Abstract
The organic reactions in water, as the most significant green solvent, have attracted much attention due to their unique properties over conventional organic ones. In the catalytic area, heteropoly acids (HPAs) are also promising green solid acids to replace environmentally harmful liquid acid catalysts. Herein, we wish to report the organic reactions catalyzed by HPAs, their salt, and polyoxometalates (POMs) in aqueous systems.
1. Introduction
One of the most striking concepts in chemistry for sustainability is Green Chemistry, and it is the utilization of a set of principles that reduces or eliminates the use or generation of hazardous substances in the design, manufacture, and applications of chemical products (Citation1). It has been specifically concerned with feedstock, reactions, solvents, and separations in synthetic processes in general (Citation2–Citation4).
1.1. Water as an alternative solvent
As far as the largest amount of “auxiliary waste” in most chemical processes is associated with solvent usage, accordingly, a part of Green Chemistry connects to the elimination of volatile organic solvents or their replacement by nonvolatile, nonflammable, nontoxic, and inexpensive green solvents. In this regard, water can best meet the requirements (Citation3, Citation5). The types of organic reactions in water are as diverse as those in nonaqueous conditions, and many remarkable reviews have been published so far on specific topics in this field (Citation1, Citation5–Citation9).
In fact, the use of water in organic reactions has developed not only the issues and aspects of the reactions from the viewpoint of green and sustainable properties but also the synthetic competence by reducing the number of steps, stabilizing the catalyst, facilitating product isolation, changing the reaction selectivity, even when the reactants are sparingly soluble or insoluble in this medium. Although “on-water” techniques have provided excellent solutions for some situations, there will be cases, such as using heteropoly acids (HPAs), where complete solubility in water is much desired (Citation3, Citation5, Citation6, Citation10).
1.2. HPAs
In a recent decade, using efficient industrial catalysts which are also ecofriendly, green, and simply recyclable have been under great considerations and attentions.
HPAs are promising solid acids to supplant environmentally unsafe and hazardous liquid acid catalysts (Citation11–Citation13). The catalytic application of HPAs, polyoxometalates (POMs) (Citation12, Citation14–Citation17) as efficient homogeneous or heterogeneous solid acids catalysts have been recognized and established both by successful large-scale applications in industry and promising laboratory results (Citation12, Citation18–Citation20).
There are commonly four types of HPAs including Anderson HxAy[BD6O24].zH2O, Keggin HxAy[BD12O40].zH2O, Well-Dawson HxAy[B2D18O62].zH2O, and Preyssler HxAy[B5D30O110].zH2O. In the mentioned formula, on the whole, A represents Li, Na, K, Rb, Cs, Mg, Ca, Al, NH4, an ammonium salt, or a phosphonium salt. B is a symbol of P, Si, As, or Ge and D represents at least one element such as Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Ga, Zr, Nb, Mo, Tc, Rh, Cd, In, Sn, Ta, W, Re, and Tl (Citation21).
Depending on the reaction conditions, the activity/mole proton of HPAs may be higher, 3–100 times, than conventional organic and inorganic acids (Citation22). For that reason, they are considered as super-acids (Citation23).
The problem of their acid strength determination in terms of pKa and the number of acid sites has constantly been a challenge to search for trustworthy experimental methods (Citation24–Citation28).
According to data, generally, HPAs are much stronger than H3PO4 and also than the usual inorganic acids (HCl, H2SO4, HNO3, and HBr) and even such strong acids as HClO4 and CF3SO3H. It is notable that sulfuric acid ranks 2–5 units of pKa below HPAs (Citation29).
For example, the initial pH values of the Keggin-type solutions are in the range of 0.35–0.40 (Citation30) or the acid strengths of Cs2.5 −salt and H3PW12O40 measured by Hammett indicators are similar in pKa values ranging from −5.6 to −14.5 (Citation31).
It is worth mentioning that the catalyst may be reused without significant loss of activity. At the end of the reaction, the catalyst can be filtered, separated by centrifugation, or evaporation, and then washed with proper solvent, dried, and reused in another reaction. They are usable even after three to five runs, depending on the catalyst. The examples are the recovery of 90–95% for catalyst H7[PMo8V4O40] (Citation32), 78–94% for H14[NaP5W30O110] (Citation33), 90–95% for 20% PdMPA/SiO2 (Citation34), 80–87% for H6P2W18O62.18H2O (Citation35). The catalytic activity of Cs2.5H0.5PW12O40 can also be retained well in the second and third runs (Citation36, Citation37).
1.3. Mechanism
Generally, HPA-catalyzed reactions may be presented by the conventional mechanism of Brønsted acid catalysis. The mechanism is started with protonation of the substrates and followed by conversion of the ionic intermediates to the related products. Misono and co-workers introduced two types of surface and bulk mechanism for heterogeneous catalysis (Citation38). In surface type catalysis, the reactions occurred on the surface of bulk or supported heteropoly compounds and the catalytic activity frequently depended on the surface acidity of HPA. Here, the reaction rate and yield are parallel to the number and potency of the accessible surface acid sites. The bulk type mechanism is chiefly related to reactions of polar substrates on bulk heteropoly compounds. These substrates are capable of absorbing into the catalyst bulk, and as a result, all protons either in the bulk or on the surface are suggested to be included in a catalytic reaction (Citation23, Citation39).
1.4. HPA–water system
HPAs contain as much as 20 to nearly 30 water molecules per one anion. Anhydrous HPA loses its water of constitution (dehydroxylation) at 350–500 °C (Citation20).
HPAs are insoluble in nonpolar solvents but greatly soluble in polar solvents as well as being fully dissociated in aqueous solution which gives the strong acidity (Citation40). The primary structure of HPAs (heteropoly anions) is rather stable unlike the rigid network structure of most solid acids. The secondary structures of heteropoly anion (heteropoly anion together with protons) are rather transferable on account of the interaction with polar molecules. For such systems, water as a polar organic molecule plays a key role which is absorbed into the bulk of crystallites of HPAs and protonated clusters and influences their acidity and absorption properties (Citation13, Citation27, Citation28, Citation40, Citation41).
Most solid acids lose their catalytic activities in aqueous solutions due to rigorous poisoning of the acid sites. Heteropoly compounds are good water-tolerant hydrophobic solid acids as well as having biphasic reaction systems containing an aqueous phase (Citation7).
HPAs in water have also been used in inorganic chemistry for the synthesis of hybrid nanocomposite membranes (Citation42, Citation43) and many new and diverse catalysts (Citation44–Citation52). HPAs have been extensively used in analytical chemistry for determination and removal of metal ions like phosphates (Citation49, Citation53–Citation55), arsenates (Citation55–Citation58), arsenic (Citation57–Citation64), silicates (Citation65–Citation68), and many other ions (Citation69, Citation70) from aqueous solutions. They have also other applications in aqueous systems (Citation71).
Consequently, these advantages and benefits make this striking strategy the protocol of choice for acid catalyzed organic reactions especially for the synthesis of the solid and water insoluble products.
In the following sections, we wish to report the advances in application of HPAs, their salts, and POMs in organic synthesis in aqueous systems.
2. Condensation reactions
HPAs are efficient catalysts for the cyclocondensation reaction. An example is the condensation of anthranilamide with aldehydes in water at ambient temperature and afforded the corresponding 2,3-dihydro-4(1H)-quinazolinone compounds in good to excellent yields (79–97%) (). Among four HPAs, H3PW12O40 was the best catalyst. Hence, in this reaction, H3PW12O40 was much more reactive in water than in other organic solvents (). Furthermore, it was shown that only 0.10 mol% of the catalyst was sufficient for the completion of the reaction in high yields at room temperature. A series of aldehydes with either electron-donating or electron-withdrawing groups attaching to aromatic ring were investigated. The substitution groups on the aromatic ring had no obvious effect on the yield. When aromatic aldehydes were replaced by aromatic heterocyclic aldehydes, the corresponding products were obtained with high yields as well (Citation72, Citation73).
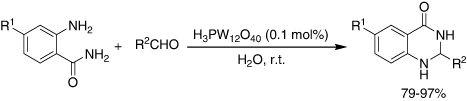
Table 1. Reactivity of HPAs in different solvents for the synthesis of 2,3-dihydro-4(1H)quinazolinone compounds.
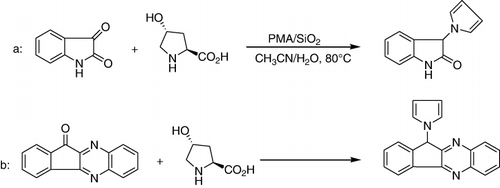
Isatins react capably with 4-hydroxyproline in the presence of 10 mol% of phosphomolybdic acid supported on silica gel (PMA/SiO2)in acetonitrile-water (4:1) under reflux conditions to furnish N-substituted pyrroles in excellent yields (a). Similarly, 11H-indeno[1,2-b] quinoxalin-11-ones also participated in this reaction (b). Among diverse HPAs, PMA was one of low cost and commercially available catalysts. In the absence of water, the reaction took a long time, and the products were obtained in low yields. The presence of electron-withdrawing groups on isatin derivatives reduced the yields of products to some extent, whereas electron-rich isatins gave higher yields (Citation74).
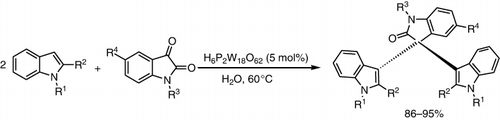
Keggin-type HPA was found to be an efficient and reusable catalyst for the synthesis of biologically active quinoxaline derivatives from the condensation of 1,2-diamine with 1,2-dicarbonyl compounds in excellent yields in water (). This method afforded a new and efficient protocol in terms of a small quantity of catalyst, a wide scope of substrates, and a simple work-up procedure (Citation75).
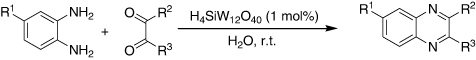
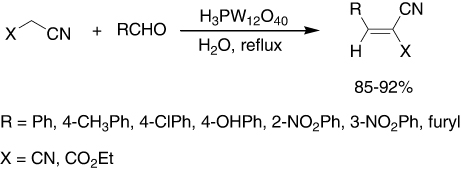
12-tungstphosphoric acid was also a good catalyst for Knoevenagel condensation of malononitrile and ethylcyanoacetate with various aldehydes for synthesis of derivatives (). The effect of different solvents on this reaction was studied. The results established that water was the best solvent for this reaction (Citation12).
Palladium exchanged molybdophosphoric acid supported on silica was reported as a highly effective catalyst for direct reductive amination of carbonyl compounds. A diversity of secondary and tertiary amines synthesized over this catalyst in excellent yields under mild reaction conditions ().
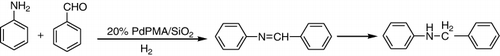
It is noteworthy to mention that the present catalyst was active in many solvents including water. It was found that dimethyl formamide (DMF) was best solvent as it gave a high yield compared to other solvents. Aromatic aldehydes with electron-donating groups smoothly underwent reductive amination to give their corresponding N-phenyl amines in a good yield without affecting the functional groups. Aromatic aldehydes and amines with electron-withdrawing group such as NO2 gave a low yield. This might be due to the reduction of NO2 group during the reaction (Citation34).
A novel, highly efficient and entirely green protocol for Friedel–Crafts alkylation of indole (a) and pyrrole (b) was established with several electron-deficient olefins in water at room temperature with good to excellent yields. Interestingly, the Michael addition between indole and methyl vinyl ketone in the presence of catalysts, such as CeCle3·7H2O, FeCl3·6H2O, ZrCl4, WCl6, and H3PO4, proceeded efficiently, furnishing moderate to excellent yields of the desired products in water. HPAs such as H3PMo12O40 and H3PW12O40 were also effective in this reaction and showed the same catalytic activity in a short reaction time in water. By using substituted indoles such as N-methylindole, 2-methylindole, and 5-bromoindole, good to excellent yields (74–97%) were obtained. The substituents did not affect the reactivity of indoles significantly. The only significant difference in reactivity was observed for N-methylindole, which gave a higher yield in comparison with indole.
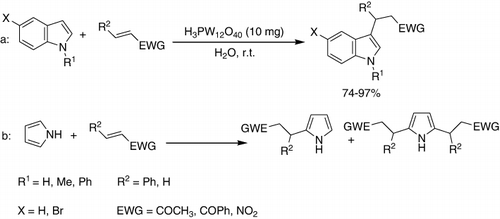
Considering Michael acceptors, the reactions proceeded smoothly with electron-deficient olefins such as methyl vinyl ketone, chalcone, chlorochalcone, and b-nitrostyrene. In all cases, the reactions proceeded at room temperature with high selectivity. To extend the studies, the Friedel–Crafts alkylation was also investigated in water and other organic solvents, using different catalysts () (Citation76).
Table 2. Investigation of Friedel-Crafts alkylation in different solvent and catalysts.
3. Carbonylation reactions
Methyl glycolate (MG) was synthesized effectively from the carbonylation of HCHO, using HPAs as catalysts, followed by esterification with methanol (). The yields above 89% could be obtained when H3PW12O40 and sulfolane were employed as a catalyst and a solvent, respectively. Water acted not only as crystal water of HPAs, but also as a reactant for development of glycolic acid. The results indicated that the excess water would act partly as a solvent, which was unfavorable for the formation of glycolic acid (Citation77).
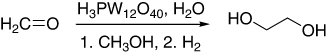
Oxometallic acids, HPAs of Mo, W, and their sodium salts were investigated as promoters for homogeneous rhodium catalyzed methanol carbonylation. The experiments were carried out under low water conditions. All the catalysts with the promoters demonstrated better catalytic activity than the pristine Monsanto catalyst. The promoting effects of acids upon reaction rates were generally superior than the salts. The catalytic rate was efficiently enhanced by increasing the content of the promoters, especially at low content level (Citation78).
4. Hydrolysis reactions
Hydrolysis reactions of esters, ethyl acetate, cyclohexyl acetate (Citation79, Citation80), 2-methylphenyl acetate (Citation81, Citation82) (Citation83), and 2-nitrophenyl acetate (Citation81, Citation82) in the presence of a large excess of water were examined by using various solid acids (). Inorganic solid acids such as an acidic cesium salt of 12-tungstophosphoric acid CS2.5H0.5PWI2O40, H-ZSM-5, SO42−/ZrO2, and Nb2O5, and polymer resins, Amberlyst-15, Amberlite-200C, and Nafion-H catalyzed the reactions, whereas H-Y, H-mordenite, SiO2−Al2O3, and γ-Al2O3 were inactive. Among inorganic solid acids, Cs2.5H0.5PWl2O40 was most active for all reactions in the unit of the activity per catalyst weight (Citation79).
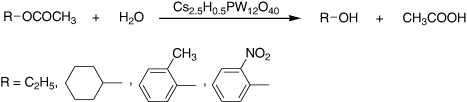
The hydrolysis of esters in large excess water catalyzed by a range of solid and liquid acids was studied. It was found that a solid CS2.5H0.5PW12O40 was remarkably active for the hydrolysis of 2-methylphenylacetate in excess water, while other inorganic solid acids, such as H-ZSM-5, Nb2O5, or SO42 −/ZrO2, were almost inactive. Among the acidic salts, CS2.5H0.5PW12O40 displayed the highest activities for various reactions due to the high surface acidity. Furthermore, it was noted that heteropoly compounds supported on SiO2, catalyzed the hydrolysis of ethyl acetate, and a stoichiometric Cs salt of H3PMo12O40 had hydrophobic nature (Citation84).
Acid-catalyzed esterification, which produced various esters, such as n-butyl acetate, is one of the most important reactions in this category. The reaction is a reversible process and acids can catalyze both esterification and hydrolysis. The esterification between n-butanol and acetic acid was catalyzed by SBA-15 with a HPA fixed in the channels, which was synthesized with different hydrophilic/hydrophobic property. The H3PW12O40-loaded catalyst modified with amino showed the highest yield (96%) of n-butyl acetate with 100% selectivity. The counterreaction of hydrolysis was then suppressed. The probability that water and n-butyl acetate encountered at the acid sites were less than at acetic acid and n-butanol sites, both of which were hydrophilic, permitted the equilibrium to shift toward the product side. On the other hand, the water concentration in the reaction system influences the composition of the reaction system, so there was also a retarding effect in the activity due to the cumulation of the hydrophilic water without rapid desorption as it was formed (Citation85).
The hydrolysis of bistrimethylol propane monoformal in water at 348 K was done using HPAs [H3PW12O40 (HPW), H4SiW12O40 (HSiW), H4GeW12O40 (HGeW), H3PMo12O40 (HPMo)] and their acidic Cs salts (). Cs2.5H0.5PW12O40 (Cs2.5PW) was the most active per proton in the solution or in the bulk of solid catalyst. The Cs2.2H0.8PW12O40 (Cs2.2PW) was the most active when supposedly compared with per surface proton and per weight of catalyst. Dissolved acids showed the activity order of HPW > HSiW > HGeW > HPMo >> H2SO4, HCl. Since the protons on the surface were only a part of the total protons in the solid bulk, Cs2.2PW and Cs2.5PW had much higher turnover numbers than dissolved acids (Citation86).
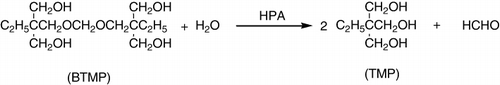
Cs2.5 was highly active for organic reactions such as hydrolysis of esters and oligosugars in water as shown in . The reaction was used as a test for the water-tolerant catalysts (Citation82).
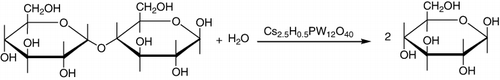
The direct hydrolysis of cellulose to glucose, hydroxymethylfurfural (HMF), and other soluble by-products in water at 190 °C, using zeolites (H-BEA and H-MOR), sulfated zirconia supported over mesoporous silica (SBA-15), Amberlyst-15, HPAs, and AlCl3·6H2O as acid catalysts was studied. HPAs (H3PW12O40 and H4SiW12O40) and their salts with metal cations acted as efficient homogeneous catalysts in an aqueous phase. The high activity but low formation of glucose was observed in the case of AlCl3.6H2O and phosphotungstic acid (PTA), while better yields of glucose were observed in the case of phosphomolybdic acid (PMA) which is a weaker acid than PTA. This lower acidity restricted the secondary reactions and resulted in higher formation of glucose (Citation87).
The hydrogenolysis of cellulose was reported including hydrolysis to glucose and further hydrogenation, hydrogenolysis, and dehydration reactions to sorbitol, sorbitan, and isosorbide together with erythritol, xylitol, glycerol, propylene and ethylene glycol, and methanol (n = 1–3) (). Noted that the presented yields in the case of recycling experiments were based on the freshly added substrate to allow comparability with reference experiments (Citation88).
A series of HPA-ionic liquids [C4H6N2(CH2)3SO3H]3nHnPW12O40([MIMPSH]nH3nPW12O40, n = 1, 2 3, abbreviated as [MIMPSH]nH3nPW), was also utilized to catalyze one-pot depolymerization of cellulose into glucose. Their performances were much better than those of the previously reported HPAs, such as H3PW12O40, Cs2.5H0.5PW12O40. Besides cellulose, the HPA ionic liquids were able to catalyze the conversion of sucrose and starch into glucose. In addition, one-pot synthesis of levulinic acid (LA) directly from cellulose was realized by HPA-ionic liquid catalysts in a water–methyl isobutyl ketone (MIBK) biphasic system. The separation of the products and catalysts was simple, and the retrieved [MIMPSH]nH3nPW could be repeatedly used without noticeable loss of performance (Citation89).
5. Hydration reactions
The acidic Cs salt, Cs2.5, was found to be an efficient water-tolerant solid acid for hydration of alkene in excess water () (Citation82, Citation83). Among the heteropoly compounds, an insoluble acidic salt Cs2.5H0.5PW12O40 exhibited very high activities for various reactions because of its superacidity and high surface area. The acidic salt, however, formed colloidal suspension in water, and recovering of the catalysts was not easy. To solve this problem, immobilization of Cs2.5 on oxide supports was attempted (Citation83)
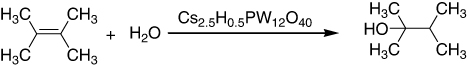
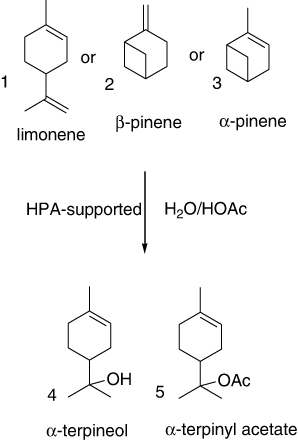
The liquid-phase hydration and acetoxylation of limonene (1), β-pinene (2), and α-pinene (3) catalyzed by dissolved or silica-supported HPA H3PW12O40 (PW) in acetic acid and acetic acid/water solutions were studied (). All three substrates gave α-terpineol (4) as the major product along with α-terpinyl acetate (5). The reaction rate raised in the order: limonene < α-pinene < β-pinene. At room temperature under optimized conditions, 2 and 3 formed a mixture of 4 and 5 with 85% selectivity at 90% substrate conversion. The PW showed a much higher catalytic activity than conventional acid catalysts such as H2SO4 and Amberlyst-15 (Citation90, Citation91).
H3PMo12O40–PPO composite catalyst coated on Al2O3 was also prepared as heterogeneous catalysts for the liquid-phase tert-butanol (TBA) synthesis from isobutene and water. It was found that all the composite film catalysts illustrated higher catalytic activities than homogeneous H3PMo12O40. Among the composite film catalysts, H3PMo12O40–PPO showed the best catalytic performance. The H3PMo12O40–PPO/Al2O3 also showed a higher activity than a homogeneous solution of H3PMo12O40 (Citation92).
Catalytic utilities of Cs2.5H0.5PW12O40, an acidic salt of dodecatungstophosphoric acid, were investigated for two types of reactions in large excess water containing hydration of olefins, 2,3-dimethyl-2-butene, and cyclohexene. Hydration of 2,3-dimethyl-2-butene was efficiently catalyzed by the fine particles at 343 K (Citation37).
6. Oxidation reactions
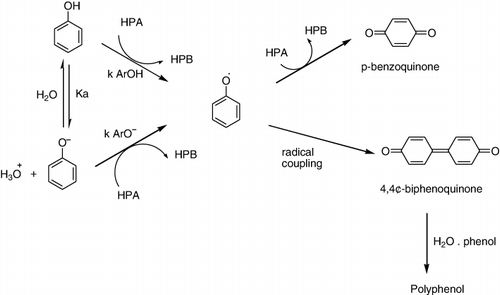
Oxidation of phenol is an important reaction in both industrial and biological processes. This reaction can proceed through either polar or free radical mechanisms. The kinetics of oxidation of phenol and a few ring-substituted phenols by heteropoly 11-tungstophosphovanadate (V), [PVvW11O40]4- (HPA) was studied spectrophotometrically in aqueous acidic medium containing perchloric acid and also in acetate buffers of several pH values at 25° C (). In the aqueous medium, in the absence of bases, the proton is transferred to water and the electron was transferred to the oxidant from ArOH. The one-electron oxidation of phenol by [PVvW11O40]4 − (HPA) generated phenoxyl radical (ArO·) and one-electron reduced heteropoly blue [PVIvW11O40]4 −. The resultant V(V)-substituted heteropolyoxometalates could act as one or two electron or even several electron acceptors. In the acidic medium, ArOH was the reactive species, and the ArOH-HPA reaction was carried out through a separated-concerted proton-electron transfer (CPET) mechanism in which water acted as a proton acceptor (Citation93).
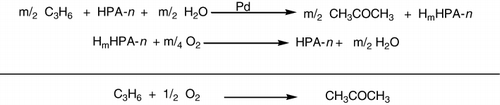
Oxidation of propene to acetone in water solutions in the presence of homogeneous catalysts (Pd2 + + HPA-n, where HPA-n= H3 + nPVnMo12 − nO40, n= 1–4) was studied (). The kinetics of the highly selective oxidation of propene to acetone in the presence of homogeneous catalysts Pd2+ + HPA-n (n =1–4) did not depend on the number x of vanadium atoms in the HPA-n molecule. The rate did not depend on the HPA-n concentration and the acidity of the catalyst solution (Citation94, Citation95).
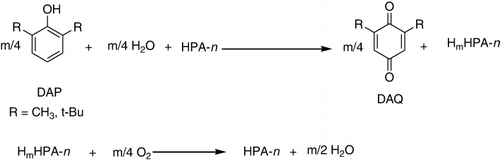
A new method was proposed for obtaining 2,6-dialkyl-1,4-benzoquinones by oxidation of 2,6-dimethyl and 2,6-ditertbutylphenols by molecular oxygen in a two-phase “water–organic” system and in the presence of P–Mo–V HPAs (). Unlike one-phase systems, two-phase catalytic systems, where HPA-n catalyst was in the water phase, and a substrate and the product were in the organic phase, and it had the advantages of simple separation of the reaction products and increased catalyst selectivity because of lessening instantaneous concentration of the substrate interacting with HPA-n. For reaction (2), the highest activity was exhibited by HPA-n where n = 6. On addition of cations to HPA-n, the selectivity of DAP oxidation decreased because of the decreasing oxidative potential of HPA-n (Citation96).
Vitamin K3, 2-methyl-1,4-naphthoquinone (menadione, MD), is the starting reagent in the synthesis of all vitamins of the K-group. Mo-V-phosphoric HPAs of the Keggin structure H 3 + nPMo12-nVnO40 (HPA-n) and their acidic salts were found to be efficient catalysts for 2-methyl-1-naphthol (MN) oxidation by dioxygen to 2-methyl-1,4-naphthoquinone(menadione or vitamin K3) (). Among all HPA-n, HPA-2 had the maximum selectivity, whereas the maximum reaction rate occurs for HPA-4. The most favorable molar ratio was found to lie within 3 < [HPA-n]:[MN] < 4. This mechanism assumed that HPA-n, similar to the VO2-ion, was a single-electron oxidant. The complete MN oxidation to MD required four electron transfers from MN to HPA-n (Citation97).
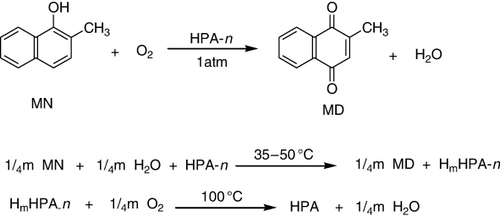
Molybdo(vanado)phosphoric HPAs of Keggin structure supported on oxide supports (SiO2, TiO2, and Al2O3) were used as catalysts for ethane to acetic acid oxidation in the range of reaction temperature from 250 to 400 °C. Vanadium atoms introduced into Keggin structure improved oxidative activity of catalytic system, while vanadyl groups exchanged into cationic position diminished ethane conversion. The nature of support (acidic or base centers on the surface) influenced both ethane conversion and distribution of products. Ethane oxidation over silica- and titania-supported HPMoVn was due to the presence of regular or defected Keggin structure, while low catalytic performance on alumina-supported samples was recognized to mix Mo–V–P oxides formed as a result of HPMoVn decomposition. Presence of water vapors in the reaction mixture was indispensable for both catalysts surface modification and for acetic acid desorption (Citation98).
Molybdovanadophosphoric acid (H3 + nPMo12 − nVnO40; n = l–2) and its salts were also found to be effective for the oxidative dehydrogenation of isobutyric acid (IBA) to methacrylic acid (MAA). V2O5-P2O5 binary oxides were established to be effective, much like vanadium-containing heteropoly compounds. The products were MAA, acetone, propylene, and carbon oxides. The formation of propylene was much greater than in the case of the H5PMo10V2O40 catalyst. The reaction was carried out in the presence of 16% water vapor. Oxidation activity decreased by one-fourth, but the selectivity remained unchanged (Citation99).
The oxidation reaction was done utilizing 34% of hydrogen peroxide in water catalyzed by some W- and Mo-based heteropolyoxometalates. Findings showed that dodecatungstophosphoric acid (K4SiW9Mo2O39), H3PW12O40, was the most efficient catalyst. This methodology might be an alternative for ecofriendly green oxidation. Water turned out to be the best solvent for these oxygenation transformations.
Other organic solvents such as chloroform, ethanol, and acetone were not suitable for the oxygenation system. These results showed that phenylenediamine was the most reactive substrate, whereas electron deficient 4-NO2-aniline led to only 32% of the nitroso product (Citation100, Citation101).
Partially reduced multicomponent heteropoly compound catalysts with the Keggin structure were prepared and applied for the selective oxidation of propane. The influence of the pore diameter of the support on the reaction was also tested. Results showed that the partially reduced HPA catalyst exhibited the highest activity for the selective oxidation of propane under the optimum reaction conditions: T = 390 °C, C3H8:O2:H2O:N2 = 1:2:2:5 (mol/mol) and space velocity = 1500 h−1. The maximum conversion of propane and the maximum yield of acrylic acid were 38 and 14.8%, respectively (Citation94).
7. Multicomponent reactions (MCRs)
Among a series of insoluble salts of Keggin heteropoly compounds for the Mannich-type reaction of benzaldehyde, aniline, and cyclohexanone, Cs2.5H0.5PW12O40 showed excellent catalytic activity (a). It afforded structurally divers ß-amino ketones with major anti-diastereoselectivity. This salt was prepared by exchanging HPA protons with large alkali metal cations such as K↰, Cs↰, or [(n-Bu)4N]↰ to get insoluble salts (Citation102).
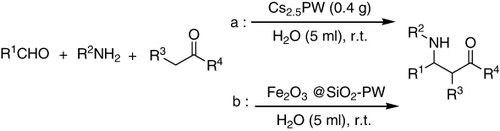
A series of insoluble salts of Keggin heteropoly compounds were prepared and used as catalysts for Mannich-type reaction of benzaldehyde, aniline, and cyclohexanone in water. Among them, Cs2.5H0.5PW12O40 showed excellent catalytic activity. This fast procedure afforded structurally divers β-amino ketones with major anti-diastereoselectivity (Citation102).
In the presence of cationic surfactants, cetylpyridinium bromide (CPB) and cetyltrimethylammonium bromide (CTAB), the product yield was declined. It may be due to the positively charged micellar interface, which afforded an unfavorable electrostatic interaction with protonated imine present in water solution. Anionic surfactants gave the desired product in a modest yield. Additionally, these surfactants produced colloidal dispersion by emulsion formation as a result of which, phase separation was difficult (Citation102).
Several electron-rich aromatic aldehydes led to the desired products in good yield. Though, under the same reaction conditions aliphatic aldehydes, such as isobutyaldehyde, gave a mixture due to enamine formation; the desired product was achieved in low yield amines with electron-withdrawing groups, such as 4-chloroaniline and 3,4-dichloroaniline, that gave the desired product in good yields (Citation18).
Biginelli three-component condensation of an aldehyde, β-keto ester, and urea proceeded smoothly on the surface of the silver salt of HPA, i.e., Ag3PW12O40 in water afforded the corresponding 3,4-dihydropyrimidinones in high-to-quantitative yields under mild conditions (a). The heterogeneous solid acid provided ease of separation of the catalyst and isolation of the products. Most importantly, aromatic aldehydes carrying either electron-donating or -withdrawing substituents reacted well under the reaction conditions to give the corresponding dihydropyrimidinones in high-to-quantitative yields with high purity. This method was applicable to a wide range of substrates, including aromatic, aliphatic, α,β-unsaturated, and heterocyclic aldehydes, and provided a variety of biologically relevant dihydropyrimidinones in high to quantitative yields after short reaction times (Citation103).
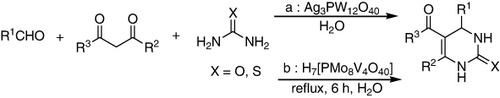
The use of HPAs as a catalyst for the synthesis of various substituted 3,4-dihydropyrimidin-2(1H)-ones using Biginelli protocol in water was reported. The best conditions to prepare the dihydropyrimidinones were achieved when both urea (or thiourea), ethylaceto acetate, and aldehyde were heated under reflux conditions in the presence of 0.03 mmol of HPA catalyst and water as green solvent for 6 h. This method was effective with a variety of substituted aromatic aldehydes independent of the nature of the substituents in the aromatic ring, representing an improvement to the classical Biginelli's methodologies. Substituents with electron-withdrawing groups gave relatively higher yields. The yields of the reaction in the presence of water were greater, and the reaction times were generally shorter than the conventional methods. The HPAs of the series H3 + nPMo12-nVnO40 (n=1–4) showed good to excellent catalytic behaviors (Citation32).
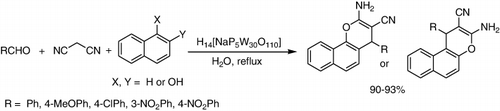
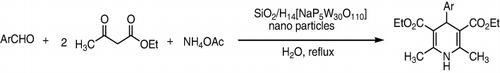
H14[NaP5W30O110] was used successfully in reaction of aldehydes, malononitrile alfa, and beta naphtoles for the synthesis of 2-amino-4H-chromenes in water. The products were obtained in very good yields (90–93%) () (Citation104).
A highly efficient one-pot synthesis of 1,8-dioxo-octahydroxanthenes from dimedone and various aromatic aldehydes under reflux conditions in water, catalyzed by silica-supported preyssler nano particles (SPNP) was reported. As seen, ethanol and water were favorable solvents for this synthesis (). As it was observable, by using this nanocatalyst, the aromatic aldehydes containing both electron-donating and electron-withdrawing groups afforded the products of excellent yields. Although, electron-withdrawing groups were slightly better. The effect of various solvents on the rate of the reaction was also studied and shown in (Citation105).
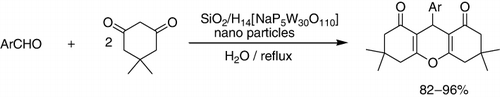
Table 3. The effect of solvent on the rate of one-pot synthesis of 1,8-dioxooctahydroxanthenes.
Synthesis of 1,4-dihydropyrano[2,3-c] pyrazole and pyrano[2,3-d]pyrimidine derivatives was also reported via three-component, one-pot condensation of 3-methyl-1-phenyl-1H-pyrazol-5(4H)-one or barbituric acid, aldehydes, and malononitrile in the presence of a catalytic amount of Preyssler-type HPA in water or ethanol under refluxing conditions (). It was found that there was a correlation between reaction rate and the temperature, and the best results were obtained in refluxing water and ethanol () (Citation106).
![Scheme 22. Synthesis of 1,4-dihydropyrano[2,3-c] pyrazole and pyrano[2,3-d]pyrimidine derivatives via three-component condensation.](/cms/asset/c8116fe0-ed1b-4580-a5b0-bbe89ee00f02/tgcl_a_846415_f0022_b.jpg)
Table 4. The correlation between reaction rate and temperature in the synthesis of pyrazole and pyrimidine derivatives.
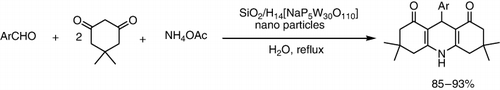
SPNP was used as a reusable and green solid acid catalyst for the synthesis of 1,8-dioxodecahydroacridines via three component reaction of aryl aldehydes, dimedone, and ammonium acetate in water and reflux conditions. Ethanol and water were favorable solvents. But water was chosen as a greener solvent. It could be seen that by using this nanocatalyst, the aromatic aldehydes containing electron-donating and electron-withdrawing groups afforded the products with excellent yields () (Citation107).
A variety of fused 1,4-dihydropyridine derivatives were synthesized from the reaction of different aryl aldehydes, ethyl acetoacetate, and ammonium acetate in water in the presence of a catalytic amount of Preyssler HPA. SPNP was also found to be effective for this synthesis () (Citation108).
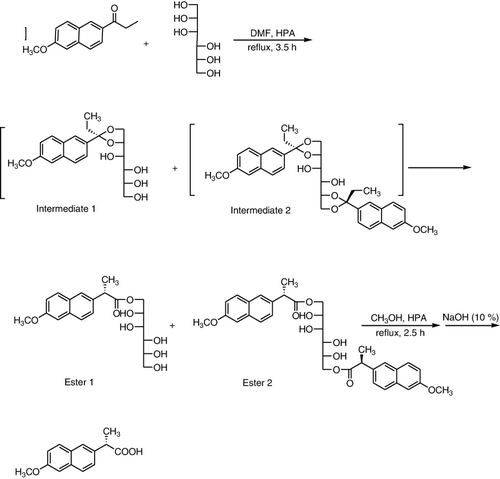
A simple, clean, and environmentally benign route to the enantioselective synthesis of (S)-2-(6-methoxynaphtalen-2-yl)propanoic acid, (S)-Naproxen was described by using Preyssler HPA, H14[NaP5W30O110], as a green and reusable catalyst in water and in the presence of 1-(6-methoxynaphthalen-2-yl)propan-1-one, D-mannitol. The products were obtained in very good yields () (Citation109).
The three-component synthesis of 2-amino-5-oxo-dihydropyrano[3,2-c]chromene derivatives was reported by condensing 4-hydroxycoumarin, aldehydes, and alkylnitriles using a catalytic amount of H6P2W18O62.18H2O in aqueous ethanol under heating conditions (). Compared with H6P2W18O62.18H2O, using H14[NaP5W30O110] or NH2SO3H as catalyst in the model reaction under refluxing conditions in aqueous ethanol (50:50) afforded less yields (Citation35).
![Scheme 26. Three-component synthesis of 2-amino-5-oxo-dihydropyrano[3,2-c]chromene derivatives.](/cms/asset/c3db21d4-bfd6-4d03-b85d-f34696b9d8b3/tgcl_a_846415_f0026_b.jpg)
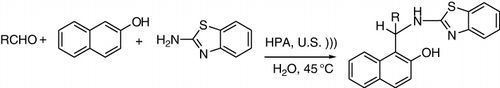
A one-pot condensation of 2-aminobenzothiazole, 2-naphthol, and appropriate aldehydes catalyzed by HPA in an aqueous medium afforded the Mannich adduct 2′-aminobenzothiazolomethylnaphthols at 45 °C under ultrasound irradiation (). The reaction worked well with electron-withdrawing (NO2, Cl, CN) as well as electron-donating (Me, MeO) groups, giving various derivatives in 80–92% yields. The results of the extension of the reaction under the ultrasonic irradiation showed that in the case of 3-amino-1,2,4-triazole, the reaction did not proceed, and 2-aminobenzimidazole provided a very low yield after 24 h and increasing the temperature from 45 °C to 90 °C (Citation110).
An improved procedure for the synthesis of oxindoles derivatives was developed via the electrophilic substitution reaction of indoles with various isatins in the presence of a Wells–Dawson tungsten HPA, H6P2W18O62 (5mol%), in water (). It gave 3,3-di(indolyl)-oxindole in 95% yield for 30 min. In all conversions, electrophilic activation occurred only at the carbonyl of the 3-position. The carbonyl at the 2-position was unreactive, and this might be due to stabilization by the indole nitrogen (Citation111).
HPAs catalyzed the electrophilic substitutions of indole and substituted indole with a variety of aldehydes and ketones in water to afford bis(indolyl)methanes at room temperature with excellent yields. This method was also highly chemoselective for aldehydes (). In addition, as shown in , H3PMo12O40 and H3PW12O40 were found out to have the same results for this reaction in water and other solvents (Citation112).
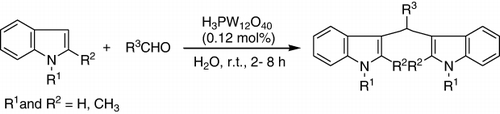
Table 5. Effect of different HPAs and solvents on electrophilic substitution of indoles.
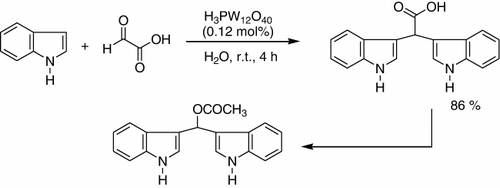
The electreophilic reaction of indole was carried out with glyoxylic acid (50% solution in water), and the expected product was obtained in good yields under these reaction conditions () (Citation112).
8. Miscellaneous
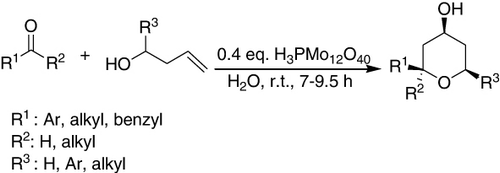
H3PMo12O40 was found to catalyze the Prins cyclization of homoallylic alcohols with aldehydes at room temperature to provide tetrahydropyran-4-ol derivatives in 80–90% yields with all cis-selectivity (). Only cyclic ketones could give spirocyclic products (Citation113).
HPA was found to be an effective catalyst for ring opening reaction of epoxides with various aromatic amines to produce the corresponding β-amino alcohols in moderate to excellent yields in water (). Aniline, 4-methoxy aniline, 4-chloroaniline, 4-bromoaniline, and 4-isopropyl aniline reacted well with aliphatic epoxide such as glycidyl phenyl ether, glycidyl isopropyl ether, 1,2-epoxy butane, 1,2-epoxy propane, and allyl 2,3-epoxypropyl ether to give the corresponding amino alcohols in good yields (Citation114).
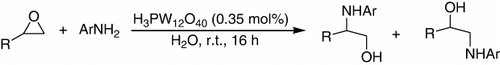
Cyclohexene oxide was treated with 1 equivalent of aniline at room temperature, and the desired product obtained in 54% yield within 48 h in water as the sole solvent. The best results were obtained when using 0.01 mol% of HPAs such as H3PMo12O40 (, entry 14) and H3PW12O40 (, entry 15) at room temperature for 2 h (Citation114).
Table 6. Effect of different catalysts and solvents in ring opening reaction of epoxides with various aromatic amines.
Interestingly, addition of CTAB was not effectual, but addition of sodium dodecyl sulfate (SDS) enhanced the yield of the reaction (Citation114).
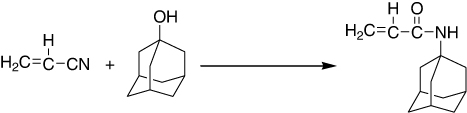
The catalytic synthesis of N-adamantylacrylamide from acrylonitrile and 1-adamantanol was studied over various solid and liquid acids. Solid acids such as Cs2.5H0.5PW12O40, Amberlyst 15, Nafion, and Nafion–SiO2 composite gave yields higher than 97% at 373 K and were superior in yield to liquid acids like p-toluenesulfonic acid, H3PW12O40, and H2SO4 (). It was further verified that Cs2.5H0.5PW12O40 exhibited the highest catalytic performance for this reaction in the presence of excess water. Together with the hydrophobicity, the strong acidity of Cs2.5 was responsible for the high catalytic act for the Ritter-type reaction in the presence of excess water (Citation115).
The decomposition of hydroperfluorocarboxylic acids [H-PFCAs; HCnF2nCOOH (n = 4 and 6)] induced by HPA photocatalyst H4SiW12O40 in water was investigated. H-PFCAs were not decomposed by irradiation with ultraviolet (UV)–visible light (>290 nm) in the absence of H4SiW12O40. In contrast, UV–visible light irradiation of H-PFCAs in the presence of H4SiW12O40 efficiently decomposed H-PFCAs to F − and CO2, at relatively high pH (up to 5.2), at which pH, the conventional HPA photocatalyst H3PW12O40 couldn't be used (Citation116).
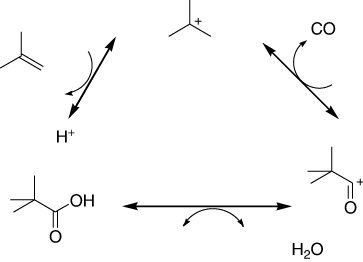
Tertiary carboxylic acids were manufactured by hydrocarboxylation of branched alkenes in the presence of CO, water, and a corrosive homogeneous acid. Acidic ion-exchange resins also catalyzed the hydrocarboxylation reaction when operated under specific conditions. The best resins included the Nafion NR50 as well as styrene-based sulfonated ones with high content of acid sites (>4.5 mmol/g) and moderate divinyl-benzene content (8–12%), such as the Amberlyst 15 and 36. Unlike, inorganic solid acids, such as zeolites, sulfated zirconia or HPAs showed minor activity () (Citation117).
The synthesis of diphenylmethane (DPM) from benzene and formalin (a mixture of formaldehyde and water) or paraformaldehyde was studied using a range of solid acids. The reaction between benzene and paraformaldehyde (oligomers offormaldehyde) went on readily at 140 °C on typical solid acids. The activity of a silica-composite of polymer resin, Aciplex-SiO2, was found to be greater to that of other solid acids such as zeolites (HY, H-ZSM-5, and beta-zeolite), heteropoly compounds (H3PW12O40 and Cs2.5H0.5PW12O40), and the other polymer resins (Nafion-H, Nafion-SiO2, and Amberlyst). The addition of water to the reactant mixture decreased the activity of Aciplex-SiO2 greatly (Citation118).
One-step synthesis of methylmethacrylate (MMA), using heteropoly compounds as catalysts, was demonstrated by feeding a mixture of methacrolein (MAL), air, water, nitrogen, and MeOH. The HPCs with different counterions, such as H1.7Cs1.5Cu0.25As0.1PMo11VO40 (CsPMo11VO40), H1.7La0.7Cu0.25As0.1PMo11VO40 (LaPMo11VO40) and H1.7K1.5Cu0.25As0.1PMo11VO40 (KPMo11VO40), were studied in oxidation of MAL, esterification of MAA and one-step synthesis of MMA from MAL, correspondingly. The HPCs catalysts were not only applied to the both oxidation and esterification reactions but could also be used in the coupling reactions from MAL to MMA. When the reaction temperature was at 300 °C, space velocity was 400 h −1, and MAL/air/N2/H2O/MeOH was equal to 5/50/22.5/10/12.5, the 44.6% selectivity of MMA and 45.7% selectivity of MAA with 93.3% conversion of MAL could be achieved over CsPMo11VO40 in coupling reaction. Cs-containing HPAs showed better structural stability and catalytic performances (Citation119).
![]()
Methylmethacrylate (MMA), Methacrolein (MAL)

Methacrylic acid (MAA)
The solid catalyst which was prepared by noncovalent fixing of the tungstophosphate anion on chemically modified hydrophobic mesoporous silica gel was found to catalyze the selective epoxidation of a variety of olefins by 15% aqueous H2O2 without using organic solvents (). It implied that different types of silanol groups on the silica gel surface had different reactivities with either Ph3SiOEt or Me2NCH (OCH2Ph)2. Catalyst surface could play an essential role in the efficient and selective epoxidation. Most probably, high hydrophobicity of both the silica gel surface and the counter cation [R] might favor strong binding of the catalysis center [PW12O40]3- on the silica gel surface and also easy access of the hydrophobic reactant 1-octene to the catalysis center. Additional improvements of the solid catalyst could be achieved by fine tuning both the silica-surface morphology (e.g., porosity) and the surface-modification agents (Citation120).
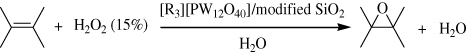
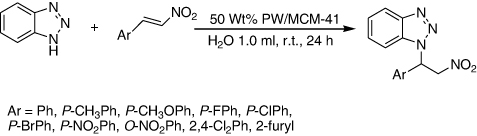
MCM-41 supported HPAs catalysts were synthesized, characterized, and their catalytic activity was evaluated in an aza-Michael addition reaction between nitroolefins and benzotriazole in water at room temperature. 50 wt% PW/MCM-41 showed the highest activity (up to 96% yield) (). The best result was achieved when using 50 wt% PW/MCM-41 (0.005g) in water at room temperature for 24 h, and the yield was up to 93%. The electron-donating as well as electron-withdrawing substituents on the aromatic ring were endured under these conditions and the reactions proceeded in good yields of 71–96%. This method had the advantages of simple manipulation and high turnover, which could be useful for the syntheses of the nitrogen-containing heterocycles (Citation121).
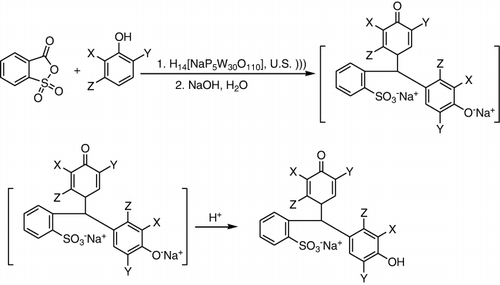
A new synthesis of hydroxytriarylmethane derived from the reaction of 2-sulfobenzoic anhydride and phenols in the presence of HPAs under ultrasonic irradiation was reported (). The highest yield of the products was achieved when H14[NaP5W30O110] was used as the catalyst. Comparison of the catalyst showed that, in all cases, the supported Preyssler catalyst was less active than the nonsupported one. In supported type, there were poly anion-support interactions of an acid-base nature. Some protons of the poly acid and some basic sites of the support (e.g., hydroxyl groups) could interact. This would lead to lessened availability of hydrogens because of this extra ion interaction. The catalytic activity was increased in the following order: H14[NaP5W30O110] > H3[PW12O40] > H4[SiW12O40]. Phenols with electron-withdrawing groups gave hydroxytriarylmethane derivatives in low yields. As ultrasound generated strong turbulence and microscale liquid circulation currents, uniform mixing at microlevel occurred and the mass transfer resistances was eliminated. This explanation could justify the shorter reaction time observed for reactions under ultrasonic condition (Citation122).
The H5PV2Mo10O40 catalyzed the oxidation of thiols to their homodisulfides using hydrogen peroxide as the oxidant under mild conditions. In EtOH + H2O, the reaction was fulfilled within 2 h (). The other solvents provided moderate yields with longer reaction times, except CH3CN and CH3NO2, in which the yields were very low even after 5 h. One heteroaromatic thiol, i.e., pyridine-2-thiol, was successfully oxidized in good yield as well as benzylthiol as a benzylic aliphatic representative. In general, the yields were very good (>75%) to excellent (>90%) with no noticeable relationship between the aromatic substituent and yield (Citation123).
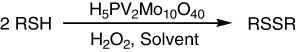
The direct synthesis of hydrogen peroxide from molecular H2 and O2, signified a green and economic alternative to the current anthraquinone process applied for the industrial production of H2O2. It was shown that Au–Pd-exchanged and supported Cs-containing HPA catalysts, with the Keggin structure considerably more effective in attaining high H2O2 yields in the absence of acid or halide additives than formerly reported catalysts. The Au–Pd-exchanged Cs-HPA catalysts also confirmed superior H2O2 synthesis activity under challenging conditions (ambient temperature, water-only solvent, and CO2-free reaction gas). Au had a crucial role in achieving the superior performance of those catalysts. The HPAs restricted the subsequential hydrogenation/decomposition of H2O2 (Citation124).
9. Conclusion
HPAs were applied as catalysts in aqueous media to catalyze diversity of organic reactions consisting condensation, carbonylation, hydrolysis, hydration, oxidation, multicomponent, and many more reactions competently. As reported here, water can have the role of substrate (hydration of olefins) or product (dehydration of alcohols). Sometimes only the presence of water, without the direct participation in the reaction, as it was observed in the etherification, is enough to modify the mechanism of reaction by influencing the secondary structure of HPAs. Unlike many solid acids whose catalytic activities are usually suppressed significantly in the presence of water, HPAs are water-tolerant as well as having very high solubilities in polar solvents such as water, lower alcohols, ketones, ethers, esters, etc. The catalytic activity of HPAs may be enhanced by different support. It was demonstrated that in contrast to highly water soluble acidic forms, some acidic salts of HPAs are very water-tolerant catalysts due to their hydrophobic nature and used for hydration of olefins and hydrolysis of esters, etc. Moreover, as far as most organic products are insoluble in water, a very easy workup can be realized that does not require more organic solvents.
Summarizing, water plays the important and sometimes unorthodox role in many organic reactions with contribution of HPA as the catalysts.
References
- Herrerías, C.I.; Yao, X.; Li, Z.; Li, C.-J. Chem. Rev. 2007, 107, 2546–2562. 10.1021/cr050980b
- Anastas, P.; Eghbali, N. Chem. Soc. Rev. 2009, 39, 301–312. 10.1039/b918763b
- Li, C.-J.; Trost, B.M. Proc. Natl. Acad. Sci. 2008, 105, 13197–13202. 10.1073/pnas.0804348105
- Anastas, P.T.; Kirchhoff, M.M. Acc. Chem. Res. 2002, 35, 686–694. 10.1021/ar010065m
- Li, C.-J.; Chen, L. Chem. Soc. Rev. 2005, 35, 68–82. 10.1039/b507207g
- Li, C.-J. Chem. Rev. 2005, 105, 3095–3166. 10.1021/cr030009u
- Okuhara, T. Chem. Rev. 2002, 102, 3641–3666. 10.1021/cr0103569
- Skouta, R. Green Chem. Lett. Rev. 2009, 2, 121–156. 10.1080/17518250903230001
- Li, C.J. Chem. Rev. 1993, 93, 2023–2035. 10.1021/cr00022a004
- Gu, Y. Green Chem. 2012, 14, 2091–2128.
- Zong, Y.X.; Zhao, Y.; Luo, W.C.; Yu, X.H.; Wang, J.K.; Pan, Y. Chin. Chem. Lett. 2010, 21, 778–781. 10.1016/j.cclet.2010.03.022
- Heravi, M. M.; Sadjadi, S. J. Iran. Chem. Soc. 2009, 6, 1–54. 10.1007/BF03246501
- Izumi, Y.; Hasebe, R.; Urabe, K. J. Catal. 1983, 84, 402–409. 10.1016/0021-9517(83)90011-8
- Katsoulis, D.E. Chem. Rev. 1998, 98, 359–388. 10.1021/cr960398a
- Bamoharram, F.F.; Ahmadpour, A.; Heravi, M.M.; Ayati, A.; Rashidi, H.; Tanhaei, B. Synth. React. Inorg. Met.-Org. Nano-Met. Chem. 2012, 42, 209–230. 10.1080/15533174.2011.609849
- Klemperer, W.G.; Wall, C.G. Chem. Rev. 1998, 98, 297–306. 10.1021/cr9603993
- Weinstock, I.A. Chem. Rev. 1998, 98, 113–170. 10.1021/cr9703414
- Azizi, N.; Torkiyan, L.; Saidi, M.R. Org. Lett. 2006, 8, 2079–2082. 10.1021/ol060498v
- Firouzabadi, H.; Jafari, A.A. J. Iran. Chem. Soci. 2005, 2, 85–114. 10.1007/BF03247201
- Micek-Ilnicka, A. J. Mol. Catal. 2009, 308, 1–14. 10.1016/j.molcata.2009.04.003
- Yamada, I.; Saito, S.; Watanabe, H.; Kubota, T.; Google Patents 2011.
- Mizuno, N.; Misono, M. Chem. Rev. 1998, 98, 199–217. 10.1021/cr960401q
- Okuhara, T.; Mizuno, N.; Misono, M. Adv. Catal. 1996, 41, 113–252.
- Reddy, K.M.; Lingaiah, N.; Prasad, P.S.S.; Suryanarayana, I. J. Sol. Chem. 2005, 35, 407–423. 10.1007/s10953-006-9025-1
- Zhang, F.; Yuan, C.; Wang, J.; Kong, Y.; Zhub, H.; Wang, C. J. Mol. Catal. A: Chem. 2006, 247, 130–137. 10.1016/j.molcata.2005.11.036
- Ghanbari-Siahkali, A.; Philippou, A.; Dwyer, J.; Anderson, M.W. Appl. Catal., A. 2000, 192, 57–69. 10.1016/S0926-860X(99)00333-6
- Ghosh, A.K.; Moffat, J.B. J. Catal. 1986, 101, 238–245. 10.1016/0021-9517(86)90249-6
- Matachowski, L.; Zie, A.; Zembala, M.; Drelinkiewicz, A. Catal. Lett. 2009, 133, 49–62. 10.1007/s10562-009-0149-y
- Timofeeva, M.N. Appl. Catal., A. 2003, 256, 19–35. 10.1016/S0926-860X(03)00386-7
- Odyakov, V.F.; Zhizhina, E.G.; Maksimovskaya, R.I.; Matveev, K.I. Kinet. Catal. 1995, 36, 733–738.
- Okuhara, T.; Nishimura, T.; Watanabe, H.; Misono, M. J. Mol. Catal. 1992, 74, 247–256.
- Gharib, A.; Jahangir, M.; Roshani, M.; Scheeren, J.H.W.; Moghadaszadeh, S.; Rezaee, A. GU J. Sci. 2012, 25, 823–833.
- Heravi, M.M.; Ghodsa, A.; Derikvanda, F.; Bakhtiaria, K.; Bamoharram, F.F. J. Iran. Chem. Soc. 2010, 7, 615–620. 10.1007/BF03246049
- Srivani, A.; Prasad, P.S.S.; Lingaiah, N. Catal. Lett. 2012, 142, 389–396. 10.1007/s10562-012-0778-4
- Heravi, M.M.; Jani, B.A.; Derikvand, F.; Bamoharram, F.F.; Oskooie, H.A. Catal. Commun. 2008, 10, 272–275. 10.1016/j.catcom.2008.08.023
- Javid, A.; Heravi, M.M.; Bamoharram, F.F. E-J. Chem. 2011, 8, 910–916. 10.1155/2011/980242
- Nakato, T.; Toyoshi, Y.; Kimura, M.; Okuhara, T. Catal. Today 1999, 52, 23–28. 10.1016/S0920-5861(99)00059-0
- Dastan, A.; Kulkarni, A.; Torok, B. Green Chem. 2012, 14, 17–37. 10.1039/c1gc15837f
- Kozhevnikov, I.V. Appl. Catal. A: General. 2003, 256, 3–18. 10.1016/S0926-860X(03)00406-X
- Kozhevnikov, I.V. Chem. Rev. 1998, 98, 171–198. 10.1021/cr960400y
- Klemperer, W.G.; Wall, C.G. Chem. Rev. 1998, 98, 297–306. 10.1021/cr9603993
- Lakshminarayana, G.; Nogami, M. Electrochim. Acta. 2009, 54, 4731–4740. 10.1016/j.electacta.2009.04.007
- Safronova, E.Y.; Stenina, I.A.; Yaroslavtsev, A.B. Russ. J. Inorg. Chem. 2010, 55, 13–17. 10.1134/S0036023610010031
- Mukai, S.R.; Masuda, T.; Ogino, I.; Hashimoto, K. Appl. Catal. A. 1997, 165, 219–226.
- Vázquez, P.; Blanco, M.; Cáceres, C. Catal. Lett. 1999, 60, 205–215. 10.1023/A:1019071410838
- Wang, J.-P.; Zhao, J.-W.; Li, S.-Z.; Niu, J.-Y. J. Coord. Chem. 2006, 59, 597–605. 10.1080/00958970500361189
- Caliman, E.; Dias, J.A.; Dias, S.C.L.; Prado, A.G.S. Catal. Today. 2005, 107–108, 816–825. 10.1016/j.cattod.2005.07.102
- Chen, L.; Wang, X.; Guo, X.; Guo, H.; Liu, H.O.; Chen, Y. Chem. Eng. Sci. 2007, 62, 4469–4478. 10.1016/j.ces.2007.05.013
- Pai, Z.P.; Kochubey, D.I.; Berdnikova, P.V.; Kanazhevskiy, V.V.; Prikhod'ko, I.Y.; Chesalov, Y.A. J. Mol. Catal., A. 2007, 332, 122–127.
- Botella, P.; Solsona, B.; López Nieto, J.M.; Concepción, P.; Jordá, J.L.; Doménech-Carbó, M.T. Catal. Today. 2010, 158, 162–169. 10.1016/j.cattod.2010.05.024
- Zhen, B.; Li, H.; Jiao, Q.; Li, Y.; Wu, Q.; Zhang, Y. Ind. Eng. Chem. Res. 2012, 51, 10374–10380. 10.1021/ie301453c
- Sunita, G.; Devassy, B.M.; Vinu, A.; Sawant, D.P.; Balasubramanian, V.V.; Halligudi, S.B. Catal. Commun. 2008, 9, 696–702. 10.1016/j.catcom.2007.08.007
- Yaqoob, M.; Nabi, A.; Worsfold, P.J. Anal. Chim. Acta. 2004, 510, 213–218. 10.1016/j.aca.2003.12.070
- Xue, Y.; Zheng, X.; Li, G. Talanta. 2007, 72, 450–456. 10.1016/j.talanta.2006.11.003
- López Carreto, M.; Sicilia, D.; Rubio, S.; Pérez-Bendito, D. Anal. Chim. Acta. 1993, 283, 481–488. 10.1016/0003-2670(93)85260-Q
- Abbas, D.; Noorel, M. Anal. Lett. 2003, 36, 1231–1244. 10.1081/AL-120020155
- Metelka, R.; Slavíková, S.; Vytřas, K. Talanta. 2002, 58, 147–151. 10.1016/S0039-9140(02)00263-1
- Dasgupta, P.K.; Huang, H.; Zhang, G.; Cobb, G.P. Talanta. 2002, 58, 153–164.
- Paul, J. Anal. Chim. Acta. 1966, 35, 200–205. 10.1016/S0003-2670(01)81651-2
- Kanke, M.; Kumamaru, T.; Sakai, K.; Yamamoto, Y. Anal. Chim. Acta 1991, 247, 13–18. 10.1016/S0003-2670(00)83046-9
- Tikhomirova, T.I.; Kuznetsov, M.V.; Dubovik, D.B.; Tsizin, G.I.; Zolotov, Y.A. J. Anal. Chem. 2000, 55, 846–850. 10.1007/BF02757847
- Attiq-Ur-Rehman; Yaqoob, M.; Waseem, A.; Nabi, A. Int. J. Environ. Anal. Chem. 2008, 88, 603–612. 10.1080/03067310801912103
- Shemirani, F.; Baghdadi, M.; Ramezani, M. Talanta. 2005, 65, 882–887. 10.1016/j.talanta.2004.08.009
- Yamamoto, Y.; Kumamaru, T.; Hayashi, Y.; Kanke, M.; Matsui, A. Talanta. 1972, 19, 1633–1638. 10.1016/0039-9140(72)80236-4
- Yaqoob, M.; Nabi, A.; Worsfold, P.J. Anal. Chim. Acta. 2004, 519, 137–142. 10.1016/j.aca.2004.05.076
- Ramachandran, R.; Gupta, P.K. Anal. Chim. Acta. 1985, 172, 307–311. 10.1016/S0003-2670(00)82621-5
- Medvetskii, A.V.; Tikhomirova, T.I.; Smolenkov, A.D.; Shapovalova, E.N.; Shpigun, O.A. J. Anal. Chem. 2007, 62, 213–218. 10.1134/S1061934807030033
- Linares, P.; De Castro, M.D.L.; Valcarcel, M. Talanta. 1986, 33, 889–893. 10.1016/0039-9140(86)80218-1
- Nabi, S.A.; Bushra, R.; Shahadat, M. Toxicol. Environ. Chem. 2012, 94, 468–481. 10.1080/02772248.2011.651084
- Peretrukhin, V.F.; Maslennikov, A.G.; Tsivadze, A.Y.; Delegard, C.H.; Yusov, A.B.; Shilov, V.P.; Bessonov, A.A.; German, K.E.; Fedoseev, A.M.; Kazanskii, L.P.; Budanova, N.Y.; Kareta, A.V.; Gogolev, A.V.; Gedgovd, K.N.; Bulatov, G.S. Prot. Met. 2008, 44, 211–232. 10.1134/S0033173208030016
- Bogoslovskii, V.V.; Pizelkin, I.P. J. Anal. Chem. 2006, 61, 1121–1123. 10.1134/S1061934806110141
- Jalil, P.A. J. Anal. Appl. Pyrolysis. 2002, 65, 185–195. 10.1016/S0165-2370(01)00193-0
- Zong, Y.X.; Zhao, Y.; Luo, W.C.; Yu, X.H.; Wang, J.K.; Pan, Y. Chin. Chem. Lett. 2010, 21, 778–781. 10.1016/j.cclet.2010.03.022
- Kumar, M.A.; Krishna, A.B.; Babu, B.H.; Reddy, C.B.; Reddy, C.S. Syn. Commun. 2008, 38, 3456–3464. 10.1080/00397910802157843
- Huang, T.K.; Shi, L.; Wang, R.; Guo, X.Z.; Lu, X.X. Chin. Chem. Lett. 2009, 20, 161–164. 10.1016/j.cclet.2008.10.048
- Azizi, N.; Arynasab, F.; Saidi, M.R. Org. Biomol. Chem. 2006, 4, 4275–4277. 10.1039/b610263h
- Sun, Y.; Wang, H.; Shen, J.; Liu, H.; Liu, Z. Catal. Commun. 2009, 10, 678–681. 10.1016/j.catcom.2008.11.015
- Qian, Q.; Zhang, S.; Yuan, G. Catal. Commun. 2007, 8, 483–487. 10.1016/j.catcom.2006.07.025
- Kimura, M.; Nakato, T.; Okuhara, T. Appl. Catal., A. 1997, 165, 227–240. 10.1016/S0926-860X(97)00204-4
- Okuhara, T.; Kimura, M.; Kawai, T.; Xu, Z.; Nakato, T. Catal. Today. 1998, 45, 73–77. 10.1016/S0920-5861(98)00251-X
- Kimura, M.; Nakato, T.; Okuhara, T. Appl. Catal. A., Gen. 1997, 165, 227–240. 10.1016/S0926-860X(97)00204-4
- Okuhara, T. Catal. Today. 2002, 73, 167–176. 10.1016/S0920-5861(01)00509-0
- Okuhara, T.; Kimura, M.; Kawai, T.; Xu, Z.; Nakato, T. Catal. Today. 1998, 45, 73–77. 10.1016/S0920-5861(98)00251-X
- Okuhara, T.; Kimura, M.; Nakato, T. Appl. Catal., A. 1997, 155, 9–13. 10.1016/S0926-860X(97)00062-8
- Liu, H.; Xue, N.; Peng, L.; Guo, X.; Ding, W.; Chen, Y. Catal. Commun. 2009, 10, 1734–1737. 10.1016/j.catcom.2009.05.023
- Misono, M.; Ono, I.; Koyano, G.; Aoshima, A. Pure Appl. Chem. 2000, 72, 1305–1311. 10.1351/pac200072071305
- Lanzafame, P.; Temi, D.M.; Perathoner, S.; Spadaro, A.N.; Centi, G. Catal. Today. 2012, 179, 178–184. 10.1016/j.cattod.2011.07.018
- Palkovits, R.; Tajvidi, K.; Ruppert, A.M.; Procelewska, J. Chem. Commun. 2011, 47, 576–578. 10.1039/c0cc02263b
- Sun, Z.; Cheng, M.; Li, H.; Shi, T.; Yuan, M.; Wang, X.; Jiang, Z. RSC Adv. 2012, 2, 9058–9065. 10.1039/c2ra01328b
- Robles-Dutenhefner, P.A.; da Silva, K.A.; Siddiqui, M.R.H.; Kozhevnikov, I.V.; Gusevskaya, E.V. J. Mol. Catal., A. 2001, 175, 33–42. 10.1016/S1381-1169(01)00217-5
- Izumi, Y.; Urabe, K. In Studies in Surface Science and Catalysis; Hideshi Hattori, M.M.; Yoshio, O., Eds.; Elsevier, 1994; pp 1–8.
- Lim, S.S.; Kim, Y.H.; Park, G.I.; Lee, W.Y.; Song, I.K.; Youn, H.K. Catal. Lett. 1999, 60, 199–204. 10.1023/A:1019031729493
- Vairalakshmi, M.; Raj, V.; Sami, P.; Rajasekaran, K. Transition Met. Chem. 2011, 36, 875–882. 10.1007/s11243-011-9544-5
- Jiang, H.-S.; Mao, X.; Xie, S.-J.; Zhong, B.-K. J. Mol. Catal., A. 2002, 185, 143–149. 10.1016/S1381-1169(01)00449-6
- Zhizhina, E.G.; Simonova, M.V.; Odyakov, V.F.; Matveev, K.I. React. Kinet. Catal. Lett. 2006, 89, 157–166. 10.1007/s11144-006-0098-z
- Kolesnik, I.G.; Zhizhina, E.G.; Matveev, K.I. J. Mol. Catal., A. 2000, 153, 147–154. 10.1016/S1381-1169(99)00328-3
- Matveev, K.I.; Odyakov, V.F.; Zhizhina, E.G. J. Mol. Catal., A. 1996, 114, 151–160. 10.1016/S1381-1169(96)00313-5
- Sopa, M.; Wącław-Held, A.; Grossy, M.; Pijanka, J.; Nowińska, K. Appl. Catal., A. 2005, 285, 119–125. 10.1016/j.apcata.2005.02.013
- Ai, M. J. Catal. 1986, 98, 401–410. 10.1016/0021-9517(86)90329-5
- Tayebee, R.; Alizadeh, M.H. Chin. J. Chem. 2007, 25, 1340–1343. 10.1002/cjoc.200790249
- Tayebee, R.; Alizadeh, M.H. Monatsh. Chem. 2007, 138, 763–769. 10.1007/s00706-007-0660-z
- Rafiee, E.; Eavani, S.; Nejad, F.K.; Joshaghani, M. Tetrahedron. 2010, 66, 6858–6863. 10.1016/j.tet.2010.06.052
- Yadav, J.S.; Reddy, B.V.S.; Sridhar, P.; Reddy, J.S.S.; Nagaiah, K.; Lingaiah, N.; Saiprasad, P.S. Eur. J. Org. Chem. 2004, 2004, 552–557. 10.1002/ejoc.200300559
- Heravi, M.M.; Bakhtiari, K.; Zadsirjan, V.; Bamoharram, F.F.; Heravi, O.M. Bioorg. Med. Chem. Lett. 2007, 17, 4262–4265. 10.1016/j.bmcl.2007.05.023
- Javid, A.; Heravi, M.M.; Bamoharram, F.F.; Nikpour, M. E-J. Chem. 2011, 8, 547–552. 10.1155/2011/980546
- Heravi, M.M.; Ghods, A.; Derikvand, F.; Bakhtiari, K.; Bamoharram, F.F. J. Iran. Chem. Soc. 2010, 7, 615–620. 10.1007/BF03246049
- Javid, A.; Khojastehnezhad, A.; Heravi, M.; Bamoharram, F.F. Synth. React. Inorg. Met.-Org. Nano-Met. Chem. 2012, 42, 14–17. 10.1080/15533174.2011.609221
- Gharib, A.; Jahangir, M.; Roshani, M.; Scheeren, J.W. Synth. Commun. 2012, 42, 3311–3320. 10.1080/00397911.2011.581405
- Gharib, A.; Daneshtalab, M.; Scheeren, J.; Bamoharram, F.; Roshani, M.; Jahangir, M. Pol. J. Chem. Tech. 2009; p 8.
- Javanshir, S.; Ohanian, A.; Heravi, M.M.; Naimi-Jamal, M.R.; Bamoharram, F.F. J. Saudi Chem. Soc. 2011.
- Alimohammadi, K.; Sarrafi, Y.; Tajbakhsh, M. Monatsh. Chem. 2008, 139, 1037–1039. 10.1007/s00706-008-0885-5
- Azizi, N.; Torkian, L.; Saidi, M.R. J. Mol. Catal., A. 2007, 275, 109–112. 10.1016/j.molcata.2007.05.024
- Yadav, J.S.; Reddy, B.V.S.; Kumar, G.G.K.S.N.; Aravind, S. Synthesis. 2008, 3, 395–400. 10.1055/s-2007-1000932
- Azizi, N.; Saidi, M.R. Tetrahedron. 2007, 63, 888–891. 10.1016/j.tet.2006.11.045
- Matsuda, H.; Okuhara, T. Catal. Lett. 1998, 56, 241–243. 10.1023/A:1019085818706
- Hori, H.; Ishida, K.; Inoue, N.; Koike, K.; Kutsuna, S. Chemosphere. 2011, 82, 1129–1134. 10.1016/j.chemosphere.2010.11.038
- Lange, J.P.; Petrus, L. Appl. Catal., A. 2001, 216, 285–294. 10.1016/S0926-860X(01)00559-2
- Hou, Z.; Okuhara, T. Appl. Catal., A. 2001, 216, 147–155. 10.1016/S0926-860X(01)00546-4
- Guo, X.; Huang, C.; Chen, B. Korean J. Chem. Eng. 2008, 25, 675–680. 10.1007/s11814-008-0111-5
- Sakamoto, T.; Pac, C. Tet. Lett. 2000, 41, 10009–10012. 10.1016/S0040-4039(00)01787-1
- Xie, S.-L.; Hui, Y.-H.; Long, X.-J.; Wang, C.-C.; Xie, Z.-F. Chin. Chem. Lett. 2013, 24, 28–30. 10.1016/j.cclet.2012.12.009
- Hamidian, H. Org. Chem. Int. 2013, 2013, 5.
- Shojaei, A.F.; Rezvani, M.A.; Heravi, M. J. Serb. Chem. Soc. 2011, 76, 955–963. 10.2298/JSC100904086S
- Ntainjua, E.N.; Piccinini, M.; Freakley, S.J.; Pritchard, J.C.; Edwards, J.K.; Carley, A.F.; Hutchings, G.J. Green Chem. 2012, 14, 170–181. 10.1039/c1gc15863e