Abstract
Chemiluminescent acridinium dimethylphenyl esters containing hydrophilic N-sulfopropyl groups in the acridinium ring are used as labels in automated immunoassays for clinical diagnostics. Introduction of the N-sulfopropyl group in these labels is normally accomplished by N-alkylation of the corresponding, nonchemiluminescent acridine ester precursors with the toxic carcinogen 1,3-propane sultone. In the current study, we report that sodium 3-bromopropane sulfonate in ionic liquids (ILs) is a benign alternative to 1,3-propane sultone for introducing the N-sulfopropyl group in chemiluminescent acridinium ester labels. The sultone reagent can be eliminated in the synthesis of N-sulfopropyl acridinium dimethylphenyl ester labels by taking advantage of the increased reactivity of acridan esters toward nontoxic sodium 3-bromopropane sulfonate in [BMIM][BF4]. Sodium 3-bromopropane sulfonate in ILs is also potentially a nontoxic alternative to 1,3-propane sultone for introducing the water-soluble, three-carbon sulfobetaine moiety in other molecules as well.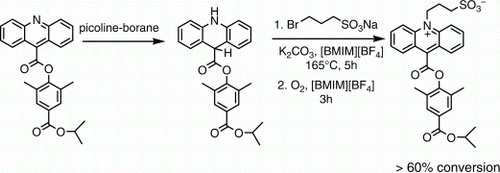
Introduction
Chemiluminescent acridinium dimethylphenyl esters () containing hydrophilic N-sulfopropyl groups in the acridinium ring are highly sensitive labels that are used in automated immunoassays for the measurement of a wide range of clinically important analytes Citation1–5. These acridinium esters are typically synthesized by N-alkylation of the nonchemiluminescent, acridine ester precursors with the toxic reagent 1,3-propane sultone Citation6–8 (). The acridine nitrogen in these esters is very unreactive toward alkylating reagents because of steric hindrance from the hydrogens at C-4 and C-5 (see for numbering system). These hydrogens are oriented in the same plane as the lone pair of electrons of the acridine nitrogen owing to the flat nature of the acridine ring (). As a consequence, harsh reaction conditions are usually employed to install the N-sulfopropyl group at the acridine nitrogen Citation1. Typically, this entails using 1,3-propane sultone as both the reactant and solvent in neat reactions and at high temperatures where sultone decomposition competes with N-alkylation of the acridine ester substrate Citation1. The formation of polysulfonated acridinium ester by-products also complicates chromatographic purification and lowers the yield of the N-sulfopropyl acridinium dimethylphenyl ester product Citation1.
Figure 1. Structures of chemiluminescent acridinium dimethylphenyl ester labels containing N-sulfopropyl groups in the acridinium ring that are used in automated immunoassays in Siemens Healthcare Diagnostics' ADVIA:Centaur® systems Citation1–3.
Figure 2. N-Alkylation of acridine methyl ester 2a with 1,3-propane sultone Citation14. The reaction proceeds via a charged transition state and is facilitated by polar solvents such as the ILs [BMIM][PF6] and [BMIM[BF4].
![Figure 2. N-Alkylation of acridine methyl ester 2a with 1,3-propane sultone Citation14. The reaction proceeds via a charged transition state and is facilitated by polar solvents such as the ILs [BMIM][PF6] and [BMIM[BF4].](/cms/asset/a86fb901-4d20-48a6-975e-235aaa9c8375/tgcl_a_768708_o_f0002g.jpg)
Despite its toxicity, 1,3-propane sultone is widely used to introduce a three-carbon sulfobetaine in various dyes and polymers to increase their water solubility Citation9. We wanted to develop less toxic alternatives to this reagent for introducing the N-sulfopropyl group in chemiluminescent acridinium esters. The reaction of acridine esters with 1,3-propane sultone proceeds via ring opening of the sultone reagent through a charged transition state and results in the formation of a zwitterionic product (). Classical Hughes-Ingold theory Citation10–13 predicts that increasing the polarity of the reaction medium will lower the activation energy of the reaction. Consistent with this prediction, in a recent study Citation14, we reported that highly polar, room temperature ionic liquids (ILs) such as1-butyl-3-methylimidazolium hexafluorophosphate [BMIM][PF6] and tetrafluoroborate [BMIM][BF4] are excellent media for the N-alkylation of acridine esters with 1,3-propane sultone. N-alkylation of a range of acridine dimethylphenyl ester substrates proceeded cleanly in these ILs and in excellent conversion with reduced quantities of the sultone reagent. Moreover, the alkylation reactions in these polar solvents proceeded with minimal polysulfonation and were amenable to scale up for the synthesis of gram quantities of hydrophilic acridinium dimethylphenyl ester labels in high yields.
The increased reactivity of acridine esters toward 1,3-propane sultone in ILs is mostly attributable to the polar nature of these unique reaction media. The empirically determined Kamlet-Taft Citation15–19parameter π* which, is a good measure of a solvent's polarity/polarizability, is typically high for ILs such as [BMIM][PF6] and [BMIM][BF4]. For example, π* values for [BMIM][PF6] and [BMIM][BF4] are 1.032 and 1.047, respectively Citation20. For comparison, Kamlet-Taft parameter π* of water is 1.09 Citation19. In our earlier study Citation9, we noted excellent correlation between the parameter π* of the solvent and the extent of conversion to N-sulfopropyl acridinium ester product in the reaction of its acridine ester precursor with limited quantities of propane sultone. However, the alkylation reaction was not particularly sensitive to either the acidity of the cation or the basicity of the anion comprising the IL as reflected by the Kamlet-Taft parameters α and β, respectively.
As we noted in our study Citation14, while a direct comparison of the toxicological properties of propane sultone with [BMIM][PF6] and [BMIM][BF4] is not available, the former is a potent carcinogen Citation6–8 due to its strong alkylating nature, whereas the latter two ILs can reasonably be expected not to be similarly toxic. However, not all ILs are as benign as once thought, and they can be quite toxic to aquatic life when discharged into the environment Citation21–28. A number of toxicological studies have investigated the relationship between IL structure and toxicity Citation21–28. Toxicity of ILs is generally attributable to the lipophilicity of the cation Citation25. The two ILs derived from the 1-butyl-3-methylimidazolium [BMIM] cation contain relatively short hydrocarbon chains and have been shown to be significantly less toxic than longer chain analogs in a range of toxicological tests Citation25. The two ILs have also been used extensively as solvents in a variety of synthetic transformations due to their excellent properties such as the ability to dissolve a wide variety of organic solutes, low vapor pressure, high thermal stability, and recyclability. An excellent review of the applications of ILs in synthesis can be found in a two-volume book edited by Wasserscheid and Welton Citation29.
Despite the increased reactivity of acridine esters toward 1,3-propane sultone in [BMIM] ILs, our previous efforts to replace this reagent with less-reactive and less-toxic alkylating reagents for the synthesis of N-sulfopropyl acridinium esters were largely unsuccessful. In the current study, we report a successful and practical approach toward the synthesis of N-sulfopropyl acridinium esters in [BMIM][BF4] without using the sultone reagent. We outline the rationale of this new green synthetic approach as well as its scope for the synthesis of these chemiluminescent labels and other potential applications.
Results and discussion
Based on our previous study Citation14, a reasonable strategy to increase the reactivity of acridine ester substrates toward propane sultone or other alkylating reagents is to identify IL solvents that are more polar than the two [BMIM]-derived ILs. Increased acridine ester reactivity would then enable further curtailment in the usage of the sultone reagent or its replacement with a less-toxic reagent such as sodium 3-bromopropane sulfonate for the synthesis of chemiluminescent acridinium dimethylphenyl esters containing N-sulfopropyl groups.
In a study reported by Poole and coworkers Citation30, who examined the properties of a series of tetra-alkylammonium sulfonates and other salts, the two ionic salts ethylammonium nitrate (π* = 1.24) and sec-butylammonium thiocyanate (π* = 1.28) were among the most polar salts. Based on their Kamlet-Taft π* values, these two ionic salts are even more polar than the two [BMIM] ILs. However, these types of salts, including the alkylammonium sulfonates, described by Poole and coworkers are unsuitable for the N-alkylation of acridine esters because the ions comprising the ionic salt are not expected to be chemically inert to alkylating reagents especially at high temperatures. Additionally, as noted by Crowhurst and coworkers Citation20, there may be little room to manipulate the structure of a particular IL to increase its polarity because to begin with most ILs tend to be quite polar. Consequently, tuning the structure of an IL such as [BMIM][BF4] to make it an even more suitable reaction medium for the N-alkylation reaction of acridine dimethylphenyl esters appears to be quite difficult and is probably impractical. Moreover, any new IL would need a comprehensive evaluation of its toxicological properties in comparison to the two [BMIM]-based ILs, before it can be implemented in a green synthetic process.
As an alternative, we envisaged that perhaps improving the reactivity of the acridine dimethylphenyl ester substrates by chemical modification may enable the use of a significantly less-toxic alkylating reagent such as sodium 3-bromopropane sulfonate in reactions in [BMIM] ILs. Recognizing that the decreased reactivity of the acridine nitrogen was due to steric hindrance from the hydrogens at C-4 and C-5 in the relatively inflexible and flat acridine ring (), we reasoned that conversion of the acridine esters to the corresponding acridan esters (, compounds 3a and 3b) might improve their reactivity. In these acridan dimethylphenyl esters, the central ring is reduced and consequently has increased conformational flexibility to allow greater access of the ring nitrogen to less-reactive alkylating reagents such as sodium 3-bromopropane sulfonate. The transition state of the acridan N-alkylation reaction also involves build up of charge and is expected to be similarly responsive to the polarity of the reaction medium as the acridine alkylation reactions. Following the N-alkylation reaction, the resulting N-sulfopropyl acridan dimethylphenyl ester would have to be oxidized to the corresponding N-sulfopropyl acridinium ester ().
Figure 3. Synthetic scheme for the synthesis of acridan esters and their N-alkylation reactions with sodium 3-bromopropane sulfonate in [BMIM][BF4]. The acridan N-alkylation reaction also has a charged transition state and is facilitated in the IL [BMIM][BF4]. Reagents: (a) Thionyl chloride, ROH; (b) p-toluenesulfonyl chloride, pyridine; (c) picoline-borane, tetrahydrofuran, 10% HCl; (d) sodium 3-bromopropane sulfonate, potassium carbonate, [BMIM][BF4]; (e) air or oxygen; (f) 1-2 M HCl; (g) TSTU, diisopropylethylamine, DMF.
![Figure 3. Synthetic scheme for the synthesis of acridan esters and their N-alkylation reactions with sodium 3-bromopropane sulfonate in [BMIM][BF4]. The acridan N-alkylation reaction also has a charged transition state and is facilitated in the IL [BMIM][BF4]. Reagents: (a) Thionyl chloride, ROH; (b) p-toluenesulfonyl chloride, pyridine; (c) picoline-borane, tetrahydrofuran, 10% HCl; (d) sodium 3-bromopropane sulfonate, potassium carbonate, [BMIM][BF4]; (e) air or oxygen; (f) 1-2 M HCl; (g) TSTU, diisopropylethylamine, DMF.](/cms/asset/8ec543d0-6604-4d12-a832-74fbcbb45a2b/tgcl_a_768708_o_f0003g.jpg)
The synthesis of acridan dimethylphenyl esters from the corresponding acridine esters entails reduction of the central ring in these compounds. The acridine dimethylphenyl esters themselves were easily synthesized by esterification of commercially available acridine-9-carboxylic acid with methyl and isopropyl 4-hydroxy-3,5-dimethylbenzoates as illustrated in (steps a and b). In our initial experiments, we observed that the reduction of these acridine esters to the corresponding acridan esters was easily achieved using sodium cyanoborohydride in a mixture of hydrochloric acid (HCl) and tetrahydrofuran (, step c). The reduction reaction proceeded cleanly and very rapidly as evidenced by the bleaching of the yellow-colored solution of the protonated acridine ester almost instantaneously upon the addition of the reducing agent. The loss of color results from disruption of the acridinium ring chromophore Citation3 by hydride reaction at C-9 in a 1,4-addition reaction that is facilitated by the positive charge on the protonated acridine nitrogen. Because sodium cyanoborohydride is generally considered to be toxic due to potential for hydrogen cyanide formation, we investigated alternate methods of reduction including catalytic hydrogenation as well as other less-toxic reducing agents such as sodium tri(acetoxy) borohydride Citation31, sodium borohydride, and picoline-borane Citation32. Catalytic hydrogenation did not afford a clean reaction, whereas the other two hydride reagents proved ineffective. Sodium borohydride reduction of acridine esters in plain methanol failed to afford any product because of reduced reactivity at C-9 of the unprotonated acridine ester substrate. In acidic methanol, sodium borohydride decomposed rapidly precluding any opportunity for reaction. Sodium tri(acetoxy)borohydride Citation31 has been advocated as a less-toxic alternative to sodium cyanoborohydride, but this reagent failed to reduce the acridine ester to the acridan ester even in acidic media. The borane complex of 2-methylpyridine (picoline-borane) is a more recent, nontoxic alternative to sodium cyanoborohydride for reductive-amination reactions Citation32 and has been used for oligosaccharide labeling Citation33 Citation34. We were pleased to note that the reduction reaction of acridine dimethylphenyl esters with picoline-borane complex in acidic media proceeded cleanly to give the corresponding acridan esters (, step c). No reduction occurred in the absence of acid. The reduction reaction could be scaled up to gram quantities using only a slight excess (1.2 molar equivalents) of the picoline-borane complex. The acridan dimethylphenyl esters 3a and 3b were formed cleanly with no by-products and required no further purification.
In our preliminary small-scale experiments, we examined the N-alkylation of acridan esters (25 mg) in [BMIM][BF4] using the reagent sodium 3-bromopropane sulfonate (, step d, ). Sodium 3-bromopropane sulfonate is commercially available, and although its toxicological properties have not been completely evaluated, it is a high melting salt with a significantly less-reactive bromide moiety (compared to propane sultone). It has low vapor pressure and low lipophilicity and is not classified as a carcinogen. Unfortunately, the utility of this benign reagent (as an alternative to propane sultone) for introducing a three-carbon sulfobetaine moiety has been limited by both its lower reactivity and poor solubility in conventional organic solvents. However, we noticed that this reagent showed significantly improved solubility in [BMIM]-ionic lquids even when compared to other polar aprotic solvents such as sulfolane. Significantly better solubility was also observed in the more hydrophilic IL [BMIM][BF4] than [BMIM][PF6] at elevated temperatures and, therefore, the former IL was used for reaction optimization. Concentrations of the acridan ester substrate and the alkylating reagent were similar to what we used previously for the N-alkylation of acridine esters with 1,3-propane sultone in ILs Citation14. All reactions were also conducted under an inert atmosphere to prevent air oxidation of the acridan dimethylphenyl ester substrate back to the inert acridine ester. Reactions were analyzed by high performance liquid chromatography (HPLC) to estimate the extent of conversion to product.
Table 1. N-Alkylation of acridan dimethylphenyl esters with sodium 3-bromopropane sulfonate in [BMIM][BF4].
In the absence of any base, the N-alkylation of acridan ester 3a with 10 equivalents of sodium 3-bromopropane sulfonate in [BMIM][BF4] at elevated temperature (160–165°C) afforded relatively poor conversion (22%) to the oxidized N-sulfopropyl acridinium ester product 5a (entry 1, , ). However, inclusion of either the organic base 2,6-di-tert-butylpyridine or the simple inorganic base potassium carbonate to neutralize the HBr formed in the substitution reaction, improved conversion to approximately 50% (entries 2–4). Even though the alkylation reactions were conducted under an inert nitrogen atmosphere, oxidation of the acridan ester substrate was observed to compete with the N-alkylation reaction. In fact, only small quantities of the N-sulfopropyl acridan dimethylphenyl ester product 4awere observed in these small-scale alkylation reactions. Instead, the oxidized N-sulfopropyl acridinium product 5a and the oxidized acridine ester starting material 2a, were the major products. We cannot rule out the possibility that adventitious oxygen in these small-scale reactions may be responsible for the undesired but facile oxidation reaction of the starting material as well as the oxidation of the N-alkylated acridan ester. Conducting the reaction under an argon atmosphere gave similar conversion (entry 5) as did increasing the amount of alkylating agent (entry 6). Increasing the reaction temperature from 160–165°C to 190°C (entry 7) also afforded a similar level of conversion to the N-sulfopropyl acridinium ester.
In addition to the oxidation side reaction, we also observed partial methyl ester cleavage (20–30%) in the N-alkylation of the acridan ester substrate 3a under all conditions. However, this side reaction could be minimized by using the acridan isopropyl ester 3b in the N-alkylation reaction with sodium 3-bromopropane sulfonate. This acridan ester substrate afforded a similar extent of conversion to the oxidized N-sulfopropyl acridinium ester product as can be noted from (entry 8) and showed less cleavage of the isopropyl ester. Retaining the ester on the phenol simplified product isolation of the acridinium ester 5b during scale up as discussed below.
In sharp contrast to the acridan ester N-alkylation reactions, attempted N-alkylation of the corresponding acridine ester 2a afforded very poor conversion to the N-sulfopropyl acridinium ester as can be noted from . In all instances, the extent of conversion to product was observed to be <20%. The poor conversion observed with acridine ester 2a also suggests that in situ formation of 1,3-propane sultone from intramolecular cyclization of sodium 3-bromopropane sulfonate is unlikely because propane sultone is a very effective N-alkylating reagent of acridine esters in ILs Citation14. A comparison of the extent of product formation in and from the acridan and the acridine esters, respectively, suggests that the former substrates are at least threefold more reactive toward sodium 3-bromopropane sulfonate.
Table 2. N-Alkylation of acridine ester 2a with sodium 3-bromopropane sulfonate in ionic liquids.
Despite the modest conversion observed in the small-scale reactions, the acridan N-alkylation reaction was useful for preparative purposes which was our main goal, and a detailed procedure is described in the experimental section. N-alkylation on a half gram to gram scale of acridan ester 3b (, steps d and e) with ten equivalents of sodium 3-bromopropane sulfonate in [BMIM][BF4] followed by oxidation led consistently to >60% conversion to the N-sulfopropyl acridinium ester 5b (, upper panel). Better conversion was observed in these larger-scale reactions. Complete oxidation of the N-sulfopropyl acridan dimethylphenyl ester 4b to the corresponding acridinium ester 5b (, step e), an intrinsically higher energy process than acridan 3b to acridine ester 2b oxidation, required molecular oxygen which was bubbled through the reaction mixture following the N-alkylation reaction. The acridinium ester product 5b and the acridine ester 2b (formed from oxidation of acridan ester starting material 3b) were precipitated from the reaction mixture by adding ice-cold water which dissolved the other reaction components (IL and salts). The mixture of product 5b and acridine ester 2b could either be extracted with a mixture of ethyl acetate and methanol or simply collected by filtration. A small quantity (~10%) of product 5b was lost in the aqueous layer by either procedure. The product 5b was further separated from the acridine ester 2b by column chromatography and was isolated typically in >40% yield and >90% purity (, middle panel). The unreacted acridine ester 2b was also isolated cleanly in near quantitative yield for recycling in the picoline–borane reduction. If we take into account the recovered acridine ester starting material the actual yield of the acridinium ester product 5b was >65% in these gram-scale reactions.
Figure 4. HPLC traces. Upper panel: analysis of the reaction mixture from N-alkylation (, steps d and e) on a gram scale of acridan isopropyl ester 3b with sodium 3-bromopropane sulfonate in [BMIM][BF4] followed by oxidation. Compound eluting at 6.4 minutes is product 5b and compound eluting at 10.2 minutes is acridine isopropyl ester 2b. Small peaks eluting at 4.4 minutes and 7.3 minutes are the corresponding carboxylic acids of 5b and 2b, respectively; middle panel: analysis of the product 5b following purification by column chromatography; lower panel: analysis of the N-sulfopropyl acridinium carboxylic acid 6 obtained by hydrolysis of the isopropyl ester 5b.
![Figure 4. HPLC traces. Upper panel: analysis of the reaction mixture from N-alkylation (Figure 3, steps d and e) on a gram scale of acridan isopropyl ester 3b with sodium 3-bromopropane sulfonate in [BMIM][BF4] followed by oxidation. Compound eluting at 6.4 minutes is product 5b and compound eluting at 10.2 minutes is acridine isopropyl ester 2b. Small peaks eluting at 4.4 minutes and 7.3 minutes are the corresponding carboxylic acids of 5b and 2b, respectively; middle panel: analysis of the product 5b following purification by column chromatography; lower panel: analysis of the N-sulfopropyl acridinium carboxylic acid 6 obtained by hydrolysis of the isopropyl ester 5b.](/cms/asset/1fb0b2c9-f995-413c-8885-7e47bd76aa82/tgcl_a_768708_o_f0004g.jpg)
Cleavage of the isopropyl ester in the product 5b to the N-sulfopropyl acridinium carboxylic acid 6 was achieved by acid hydrolysis as described previously Citation14, and 6 was isolated by a simple filtration step in >60% isolated yield and >90% purity (, lower panel). The carboxylic acid in acridinium ester 6 provides a handle for conjugation to proteins and other molecules using standard N-hydroxysuccinimide ester chemistry Citation1–3. As an alternative to the work-up procedures where the product 5b was isolated prior to hydrolysis of the isopropyl ester, the acridan N-alkylation reaction mixture could be directly subjected to acid hydrolysis. (Direct hydrolysis of acridan N-alkylation reaction mixtures containing [BMIM][BF4] is also expected to hydrolyze the tetrafluoroborate anion in the IL) Citation35. The acridinium ester 6 and the corresponding carboxylic acid of acridine ester 2b were collected by filtration. Compound 6 was converted to the N-hydroxysuccinimide ester 7 Citation14 and was purified by column chromatography in approximately 36% isolated yield.
Although not a focus of the current study, the acridan N-alkylation reaction that we have described offers the opportunity for the synthesis of other acridinium compounds containing various N-alkyl groups. Synthetic access to such compounds is very difficult owing to the poor reactivity of the acridine nitrogen. For example, Brown and coworkers Citation36 recently described the synthesis of acridinium esters containing long chain N-alkyl groups by N-alkylation of the acridine ester precursors using iodide or trifluoromethanesulfonate alkylating reagents. Typical reported yields were <15% (3–14%) using these powerful alkylating reagents and, moreover, these N-alkylation reactions were conducted in toxic solvents such as nitrobenzene or 1,2-dichloroethane. Similarly, Adamczyk Citation37 and co-workers have reported that introduction of the N-sulfopropyl group in acridine N-sulfonylcarboxamides with poor reactivity can be performed using the reagent neopentyl 3-trifloxypropane sulfonate. However, N-alkylation using even this strong alkylating reagent required 7 days to afford 34% conversion to product. Finally, the reagent 1,3-propane sultone is widely used in industry to introduce the water–soluble, three-carbon sulfobetaine moiety in a variety of molecules and polymers Citation9. Our results with acridan N-alkylation reactions suggest that sodium 3-bromopropane sulfonate in ILs may be a benign alternative to 1,3-propane sultone for such applications.
Experimental
General
Chemicals were purchased from Sigma-Aldrich (Milwaukee, Wisconsin, USA) unless indicated otherwise. Sodium 3-bromopropane sulfonate was purchased either from TCI America or Alfa Aesar. All compounds including the ILs were dried under high vacuum over P2O5.
All final compounds and intermediates were analyzed and/or purified by HPLC, using a Beckman-Coulter HPLC system. TLC analysis was performed using 250-µm analytical silica plates from EM Separations Technologies. Flash chromatography was performed using glass columns or an ‘Autoflash’ system from ISCO. Matrix-Assisted Laser Desorption Ionization-Time of Flight (MALDI-TOF) mass spectroscopy was performed using a Voyager DETM BiospectrometryTM Workstation from Perkin-Elmer. This is a benchtop instrument operating in the linear mode with a 1.2 meter ion path length, flight tube. Spectra were acquired in positive ion mode. For small molecules, α-cyano-4-hydroxycinnamic acid was used as the matrix and spectra were acquired with an accelerating voltage of 20,000 volts and a delay time of 100 nsec. For High Resolution Mass Spectra (HRMS), samples were dissolved in HPLC-grade methanol or acetonitrile and analyzed by direct-flow injection (injection volume = 5 µL) ElectroSpray Ionization (ESI) on a Waters Qtof API US instrument in the positive ion mode. Optimized conditions were as follows: capillary = 3000 kV, cone = 35, source temperature = 120°C, desolvation temperature = 350°C. NMR spectra were recorded on a Varian 500 MHz spectrometer. IR spectra of neat samples were recorded on a Bruker TENSOR37 FTIR spectrometer, ATR mode on ZnSe crystal.
Synthesis acridan esters 3a and 3b
(a) Methyl-3,5-dimethyl-4-hydroxybenzoate, compound 1a
A solution of 3,5-dimethyl-4-hydroxybenzoic acid (5 g, 0.030 mole) in methanol (75 mL) was cooled in an ice-bath and thionyl chloride (8 mL) was added dropwise (). The reaction was then warmed to room temperature and stirred for 16 hours. Solid sodium bicarbonate was then added to neutralize the acid, and the resulting suspension was concentrated under reduced pressure. The residue was partitioned between water and ethyl acetate (75 mL each). The ethyl acetate layer was separated and dried over anhydrous magnesium sulfate. It was then concentrated under reduced pressure to afford a tan powder. Yield = 5.49 g (quantitative). TLC analysis of the product using 2% ethyl acetate in chloroform showed a single spot of R f=0.4. HPLC analysis of the product was performed using a Phenomenex, C18, 10 micron, 3.9×300 mm column and a 40-minute gradient of 10 → 60% MeCN/water (each with 0.05% TFA) at a flow rate of 1.0 mL/minute and UV detection at 260 nm. Product was observed eluting at 20 minutes. δH (500 MHz, CDCl3) 2.27 (s, 6H), 3.87 (s, 3H), 7.70 (s, 2H). ν max/cm−1 3347 (OH), 1686 (CO), 1599, 1435, 1320, 1277, 1183. MALDI-TOF MS m/z 180.9 (M + H)+; HRMS m/z 181.0873 (M + H)+ (181.0865 calculated).
Compound 1b (isopropyl-3,5-dimethyl-4-hydroxybenzoate, 97% yield) was prepared similarly. δH (500 MHz, CDCl3) 1.35 (d, 6H, J=6.0), 2.27 (s, 6H), 5.27 (spt, 1H, J=6.0), 7.69 (s, 2H). ν max/cm−1 3397 (OH), 1691 (CO), 1602, 1486, 1360, 1314, 1175. MALDI-TOF MS m/z 209.3 (M + H)+; HRMS m/z 209.1188 (M + H)+ (209.1178 calculated).
(b) 2′,6′-Dimethyl-4′-methoxycarbonylphenyl acridine-9-carboxylate, compound 2a
A suspension of acridine-9-carboxylic acid (5 g, 0.0224 mole) in anhydrous pyridine (100 mL) was treated with p-toluenesulfonyl chloride (8.5 g, 0.0446 mole). The reaction was stirred under a nitrogen atmosphere for 15–20 minutes and then methyl 3,5-dimethyl-4-hydroxybenzoate (4 g, 0.0222 mole) was added. The reaction was stirred at room temperature under a nitrogen atmosphere for 16 hours and then concentrated under reduced pressure. The crude reaction mixture was dissolved in chloroform (150 mL) and washed with 5% sodium carbonate (400 mL), water (100 mL), 10% HCl (200 mL), and water (100 mL) in that order. The chloroform solution was then dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica using 70% hexanes, 25% chloroform, and 5% ethyl acetate as eluent. Yield = 5.9 g (68%); bright yellow powder. δH (500 MHz, CDCl3) 2.48 (s, 6H), 3.95 (s, 3H), 7.70 (m, 2H), 7.86 (m, 2H), 7.92 (s, 2H), 8.35 (d, 2H, J=9.0), 8.43 (d, 2H, J = 9.0). ν max/cm−1 1740 and 1716 (CO), 1437, 1321, 1223, 1187, 1132. MALDI-TOF MS m/z 386.1 (M + H)+; HRMS m/z 386.1393 (M + H)+ (386.1392 calculated).
Compound 2b (quantitative yield) was prepared similarly. δH (500 MHz, CDCl3) 1.40 (d, 6H, J=6.0), 2.49 (s, 6H), 5.28 (spt, 1H, J=6.0), 7.70 (m, 2H), 7.87 (m, 2H), 7.90 (s, 2H), 8.36 (br d, 2H), 8.43 (d, 2H, J=9.0). ν max/cm−1 1747 and 1707 (CO), 1316, 1155. MALDI-TOF MS m/z 414.3 (M + H)+; HRMS m/z 414.1689 (M + H)+ (414.1705 calculated).
(c) 2′,6′-Dimethyl-4′-isopropyloxycarbonylphenyl acridan-9-carboxylate, compound 3b
A solution of 2′,6′-dimethyl-4′-isopropyloxycarbonylphenyl acridine-9-carboxylate (0.84 g, 0.0021 mole, 2b) in tetrahydrofuran (40 mL) was mixed with 10% HCl (4 mL). The resulting yellow solution was treated with picoline-borane complex (0.27 g, 0.0025 mole mole) in one portion. The reaction was stirred at room temperature. After 2 hours, the reaction was quenched with 10% HCl (10 mL) and stirred for 3 hours. The reaction mixture was then concentrated under reduced pressure to 10–15 mL and was extracted with ethyl acetate (70 mL). The ethyl acetate layer was separated and washed twice with water (2×25 mL) followed by aqueous sodium bicarbonate (25 mL) and brine (25 mL). It was then dried over anhydrous sodium sulfate and concentrated under reduced pressure. TLC analysis (silica, 25% ethyl acetate, 75% hexanes) indicated clean product with no trace of starting material. Yield of 3b=0.84 g (quantitative, light yellow powdery solid). δH (500 MHz, CDCl3) 1.32 (d, 6H, J=6.0), 1.75 (s, 6H), 5.18 (spt, 1H, J=6.0), 5.30 (s, 1H), 6.27 (br s, 1H), 6.79 (d, 2H, J = 8.0), 6.94 (m, 2H), 7.22 (m, 2H), 7.35 (d, 2H, J=8.0), 7.61 (s, 2H). ν max/cm−1 3378 (NH), 1712 (CO), 1483, 1312, 1169. MALDI-TOF MS m/z 416.2 (M + H)+; HRMS m/z 416.1864 (M + H)+ (416.1862 calculated).
Compound 3a (quantitative yield) was prepared in a similar manner. δH (500 MHz, CDCl3) 1.75 (s, 6H), 3.84 (s, 3H), 5.30 (s, 1H), 6.26 (br s, 1H), 6.79 (dd, 2H, J=8.0, 1.0), 6.94 (m, 2H), 7.22 (m, 2H), 7.35 (d, 2H, J=8.0), 7.62 (s, 2H). ν max/cm−1 3389 (NH), 1744 and 1712 (CO), 1480, 1320, 1230, 1118. MALDI-TOF MS m/z 388.2 (M + H)+; HRMS m/z 388.1549 (M + H)+, (388.1549 calculated).
General procedure for the N-alkylation of acridan dimethylphenyl esters 3a and 3b with sodium 3-bromopropane sulfonate in ionic [BMIM[BF4]
A mixture of 2′,6′-dimethyl-4′-methoxycarbonylphenyl acridan-9-carboxylate (3a, 25 mg, 0.065 mmole), sodium 3-bromopropane sulfonate (0.146 g, 0.65 mmole), and anhydrous potassium carbonate (18 mg, 0.13 mmole) in [BMIM][BF4] (1 mL) was heated at 160–165°C under a nitrogen atmosphere for 3 hours then open to air for 1 hour ( and ). The reaction was then cooled to room temperature and dissolved in methanol (20 mL). HPLC analysis was performed using a Phenomenex C18, 10 micron, 3.9×300 mm column, and a 30-minute gradient of 10 → 100% MeCN/water (each with 0.05% trifluoroacetic acid) at a flow rate of 1.0 mL/minute and UV detection at 260 nm. 48% conversion to the product 2′,6′-dimethyl-4′-methoxycarbonylphenyl 10-sulfopropylacridinium-9-carboxylate Citation14 (5a) eluting at 17 minutes was observed. Oxidized starting material (2a) was observed to elute at 24 minutes.
General procedure for the attempted N-alkylation of acridine dimethylphenyl ester 2a with sodium 3-bromopropane sulfonate in ILs
A mixture of 2′,6′-dimethyl-4′-methoxycarbonylphenyl acridine-9-carboxylate 2a (25 mg, 0.065 mmole), sodium 3-bromopropane sulfonate (0.146 g, 0.65 mmole), and anhydrous potassium carbonate (18 mg, 0.13 mmole) in [BMIM][BF4] (1 mL) was heated at 160–165°C under a nitrogen atmosphere for 3 hours (). The reaction was then cooled to room temperature and dissolved in methanol (20 mL). HPLC analysis was performed using a Phenomenex C18, 10 micron, 3.9×300 mm column, and a 30-minute gradient of 10 → 100% MeCN/water (each with 0.05% trifluoroacetic acid) at a flow rate of 1.0 mL/minute and UV detection at 260 nm. Poor conversion to the product 2′,6′-dimethyl-4′-methoxycarbonylphenyl 10-sulfopropylacridinium-9-carboxylate 5a (~13%) eluting at 17 minutes was observed. Oxidized starting material 2a was observed to elute at 24 minutes.
Preparative procedures for the synthesis of N-sulfopropyl acridinium dimethylphenyl ester derivatives by N-alkylation of acridan dimethylphenyl ester 3b with sodium 3-bromopropane sulfonate in [BMIM[BF4]
A mixture of 2′,6′-dimethyl-4′-isopropyloxycarbonylphenyl acridan-9-carboxylate (3b, 1.00 g, 2.41 mmoles), sodium 3-bromopropane sulfonate (5.42 g, 24.1 mmoles), and anhydrous potassium carbonate (1.33 g, 9.63 mmoles) in [BMIM][BF4] (20 mL) was heated at 165°C under an argon atmosphere for 5–8 hours. It was then heated for an additional 3 hours at 165°C, and dry oxygen was bubbled through the reaction mixture. The reaction was then cooled to room temperature and a small portion was dissolved in methanol and analyzed by HPLC using a 4.0×50 mm, Phenomenex, Kinetex 2.6 micron, C18 column, and 10-minute gradient of 10 → 90% MeCN/water (each with 0.05% TFA) at a flow rate of 1 mL/minute and UV detection at 260 nm. Product 5b eluted at 6.4 min (67% conversion) and oxidized starting material 2b eluted at 10.2 min (, upper panel).
For isolation of the acridinium isopropyl ester 5b, ice-cold water (250 mL) was added to the reaction mixture, and the mixture was transferred to an Erlenmeyer flask and stored in the refrigerator at 4°C for 24 hours to complete the precipitation. The precipitated solids were collected by vacuum filtration and further dried under high vacuum. The product 5b and the acridine ester 2b were separated by flash chromatography on silica using an ISCO autoflash system. Ethyl acetate was used to elute acridine ester 2b followed by 40% methanol in ethyl acetate to elute product 5b. Yield (5b) = 0.56 g, (43%); yield (2b) = 0.35 g, (35%). δH (500 MHz, CF3COOD) 1.45 (d, 6H, J=6.0), 2.52 (s, 6H), 2.93 (br s, 2H), 3.83 (br s, 2H), 5.37 (spt, 1H, J=6.0), 5.85 (br m, 2H), 7.97 (s, 2H), 8.12 (br t, 2H), 8.53 (br t, 2H), 8.76 (d, 2H, J=9.0), 8.91 (d, 2H, J=9.0). ν max/cm−1 1749 and 1708 (CO), 1377, 1191, 1145. MALDI-TOF MS m/z 536.3 (M + H)+; HRMS m/z 536.1751 (M + H)+ (536.1743 calculated).
The isopropyl ester in compound 5b was hydrolyzed to the N-sulfopropyl acridinium carboxylic acid 6 Citation14 by acid hydrolysis. Compound 5b (0.9 g, 1.68 mmole) in 2M HCl (90 mL) was refluxed under a nitrogen atmosphere for 4 hours. The reaction was cooled to room temperature and stored in the refrigerator at 4°C for 16 hours to complete precipitation of the product. The acridinium carboxylic acid 6 was collected by filtration and dried over P2O5. Yield = 0.52 g (62%).
For N-alkylation followed by direct hydrolysis, the crude reaction mixture (0.5 g scale reaction) following alkylation and oxidation was treated with 75 mL of 2M HCl. The reaction was refluxed for 4 hours. The reaction was then cooled to room temperature, and a small portion was dissolved in methanol and analyzed by HPLC using a 4.0×50 mm, Phenomenex, Kinetex 2.6 micron, C18 column, and 10-minute gradient of 10 → 90% MeCN/water (each with 0.05% TFA) at a flow rate of 1 mL/minute and UV detection at 260 nm. Product 6 Citation14 eluted at 4.4 min (complete conversion), and the carboxylic acid corresponding to oxidized starting material 2b eluted at 7.3 min. The hot HCl solution was added slowly to ice-cold water (150 mL) and a yellow solid precipitated out. The suspension was kept in the refrigerator for 24 hours and the precipitated solids were collected by filtration. Crude 6 was converted to the N-hydroxysucinimide ester 7 Citation14 as follows. It was dissolved in DMF (15 mL) and treated with diisopropylethylamine (0.654 mL, 3.76 mmoles) followed by N,N,N′,N′-tetramethyl-O-(N-succinimidyl)uronium tetrafluoroborate (TSTU) (0.989 mg, 3.29 mmoles). The reaction was stirred at room temperature for 2 hours. HPLC analysis as described earlier showed complete conversion to the NHS ester 7 eluting at 5.1 minutes. The product 7 was purified by flash chromatography on silica using an ISCO Autoflash system. The column was first eluted with ethyl acetate followed by 40% methanol in ethyl acetate to elute product 7. Yield = 0.292 g (36%).
Conclusions
In summary, we have developed and demonstrated a practical and green N-alkylation reaction for the introduction of the hydrophilic N-sulfopropyl group in commercially useful, chemiluminescent acridinium dimethylphenyl ester labels. The new synthetic protocol takes advantage of the increased reactivity of acridan esters in ILs toward sodium 3-bromopropane sulfonate and eliminates use of the potent carcinogenic reagent 1,3-propane sultone Citation6–8. The acridan esters are easily synthesized from the corresponding acridine esters by a simple reduction reaction, using the nontoxic reducing reagent picoline-borane. The acridan N-alkylation synthetic protocol also uses a nontoxic inorganic base potassium carbonate as an acid scavenger in the N-alkylation step and, air or oxygen as the oxidant for the subsequent conversion of the N-sulfopropyl acridan ester to the chemiluminescent N-sulfopropyl acridinium ester.
TGCL 768708 Supplementary Material.doc
Download MS Word (143 KB)Related Research Data
References
- Law S-J , Leventis CS , Natrajan A , Jiang Q , Connolly PB , Kilroy JP , McCudden CR , Tirell SM. United States patent US 5,656,426 . 1997 .
- Natrajan A , Sharpe D , Costello J , Jiang Q. Zwitterionic Reagents for Labeling, Cross-Linking and Improving the Performance of Chemiluminescent Immunoassays . Anal Biochem . 2010 ; 406 : 204 – 213 . doi: 10.1016/j.ab.2010.07.025
- Natrajan A , Sharpe D , Wen D. Org Biomol Chem . 2012 ; 10 : 1883 – 1895 . doi: 10.1039/c2ob06807a
- Kricka LJ. Clinical applications of chemiluminescence . Analytica Chimica Acta . 2003 ; 500 : 279 – 286 . doi: 10.1016/S0003-2670(03)00809-2
- Roda A , Guardigli M. Analytical chemiluminescence and bioluminescence: latest achievements and new horizons . Anal Bioanal Chem . 2012 ; 402 : 69 – 76 . doi: 10.1007/s00216-011-5455-8
- Bolt HM , Golka K. J 1,3-Propane Sultone as an Extremely Potent Human Carcinogen: Description of an Exposed Cohort in Germany. Toxicol Environ Health Part A . 2012 ; 75 : 544 – 550 . doi: 10.1080/15287394.2012.675305
- Bolt HM , Golka K. 1,3-Propane sultone, an extremely potent experimental carcinogen: what should be expected in humans . Toxicol Lett . 2004 ; 151 : 251 – 254 . doi: 10.1016/j.toxlet.2003.12.073
- Ulland B , Finkelstein M , Weisburger EK , Rice JM , Weisburger JH. Carcinogenicity of Industrial Chemicals Propylene Imine and Propane Sultone . Nature . 1971 ; 230 : 460 – 461 . doi: 10.1038/230460a0
- Roberts DW , Williams DL. Sultone Chemistry . Tetrahedron . 1987 ; 43 : 1027 – 1062 . doi: 10.1016/S0040-4020(01)90041-9
- Hughes ED , Ingold CK. Mechanism of substitution at a saturated carbon atom. Part IV. A discussion of constitutional and solvent effects on the mechanism, kinetics, velocity, and orientation of substitution . J Chem Soc . 1935 ; 244 – 255 . doi: 10.1039/jr9350000244
- Hughes ED. Mechanism and kinetics of substitution at a saturated carbon atom . Trans Faraday Soc . 1941 ; 37 : 603 – 631 . doi: 10.1039/tf9413700603
- Hughes ED , Ingold CK. The mechanism and kinetics of elimination reactions . Trans Faraday Soc . 1941 ; 37 : 657 – 685 . doi: 10.1039/tf9413700657
- Cooper KA , Dhar ML , Hughes ED , Ingold CK , MacNulty BJ , Woolf LI. Mechanism of elimination reactions. Part VII. Solvent effects on rates and product-proportions in uni- and bi-molecular substitution and elimination reactions of alkyl halides and sulphonium salts in hydroxylic solvents . J Chem Soc . 1948 ; 2043 – 2049 . doi: 10.1039/jr9480002043
- Natrajan A , Wen D. Facile N-Alkylation of Acridine Esters with 1,3-Propane Sultone in Ionic Liquids . Green Chem . 2011 ; 13 : 913 – 921 . doi: 10.1039/c0gc00758g
- Kamlet MJ , Taft RW. The solvatochromic comparison method. I. The beta-scale of solvent hydrogen-bond acceptor (HBA) basicities . J Am Chem Soc . 1976 ; 98 : 377 – 383 . doi: 10.1021/ja00418a009
- Taft RW , Kamlet MJ. The solvatochromic comparison method. 2. The alpha-scale of solvent hydrogen-bond donor (HBD) acidities . J Am Chem Soc . 1976 ; 98 : 2886 – 2894 . doi: 10.1021/ja00426a036
- Yokoyama T , Taft RW , Kamlet MJ. The solvatochromic comparison method. 3. Hydrogen bonding by some 2-nitroaniline derivatives . J Am Chem Soc . 1976 ; 98 : 3233 – 3237 . doi: 10.1021/ja00427a030
- Kamlet MJ , Addoud JL , Taft RW. The solvatochromic comparison method. 6. The π* scale of solvent polarities . J Am Chem Soc . 1977 ; 99 : 6027 – 6038 . doi: 10.1021/ja00460a031
- Kamlet MJ , Addoud J-LM , Abraham MH , Taft RW. Linear Solvation Energy . J Org Chem . 1983 ; 48 : 2877 – 2887 . doi: 10.1021/jo00165a018
- Crowhurst L , Mawdsley PR , Pèrez Arlandis JM , Salter PA , Welton T. Solvent-solute interactions in ionic liquids . Phys Chem Chem Phys . 2003 ; 5 : 2790 – 2794 . doi: 10.1039/b303095d
- Pretti C , Chiappe C , Pieraccini D , Gregori M , Abramo F , Monni G , Intorre L. Acute toxicity of ionic liquids to the zebrafish (Danio rerio) . Green Chem . 2006 ; 8 : 238 – 240 . doi: 10.1039/b511554j
- Bernot RJ , Kennedy EE , Lamberti GA. Effects of Ionic Liquids on the survival, Movement and feeding Behaviour of the Freshwater Snail, physa acuta . Environ Toxicol Chem . 2005 ; 24 : 1759 – 1765 . doi: 10.1897/04-614R.1
- Couling DJ , Bernot RJ , Docherty KM , Dixon JK , Maginn EJ. Assessing the factors responsible for ionic liquid toxicity to aquatic organisms via quantitative structure–property relationship modeling . Green Chem . 2006 ; 8 : 82 – 90 . doi: 10.1039/b511333d
- Matzke M , Stolte S , Thiele K , Juffernholz T , Arning J , Ranke J , Welz-Biermann U , Jastorff B. The influence of anion species on the toxicity of 1-alkyl-3-methylimidazolium ionic liquids observed in an (eco)toxicological test battery . Green Chem . 2007 ; 9 : 1198 – 1207 . doi: 10.1039/b705795d
- Stolte S , Matzke M , Arning J , Böschen A , Pitner U , Welz-Biermann W-R , Jastorff B , Ranke J. Effects of different head groups and functionalised side chains on the aquatic toxicity of ionic liquids . Green Chem . 2007 ; 9 : 1170 – 1179 . doi: 10.1039/b711119c
- Sena DW , Kulacki KJ , Chaloner DT , Lamberti GA. The role of the cell wall in the toxicity of ionic liquids to the alga Chlamydomonas reinhardtii . Green Chem . 2010 ; 12 : 1066 – 1071 . doi: 10.1039/c000899k
- Luo Y-R , Wang S-H , Li X-Y , Yun M-X , Wang J-J , Sun Z-J. Toxicity of ionic liquids on the growth, reproductive ability, and ATPase activity of earthworm . Ecotox Environ Safe . 2010 ; 73 : 1046 – 1050 . doi: 10.1016/j.ecoenv.2010.01.017
- Pham TPT , Cho C-W , Yun Y-S. Environmental fate and toxicity of ionic liquids: A review . Water Res . 2010 ; 44 : 352 – 372 . doi: 10.1016/j.watres.2009.09.030
- Wasserscheid P , Welton T Ionic liquids in synthesis . Vols 1 and 2 . Weinheim , , Germany : Wiley-VCH Verlag GmbH & Co. KgaA ; 2008 .
- Poole SK , Shetty PH , Poole CF. Chromatographic and Spectroscopic Studies of the Solvent properties of a New series of Room-Temperature Liquid Tetraalkylammonium Sulphonates . Analytica Chimica Acta . 1989 ; 218 : 241 – 264 . doi: 10.1016/S0003-2670(00)80302-5
- Abdel-Abdel-Magid AF , Carson KG , Harris BD , Maryanoff CA , Shah RD. Reductive Amination of Aldehydes and Ketones with Sodium Triacetoxyborohydride. Studies on Direct and Indirect Reductive Amination Procedures . J Org Chem . 1996 ; 61 : 3849 – 3862 . doi: 10.1021/jo960057x
- Sato S , Sakamoto T , Miyazawa E , Kikugawa Y. One-pot reductive amination of aldehydes and ketones with a-picoline-borane in methanol, in water, and in neat conditions . Tetrahedron . 2004 ; 60 : 7899 – 7906 . doi: 10.1016/j.tet.2004.06.045
- Unterieser I , Mischnick P. Labeling of oligosaccharides for quantitative mass spectrometry . Carbohydr Res . 2011 ; 346 : 68 – 75 . doi: 10.1016/j.carres.2010.11.001
- Ruhaak LR , Steenvoorden E , Koeleman CA , Deelder AM , Wuhrer M. 2-picoline-borane: a non-toxic reducing agent for oligosaccharide labeling by reductive amination . Proteomics . 2010 ; 10 : 2330 – 2336 . doi: 10.1002/pmic.200900804
- Freire MG , Neves CMSS , Marrucho IM , Coutinho JAP , Ferandes AM. Hydrolysis of Tetrafluoroborate and Hexafluorophosphate Counter Ions in Imidazolium-Based Ionic Liquids . J Phys Chem A . 2010 ; 114 : 3744 – 3749 . doi: 10.1021/jp903292n
- Brown RC , Li Z , Rutter AJ , Mu X , Weeks OH , Smith K , Weeks I. Development and application of a novel acridinium ester for use as a chemiluminescent emitter in nucleic acid hybridisation assays using chemiluminescence quenching . Org Biomol Chem . 2009 ; 7 : 386 – 394 . doi: 10.1039/b811947c
- Adamczyk M , Chen Y-Y , Mattingly PG , Pan Y , Rege S. Neopentyl 3-Triflyloxypropanesulfonate. A Reactive Sulfopropylation Reagent for the Preparation of Chemiluminescent Labels . J Org Chem . 1998 ; 63 : 5636 – 5639 . doi: 10.1021/jo980047r