pyrano [3,2-c]pyridines with catalytic high bond-forming efficiency (BFE) good to high yields and high purity in aqueous media at room temperature. This new and efficient catalytic method is amenable to a parallel-synthesis approach to achieve a library of pyrano [3,2-c]pyridines. The investigation for finding the biological activity of the tetrahydro-4H-pyrano [3,2-c]pyridines is in progress.
Experimental
Commercially available materials were used without further purification. Melting points were determined on an Electrothermal 9100 apparatus and were uncorrected. IR spectra were obtained on an ABB FT-IR (FTLA 2000) spectrometer. 1H NMR and 13C NMR spectra were run on Bruker DRX-300 at 300 MHz for 1H-NMR, and 75 MHz for 13C-NMR. CDCl3 and DMSO-d6 were used as solvents. Mass spectra were recorded on Mass- ESI-POS (Apex Qe-FT- ICR instrument).
General procedure for the synthesis of (E)-3,5 bis (arylmethylidene)-4-piperidinones (1a–f)
A mixture of 406 mg (2 mmol) 1-phenylethyl-4-piperidinone, aromatic aldehyde (4 mmol). and NaOH (0.4 g, 1 mmol) was dissolved in 1:1 mixture of water-ethanol (25 mL). The mixture was stirred for 30 min. The yellow precipitate was separated. Further purification was done using crystallization in ethanol. Yields were between 85% and 95%.
General procedure for the synthesis of 2-amino-4-aryl-8-[(E)-arylmethylidene]-5,6,7,8-4H-pyrano [3,2-c]pyridine-3- carbonitriles (3a–f)
A mixture of 750 mg (2 mmol) 3,5-dibenzylidene-1-phenylethyl-4-piperidinone, 0.135 g (2 mmol) malononitrile and DAHP (0.026 g, 10%) or piperidine (0.017 mg, 10%)was combined with a solution of ethanol: water (1:1) (25 mL). The mixture was stirred for 4 h at room temperature. The precipitate was filtered. Yields were between 78% and 96%.
Selected data for compounds 3a–f;
2-amino-8-benzylidene-5,6,7,8-tetrahydro-6-phenethyl-4-phenyl-4H-pyrano-[3,2,c]pyridine-3-carbonitrile (3a)
Yield: 87%, melting point = 193.6–195.6°C; IR (KBr, cm−1): 3436, 3348, 2182, 1683, 1638, 1616, 1591; 1H-NMR (300 MHz, DMSO-d6) δ 2.49 (s, 4H, 2CH2), 2.71 (d, 1H, J = 15 Hz, –CH), 3.15 (d, 1H, J = 15 Hz, –CH), 3.39 (d, 1H, J = 15 Hz, –CH), 3.62 (d,1H, J = 15 Hz, –CH), 4.04 (s, 1H, H-4), 6.84 (brs, 2H, –NH2), 6.91 (brs, 1H, H–C = ), 6.98–7.01 (m, 2H, H–Ar), 7.07–7.19 (m, 3H, H–Ar), 7.21–7.29 (m, 6H, H–Ar), 7.34–7.41 (m, 4H, H–Ar); 13C-NMR (75 MHz, DMSO-d6): δ 32.8, 41.1, 51.9, 52.2, 55.8, 57.5, 112.9, 120.5, 121.6, 125.7, 127.1, 127.2, 127.6, 128.1, 128.5, 128.9, 135.9, 139.2, 139.8, 143.5, 159.8; ESI-Mass: C30H28N3O [M + H]+ found 446.22276, calc. 446.22269; C60H55N6O2 [2M + H]+ found 891.43839, calc. 891.43810.
2-amino-5,6,7,8-tetrahydro-6-phenethyl-4-thiophen-2-yl-8-((thiophen-2-yl)methylene)-4H-pyrano-[3,2-c]pyridine-3-carbonitrile (3b)
Yield: 86%, melting point = 197.6–200°C; IR (KBr, cm−1): 3428, 3322, 2196, 1682, 1644, 1596; 1H-NMR (300 MHz, DMSO-d6): δ 2.62 (s, 4H, 2CH2), 2.92 (d, 1H, J = 16 Hz, –CH), 3.21 (d, 1H, J = 16 Hz, –CH), 3.47 (d, 1H, J = 14.7 Hz, –CH), 3.74 (d, 1H, J = 14.7 Hz, –CH), 4.43 (s, 1H, H-4), 6.91 (brs, 2H, –NH2), 6.95–6.99 (m, 2H, H–Ar), 7.02 (brs, 1H, H-C = ), 7.07–7.15 (m, 5H, H–Ar, H-Thienyl), 7.19–7.24 (m, 2H, H–Ar, H-Thienyl), 7.44 (dd, 1 H, J = 4.6, 1.3 Hz, H-Thienyl), 7.61 (dd, 1H, J = 4.6, 1.3 Hz, H-Thienyl); 13C-NMR (75 MHz, DMSO-d6): δ 33.1, 36.2, 51.6, 52.3, 56.2, 57.5, 112.6, 114.4, 120.2, 124.8, 125.0, 125.5, 125.8, 126.8, 127.4, 127.9, 128.1, 128.5, 128.8, 138.7, 138.9, 139.9, 148.5, 159.6; ESI-Mass: C26H24N3OS2 [M + H]+ found 458.13559, calc:458.13553; C52H47N6O2S4 [2M + H]+ found 915.26460, calc. 915.26378.
8-(4-chlorobenzylidene)-2-amino-4-(4-chlorophenyl)-5,6,7,8-tetrahydro-6-phenethyl-4H-pyrano-[3,2-c]pyridine-3-carbonitrile (3c)
Yield: 85%, melting point = 187.6–190.1°C; IR (KBr, cm−1) 3420, 3369, 2183, 1691, 1641, 1607; 1H-NMR (300 MHz, DMSO-d6) δ 2.49 (s, 4H, 2CH2), 2.70 (d, 1H, J = 16.0 Hz, –CH), 3.15 (d, 1H, J = 16.0 Hz, –CH), 3.36 (d, 1H, J = 14.5 Hz, –CH), 3.59 (d, 1H, J = 14.5 Hz, –CH), 4.34 (s, 1H, H-4), 6.87 (brs, 1H, H–C = ), 6.90 (brs, 2H, NH2), 6.98–7.18 (m, 5H, H–Ar), 7.24 (d, 4H, J = 9.0 Hz, H–Ar), 7.93 (d, 4H, J = 9.0 Hz, H–Ar); 13C-NMR (75 MHz, DMSO-d6) δ 32.5, 40.4, 51.7, 52.1, 55.1, 56.0, 113.0, 120.3, 120.5, 125.7, 127.9, 128.0, 128.5, 128.7, 129.5, 130.7, 131.4, 134.7,139.2, 139.8, 142.5, 159.8; ESI-Mass: C30H25Cl2N3O [M + H]+ found 514.14477, calc. 514.14474, C60H51Cl4N6O2 [2M + H]+ found 1027.28221, calc. 1027.28272.
8-(4-(dimethylamino)benzylidene)-2-amino-4-(4-(dimethylamino)phenyl)-5,6,7,8-tetrahydro-6-phenethyl-4H-pyrano [3,2-c]pyridine-3-carbonitrile (3d)
Yield: 79%, melting point=(207.8–208.7)°C; IR (KBr, cm−1): 3407, 3324, 2186, 1670, 1634, 1600. 1H-NMR (300 MHz, DMSO-d6, ppm): δ 2.50(s, 4H, 2CH2), 2.72 (d, 1H, J = 16.0 Hz, –CH), 2.87 (s, 6H, -NMe2), 3.09 (d, 1H, J = 16.0 Hz, –CH), 3.38 (d, 1H, J = 14.3 Hz, –CH), 3.62 (d, 1H, J = 14.3 Hz, –CH), 3.85 (brs, 1H, H-4), 6.68 (brs, 2H, –NH2), 6.70 (d, 2H, J = 9.0 Hz, H–Ar), 6.71 (d, 2H, J = 9.0 Hz, H–Ar), 6.76 (s, 1H, H–C = ), 6.99–7.20 (m, 9H, H–Ar); 13C-NMR (75 MHz, DMSO-d6, ppm): δ 32.9, 40.2, 51.9, 52.7, 56.4, 57.7, 111.4, 111.9, 112.5, 120.7,121.5, 123.4, 123.6, 125.7, 128.0, 128.1, 128.5, 130.1, 131.2, 139.1, 140.0, 149.2, 149.5, 159.6; ESI-Mass: C34H38N5O [M + H]+ found 532.30721, calc. 532.30709; C68H75N10O2 [2M + H]+ found 1063.60745, calc. 1063.60690.
8-(3-nitrobenzylidene)-2-amino-5,6,7,8-tetrahydro-4-(3-nitrophenyl)-6-phenethyl-4H-pyrano [3,2-c]pyridine-3-carbonitrile (3e)
Yield: 95%, melting point=(190.9–192.6)°C; IR (KBr,cm−1): 3456, 3340, 2192, 1678, 1639, 1602, 1530, 1347. 1H-NMR (300MHz, DMSO-d6, ppm): δ 2.52–2.58 (m, 4H, 2CH2), 2.73 (d, 1H, J = 16.2 Hz, –CH), 3.24 (d, 1H, J = 16.2 Hz, –CH), 3.44 (d, 1H, J = 14.4 Hz, –CH), 3.64 (d, 1H, J = 14.4 Hz, CH) 4.38 (s, 1H, H-4), 6.96–7.14 (m, 8H, H–Ar, NH2, H-C = ), 7.64–7.75 (m, 4H, H–Ar), 8.02–8.19 (m, 4H, H–Ar); 13C-NMR (75 MHz, DMSO-d6,ppm): δ 32.9, 40.4, 51.5, 51.9, 54.9, 57.2, 113.5, 120.0, 120.1, 121.8, 122.0, 122.4, 123.4, 125.7, 128.0, 128.4, 129.4, 130.1, 130.5, 134.5, 135.2, 137.4, 139.5, 139.9, 145.7, 147.9, 148.0, 160.0; ESI-Mass: C30H26N5O5 [M + H]+ found 536.19293, calc. 536.19285; C60H51N10O10: [2M + H]+ found 1071.37861, calc.1071.37841.
8-(4-nitrobenzylidene)-2-amino-5,6,7,8-tetrahydro-4-(4-nitrophenyl)-6-phenethyl-4H-pyrano [3,2-c]pyridine-3-carbonitrile (3f)
Yield: 96%, melting point=(187.6–190.1)°C; IR (KBr, cm−1): 3430, 3370, 2184, 1680, 1638, 1592, 1443, 1517, 1343; 1H-NMR (300 MHz, DMSO-d6, ppm): δ 2.58 (s, 4H, 2CH2), 2.72 (d, 1H, J = 16.3 Hz, –CH), 3.23 (d, 1H, J = 16.3 Hz, –CH), 3.44 (d, 1H, J = 14.5 Hz, –CH), 3.64 (d, 1H, J = 14.5 Hz, –CH), 4.34 (s, 1H, H-4), 6.98–7.15 (m, 8H, H–Ar, NH2, H–C = ), 7.51 (t, 4H, J = 8.0 Hz, H–Ar), 8.23 (t, 4H, J = 8.0 Hz, H–Ar); 13C-NMR (75 MHz, DMSO-d6, ppm):δ, 32.8, 40.7, 51.7, 52.1, 54.7, 56.0, 57.3, 114.1, 120.0, 120.2, 123.7, 124.1, 125.8, 128.0, 128.4, 129.0, 130.1, 130.5, 139.5, 139.8, 142.7, 145.9, 150.9, 159.9; ESI-Mass: C30H26N5O5 [M + H]+ found 536.19294, calc. 536.19285; C60H51N10O10: [2M + H]+ found 1071.37830, calc.1071.37841.
Acknowledgements
Saeed Balalaie is thankful to Alexander von Humboldt Foundation for research fellowship. We are also grateful to Prof. R. Gleiter for his invaluable comments. We also thank Kimia Exir Company for financial support and donation of chemicals and Tofigh Daru Research & Engineering Co. for kind cooperation.
References
- Wnuk , S.F. ; Lewandowska , E. ; Companioni , D.R. ; Garcia , P.I. Jr. ; Secrist , J.A. Org. Biomol. Chem . 2004 , 2 , 120 – 126 .
- Cushman , M. ; Sambaiah , T. ; Jin , G. ; Illarionov , B. ; Fischer , M. ; Bacher , A. J. Org. Chem . 2004 , 69 , 601 – 612 .
- McBrien , K.D. ; Gao , Q. ; Huang , S. ; Klohr , S.E. ; Wang , R.R. ; Pirnik , D.M. ; Neddermann , K.M. ; Bursuker , I. ; Kadow , K.F. ; Leet , J.E. J. Nat. Prod . 1996 , 59 , 1151 – 1153 .
- Magedov , I.V. ; Manpadi , M. ; Ogasawara , M.A. ; Dhawan , A.S. ; Rogelj , S. ; Van Slambrouck , S. ; Steelant , W.F.A ; Evdokimov , N.M. ; Uglinskii , P.Y. ; Elias , E.M. ; Knee , E.J. ; Tongwa , P. ; Antipin , M.Y. ; Kornienko , A. J. Med. Chem . 2008 , 51 , 2561 – 2570 and references cited therein .
- Kumar , R.R. ; Perumal , S. ; Senthilkumar , P. ; Yogeeswari , P. ; Sriram , D. Bioorg. Med. Chem. Lett . 2007 , 17 , 6459 – 6462 and references cited therein .
- El-Subbagh , H.I. ; Abu-Zaid , S.M. ; Mahran , M.A. ; Badria , F.A. ; Al-Obaid , A.M. J. Med. Chem . 2000 , 43 , 2915 – 2921 and references cited therein .
- Das , U. ; Alcorn , J. ; Shirvastav , A. ; Sharma , R.K. ; De Clercq , E. ; Balzarini , J. ; Dimmock , J. R. Eur. J. Med. Chem . 2007 , 41 , 71 – 80 and references cited therein .
- Kumar , R.R. ; Perumal , S. ; Kagan , H.B. ; Guillot , R. Tetrahedron . 2006 , 62 , 12380 – 12391 .
- Kumar , R.S. ; Perumal , S. ; Kagan , H.B. ; Guillot , R. Tetrahedron: Asymmetry 2007 , 18 , 170 – 180 .
- Srinivasan , M. ; Perumal , S. Tetrahedron 2007 , 63 , 2865 – 2874 .
- Khartulyari , A. ; Maier , M.E. Eur. J. Org. Chem . 2007 , 317 – 324 .
- Ivanovic , M.D. ; Miovic , I.V. ; Vuscovic , S. ; Todorovic , Z. ; Kirokovic , V.D. ; Djorjevic , J. ; Dosen-Micovic , B. J. Serb. Chem. Soc . 2004 , 69 , 511 – 526 .
- Wang , S.-L. ; Han , Z.-G. ; Tu , S.-J. ; Zhang , X.-H. ; Wen-Juan Hao , S.Y. ; Shi , F. ; Cao , X.-D. ; Wu , S.-S. J. Heterocycl. Chem . 2009 , 46 , 828 – 831 .
- Han , Z.-G. ; Tu , S.J. ; Jiang , B. ; Yan , S. ; Zhang , X.-G. ; Wu , S.S. ; Hao , W.-J. ; Cao , X.-D. ; Shi , F. ; Zhang , G. ; Ma , N. Synthesis 2009 , 1639 – 1646 .
- Grieco , P.A. Organic Synthesis in Water ; Blackie Academic & Professional : London , 1998 .
- Li , C.-J. ; Chan , T.H. Comprehensive Organic Reactions in Aqueous Media ; John Wiley & Sons, Inc. : Hoboken , NJ , 2007 .
- Li , C.-J. Chem. Rev . 2005 , 105 , 3095 – 3166 ; Li, C.-J. Chem. Rev. 1993, 93, 2023–2035 .
- Lindström , U.M. Chem. Rev . 2002 , 102 , 2751 – 2772 .
- Pirrung , M.C. Chem.–Eur. J . 2006 , 12 , 1312 – 1317 .
- Katkar , K.V. ; Chaudhari P.S. ; Akamanchi , K.G. Green Chem . 2011 , 13 , 835 – 838 .
- Chanda , A. ; Fokin , V.V. Chem. Rev . 2009 , 109 , 725 – 748 .
- Butler , R.N. ; Coyne , A.G. Chem. Rev . 2010 , 110 , 6302 – 6337 .
- Klijn , J.E. ; Engberts , J.B.F.N. Nature 2005 , 435 , 746 – 747 .
- Hayashi , Y. Angew. Chem. Int. Ed . 2006 , 45 , 8103 – 8104 .
- Brogan , A.P. ; Dickerson , T.J. ; Janda , K.D. Angew. Chem. Int. Ed . 2006 , 45 , 8100 – 8102 .
- Chakraborti , A.K. ; Roy , S.R. J. Am. Chem. Soc . 2009 , 131 , 6902 – 6903 .
- Chakraborti , A.K. ; Rudrawar , S. ; Jadhav , K.B. ; Kaur , G. ; Chankeshwara , S.V. Green Chem . 2007 , 9 , 1335 – 1340 .
- Sharma , G. ; Kumar , R. ; Chakraborti , A.K. Tetrahedron Lett . 2008 , 49 , 4269 – 4271 .
- Khatik , G.L. ; Kumar , R. ; Chakraborti , A.K. Org. Lett . 2006 , 8 , 2433 – 2436 .
- Arya , A.K. ; Kumar , M. Green Chem . 2011 , 13 , 1332 – 1338 .
- Chouhan , M. ; Senwar , K.R. ; Sharma , R. ; Grover , V. ; Nair , V.A. Green Chem . 2011 , 13 , 2553 – 2560 , doi: 10.1039/c1gc15416h
- Khoshkholgh , M.J. ; Balalaie , S. ; Gleiter , R. ; Rominger , F. Tetrahedron 2008 , 64 , 10924 – 10929 .
- Khoshkholgh , M.J. ; Balalaie , S. ; Bijanzadeh , H.R. ; Rominger , F. ; Gross , J.H. Tetrahedron Lett . 2008 , 49 , 6965 – 6968 .
- Khoshkholgh , M.J. ; Lotfi , M. ; Balalaie , S. ; Rominger , F. Tetrahedron 2009 , 65 , 4228 – 4234 .
- Bararjanian , M. ; Balalaie , S. ; Rominger , F. ; Movassagh , B. ; Bijanzadeh , H.R. J. Org. Chem . 2010 , 75 , 2806 – 2812 .
- Bararjanian , M. ; Balalaie , S. ; Movassagh , B. ; Bijanzadeh , H.R. Tetrahedron Lett . 2010 , 51 , 3277 – 3300 .
- Lewis , R.J. Hawley's Condensed Chemical Dictionary , 13th ed. ; Von Nostrand Reinhold : New York , 1997 .
- Merck Catalogue of Chemical Reagents ; 2007 , Cat No 101206 .
- Salehi , P. ; Dabiri , M. ; Khosropour , A.R. ; Roozbehniya , P. J. Iranian. Chem. Soc . 2006 , 3 , 98 – 104 .
- Balalaie , S. ; Bararjanian , M. ; Hekmat , S. ; Salehi , P. Synth. Commun . 2006 , 36 , 2549 – 2557 .
- Bararjanian , M. ; Sheikh-Ahmadi , M. ; Hekmat , S. ; Salehi , P. Synth. Commun . 2007 , 37 , 1097 – 1108 .
- Darviche , F. ; Balalaie , S. ; Chadegani , F. Synth. Commun . 2007 , 37 , 1059 – 1067 .
- Abdolmohammadi , S. ; Balalaie , S. Tetrahedron Lett . 2007 , 48 , 3299 – 3303 .
- Balalaie , S. ; Abdolmohammadi , S. ; Bijanzadeh , H.R. ; Amani , A.M. Mol. Div . 2008 , 12 , 85 – 91 .