
Abstract
We describe a study of the gallium(III)-catalyzed 1,6-enynes cycloisomerization reaction in both homogeneous and heterogeneous conditions. With GaBr3 in homogeneous conditions, some particularities were observed in terms of selectivity compared to reported GaCl3-catalyzed reactions. The transfer of the reaction in heterogeneous conditions was realized by supporting Ga(III) salts onto montmorillonite. Both systems were compared based on reaction times, conversion, and selectivity and showed complementary activities.
Introduction
Homogeneous metal-based catalysts are occupying a prominent position in modern organic synthetic methodologies at the lab scale. A plethora of procedures could indeed be found for metal catalysts in a wide range of chemical transformations such as C–C (Citation1) and C-heteroatom (Citation2) couplings (Citation3), metathesis reactions (Citation4), electrophilic addition reactions (Citation5–9), and cycloisomerization (Citation10, Citation11), for example. When it comes to extrapolation of these reactions at larger scale, the question of metal recycling to avoid economical and environmental costs becomes crucial (Citation12). Strategies have been developed to transfer homogeneous catalysts onto solid supports and benefit from the advantages of both types of catalysis by covalent or noncovalent attachment of metal complexes or salts (Citation13–18).
In this context, gallium(III) salts (Citation19–21) and complexes (Citation22), which constitute an alternative to noble metal catalysts, seem to have been underestimated. Supported Ga catalysts have been described so far in environmental catalysis with Ga(III) oxide over alumina (Citation23–25) or in propane dehydrogenation with Ga(III) oxide over zeolites (Citation26–30). Fine chemicals could also be formed in Friedel–Crafts-type acylation reactions with Ga(NO3)3 supported on mesoporous silica (Citation31), or GaCl3 supported on polystyrene (Citation32, Citation33), in Strecker three-component reaction with Ga(III) triflate supported on Amberlyst 15 (Citation34), or in Friedel–Crafts-type alkylation reactions by Ga(III) nitrate impregnated over zeolite beta (Citation35).
In the field of 1,n-enynes cycloisomerization reactions, the use of heterogeneized gold catalysts was recently reported with gold nanoparticles supported on TiO2 (Citation36). While homogeneous Ga(III) catalysts have been reported to yield cyclic products (Citation37–39), no report to our best knowledge deals with heterogeneous Ga catalysts in this field. In the case of 1,6-enynes treated by GaCl3, a skeletal rearrangement involving the formation of fused cyclobutenes intermediates was invoked in the formation of vinylcyclopentene derivatives but with a limited substrate scope () (Citation40).
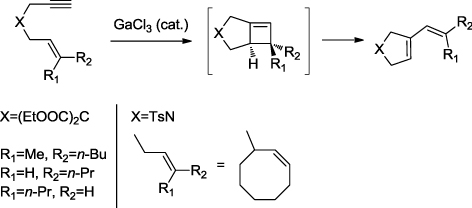
As a part of our interest for the development of sustainable methods in organic synthesis (Citation9, Citation41–45), we have chosen to evaluate the catalytic activity of Ga derivatives both as homogeneous catalyst and as a heterogeneous catalyst after impregnation on montmorillonite (MMT), a natural and cheap material with Brønsted acid properties which was never reported before as a support for such salts.
Experimental
1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on BRUCKER AC 200 (200 MHz). 1H-NMR spectra are reported as follows: chemical shift in ppm (δ) calibrated with the chemical shift of CDCl3 at 7.26 ppm, integration, multiplicities (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and b = broadened), and coupling constants (Hz). 13C NMR spectra are reported in ppm (δ) relative to CDCl3 at 77.16 ppm. Column chromatography was carried out employing silica gel (spherical, neutral, 63–200 µm, Geduran Si 60, Merck KGaA).
Gas chromatography with a flame ionization detector (GC-FID) analysis was carried out using a Varian 3900 gas chromatograph, under the following operation conditions: vector gas, N2; injector and detector temperatures, 250°C; split ratio, 1/50; constant flow, 1 mL/min; Chrompack column, polydimethylsiloxane (25 m × 0.25 mm i.d., film thickness 0.25 µm); temperature program, 80–250°C at 10°C/min and 250°C for 30 min. GC/MS analysis was performed by using a Shimadzu QP2010 gas chromatograph. (Conditions: carrier gas, He; injector and detector temperatures, 250°C; injected volume, 0.5 µl; split ratio, 1/100 (pressure, 180 kPa); SLB-5ms capillary column (thickness: 0.25 mm, length: 30 m, inside diameter: 0.25 mm); temperature program, 60–250°C at 2°C/min; and 250°C for 10 min, coupled to a mass selective detector. Mass spectra were obtained by electron ionization at 70 eV, m/z 35–400, source temperature 250°C; only the most abundant ions are given. Starting 1,6-enynes were prepared by conventional procedures of alkylation of dialkyl malonate (Citation42).
Preparation of Ga-MMT
In a flask containing lyophilized MMT (230 mg), Ga(NO3)3 (84 mg) is introduced followed by anhydrous MeOH (5 mL). The mixture is stirred with an orbital shaker during 3 hours. The supernatant is removed and the solid washed 15 times with 5 mL of MeOH. The solid obtained is dried under 1 Pa several days. Inductively coupled plasma mass spectrometry (ICP-MS) analysis was used to determine the metal content of the resulting material, after mineralization in aqueous HNO3 (69% w/w) during 4 hours at 90°C and filtration over millipore 0.22 µm. The loading was found to be 31.6 mmol/100 g.
General procedure of cyclization of 1,6-enynes with homogeneous Ga(III)
In a two-necked Schlenk flask mounted with a condenser, the substrate (0.5 mmol), GaBr3 (0.025 mmol, 8 mg), and anhydrous and degassed solvent (2 mL) are introduced under a nitrogen atmosphere and the mixture stirred at the desired temperature. After completion of the reaction, monitored by GC-FID analysis of samples withdrawn from the reaction medium, the mixture is filtered through a pad of silica gel (eluent: diethyl ether). After concentration and purification by flash chromatography, the cyclized products are obtained.
General procedure of cyclization of 1,6-enynes with heterogeneous Ga(III)
In a two-necked Schlenk flask mounted with a condenser, the substrate (0.5 mmol), Ga-MMT (0.025 mmol of metal, 79 mg), and anhydrous and degassed solvent (1.5 mL) are introduced under a nitrogen atmosphere and the mixture is stirred at the desired temperature. After reaction completion, monitored by GC-FID analysis of samples withdrawn from the reaction medium, the mixture is filtered through a pad of silica gel (eluent:diethyl ether). After concentration and purification by flash chromatography, the cyclized products are obtained.
Results and discussion
Our initial experiments were conducted on dimethyl 2-(3-methylbut-2-enyl)-2-prop-2-ynyl malonate 1a with homogeneous GaBr3 in toluene at various temperatures. The choice of GaBr3 was guided by the search for a less reactive salt, easier to handle, and less hazardous. The reaction was found to be very slow, and below 40°C, we observed the formation of the expected vinylcyclopentene 1b, together with the isomer 1c, and an undesired product formed upon the hydroarylation of 1a by toluene (). Interestingly, at 80°C and in contrast with previous work with GaCl3, the reaction outcome was dramatically modified in favor of the formation of the diene 1d. Such product formation is not surprising in itself since documented mechanisms involving nonclassical carbenium ions as intermediates could explain its formation (Citation7).
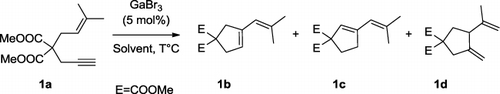
Table 1. Cycloisomerization of 1,6-enynes catalyzed by GaBr3.
While with the more active GaCl3, the reactions needed to be conducted at 0°C, GaBr3 even at 16°C only led to a moderate conversion (entry 1). At 40°C, the conversion was complete but a high mass loss was observed, since products 1b–1d were isolated with a mere 52% cumulated yield. This phenomenon was observed even at 25°C and 40°C (entries 2 and 3), presumably due to degradation (comparing for example with entry 6). At higher temperatures, the metathesis products 1b/1c became side products in favor of the formation of the cycloisomerized product 1d in 60–65% yields, depending on the solvent used (entries 4 and 6). Without catalyst, no reaction occurred (entry 7).
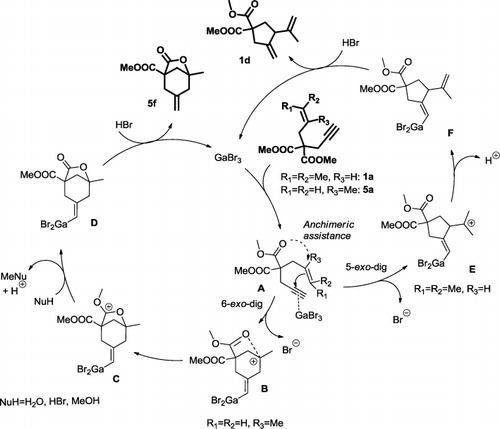
This shift of reactivity in favor of 1d was rather unexpected in Ga chemistry and prompted us to examine the behavior of other 1,6-enyne substrates in similar conditions (). With substrates bearing less substituted double bonds such as 2a–4a, the reaction did not proceed efficiently, and an important mass loss was observed, probably due to starting material degradation in the reaction conditions (entries 2–6). Substrate 2a featuring a monosubstituted olefin was cyclized in moderate yield to form 2b, further transformed into 2b′ by the addition of HBr (entries 2 and 3). The position of the bromine atom in 2b′ might seem surprising but one has to consider that the 1,3-dienic system of 2b, upon protonation, leads to a delocalized allylic cation, and the further attack of the bromide could occur at the terminal position for steric reasons. An additional H+-catalyzed isomerization of the resulting product to 2b′ would then occur. Traces of water in the organic solvent used, even after conventional drying procedure (Citation46), could indeed account for the partial hydrolysis of GaBr3 to gallium(III) hydroxide liberating HBr. The presence of a strong Brønsted acid in the reaction medium could also account for the isomerization of 1b to 1c. In toluene, the conversion was total at room temperature with an important mass loss (entry 4), while in dichloroethane (DCE), 95% of the starting material was recovered unchanged at this temperature (not shown). With substrates 3a and 4a, featuring a 1,2-disubstituted olefin, degradation occurred (entries 5 and 6). Methallyl-substituted substrate 5a, featuring a 1,1-disubstituted olefin and providing both electronic effects and limited steric hindrance at the terminal position, was selectively cyclized at room temperature to the bicyclic product 5f as the sole product, albeit with moderate conversion (entry 7). Several additional experiments at other temperatures or applying longer reaction times did not allow any improvement of the conversion. Product 5f, already described in Hg(OTf)2-catalyzed carbocyclization of 1,6-enynes (Citation47), is presumably formed upon the nucleophilic attack of the double bond on the π-activated triple bond of intermediate A via a 6-exo-dig ring-closure forming intermediate B, featuring a stabilized tertiary carbenium ion. The latter intermediate undergoes in a second step the attack an ester group of the tether leading to intermediate C further converted to D by dealkylation (Citation48–50). The hydrolysis of one ester followed by an intramolecular hydrocarboxylation could not be discarded, although no hydrolysis product was isolated during this study.
Table 2. Cycloisomerization of various 1,6-enynes catalyzed by GaBr3.
Substrate 6a, featuring a tosylamine moiety instead of a malonate, did not lead to cyclized products in these conditions (entry 8), which may be indicating a cyclization process anchimerically assisted by one of the esters in the case of 5a.
Replacing the malonate moiety by a tosylamine associated with the prenyl side chain in substrate 7a led predominantly to the formation of product 7d, as observed with 1a. The yield however was lowered by a side reaction of deprenylation yielding propargyl tosylamine (entry 9). Phenyl-substituted substrate 8a, with a phenyl group instead of the olefinic side chain, did not react and 84% of the starting material was recovered unchanged (entry 10). GaBr3 catalyst behavior was thus found to be highly dependent on the nature of the 1,6-enyne double bond. In a tentative to rationalize these results, a working mechanism is proposed in , with the cases of 1a and 5a.
To further compare with a heterogeneized catalyst, we decided to support Ga onto MMT. Such support could both ensure an easy recovery of the catalytic material from the reaction mixture and provide an additional source of activation for the reaction with the Brønsted acidity exhibited by MMT (with pK a ranging from 0 to –3 depending on the water content of the material) (Citation51). The catalyst Ga-MMT was obtained by treatment of the MMT with a solution of Ga(NO3)3 in MeOH resulting in a loading of 31.6 mmol/100 g. In these conditions, cation exchange between Na+ and Ga3+ is supposed to occur in the interlayer of the MMT, although a deposition of Ga salt at the surface could not be ruled out, affording a Ga-containing material (Ga-MMT).
A solvent screen was then performed to confirm DCE as a suitable reaction medium for the reaction and to avoid secondary reaction with the solvent ().
Table 3. Cycloisomerization of enyne 1a catalyzed by Ga-MMT: solvents screening.
Comparing with homogeneous conditions, the reaction was found to be slower. At 40°C, no product was formed in toluene, acetonitrile, refluxing dichloromethane, or diethyl ether (entries 1–4). In DCE, a conversion of 100% was observed after 96 hours at 80°C (entry 6). In refluxing methanol or at 80°C in CH3NO2, 20% and 5% conversions were observed, respectively (entries 8 and 9). In DCE at lower temperature, however, the conversion never went above 65% (entry 5). With pristine Ga-free MMT, no reaction occurred in DCE at 80°C (entry 7).
We thus decided to use these set of conditions to further investigate the reaction scope with 1,6-enynes featuring a mono-, 1,1-di-, 1,2-di-, or trisubstituted double bond. Comparing and , it seems that the Ga(III)-catalyzed cycloisomerization reaction of 1,6-enynes could be transferred in heterogeneous conditions using Ga-MMT as catalyst, but that the reactions proceed less efficiently, mainly in terms of reaction time and, in some cases, selectivity. Comparable results in terms of conversion could be obtained with 1a at the expense of 30 mol% of Ga instead of 5 mol%, but the selectivity (1b + 1c)/1d was inverted from 24/76 to 84/16 compared with homogeneous conditions (entry 2). Interestingly, with substrate 3a, cyclized products (3b + 3c) were obtained in 80% selectivity (entry 4), while in the homogeneous reaction, degradation of the starting material occurred. With substrate 5a, the cyclization to bicyclic product 5f was not observed (entry 5). Recycling studies were performed with the catalyst isolated and reengaged in the reaction of 1a (reactions conditions of ). The conversion could be maintained at 100% during four reaction cycles, but the overall yield in cyclized products dropped from 46% to 13% at fourth cycle, suggesting a substantial degradation of the starting material.
Table 4. Cycloisomerization of 1,6-enynes 1a–5a catalyzed by Ga-MMT.
Conclusions
We described a study of the Ga(III)-catalyzed 1,6-enynes cycloisomerization reaction in both homogeneous and heterogeneous conditions. With GaBr3 in homogeneous conditions, some particularities were observed in terms of selectivity compared with known Ga(III)-catalyzed reactions. The transfer of the reaction onto heterogeneous conditions with Ga-MMT was realized, and some differences of activity and selectivity were observed, comparing with the homogeneous procedure. The selectivity was changed in the case of a substrate with an electron-rich double bond. With a substrate with a phenyl-substituted double bond, the supported catalytic system allowed to obtain cyclic products instead of degradation, as observed with homogeneous conditions catalyst. The release of homogeneous Ga(III) salts in the reaction medium acting as catalyst is probably of concern here, and the development of alternative attachment strategies is thus necessary.
Acknowledgements
This work was supported by the University Nice Sophia Antipolis, the CNRS, and the ANR program CD2I (Nanocausys project, grant number 12-CDII-0010-02). We are grateful to Ms. Najiba Mahmoud for her technical assistance.
References
- Wu, X.-F.; Anbarasan, P.; Neumann, H.; Beller, M. Angew. Chem. Int. Ed. 2010, 49, (48), 9047–9050. 10.1002/anie.201006374
- Alonso, F.; Beletskaya, I.P.; Yus, M. Chem. Rev. 2004, 104, (6), 3079–3159. 10.1021/cr0201068
- Beller, M.M.; Seayad, J.; Tillack, A.; Jiao, H. Angew. Chem. Int. Ed. 2004, 43, (26), 3368–3398. 10.1002/anie.200300616
- Cossy, J.; Arseniyadis, S.; Meyer, C. Metathesis in Natural Product Synthesis: Strategies, Substrates and Catalysts; Wiley-VCH: Weinheim, 2010; p 412.
- Hashmi, A.S.K.; Hutchings, G.J. Angew. Chem. Int. Ed. 2006, 45, (47), 7896–7936. 10.1002/anie.200602454
- Ghosh, R.; Maiti, S. J. Mol. Catal. A. 2007, 264, (1–2), 1–8. 10.1016/j.molcata.2006.08.086
- Fürstner, A.; Davies, P. W. Angew. Chem. Int. Ed. 2007, 46, (19), 3410–3449. 10.1002/anie.200604335
- Yamamoto, H.; Ishihara, K. Acid Catalysis in Modern Organic Synthesis; Wiley-VCH: Weinheim, 2008; p 1136.
- Antoniotti, S.; Dalla, V.; Duñach, E. Angew. Chem. Int. Ed. 2010, 49, (43), 7860–7888. 10.1002/anie.200906407
- Michelet, V.; Toullec, P.Y.; Genêt, J.-P. Angew. Chem. Int. Ed. 2008, 47, (23), 4268–4315. 10.1002/anie.200701589
- Belmont, P.; Parker, E. Eur. J. Org. Chem. 2009, 2009, (35), 6075–6089. 10.1002/ejoc.200900790
- Torborg, C.; Beller, M. Adv. Synth. Catal. 2009, 351, (18), 3027–3043. 10.1002/adsc.200900587
- Wegener, S.L.; Marks, T.J.; Stair, P.C. Acc. Chem. Res. 2012, 45, (2), 206–214. 10.1021/ar2001342
- Hutchings, G.J. Catalysis Today. 2008, 138, (1–2), 9–14. 10.1016/j.cattod.2008.04.029
- Rangheard, C.; Fernandez, C.d. J.; Phua, P.-H.; Hoorn, J.; Lefort, L.; de, V. J. G. Dalton Trans. 2010, 39, (36), 8464–8471. 10.1039/c0dt00177e
- Gill, C.S.; Venkatasubbaiah, K.; Jones, C.W. Adv. Synth. Catal. 2009, 351, (9), 1344–1354. 10.1002/adsc.200800779
- Trilla, M.; Pleixats, R.; Man, M.W.C.; Bied, C.; Moreau, J.J.E. Adv. Synth. Catal. 2008, 350, (4), 577–590. 10.1002/adsc.200700489
- Sage, V.; Clark, J.H.; Macquarrie, D.J. J. Catal. 2004, 227, (2), 502–511. 10.1016/j.jcat.2004.08.013
- Prakash, G. K. S.; Mathew, T.; Olah, G. A., Acc. Chem. Res. 2012, 45, (4), 565–577. 10.1021/ar2002039
- Li, H.-J.; Guillot, R.; Gandon, V. J. Org. Chem. 2010, 75, (24), 8435–8449. 10.1021/jo101709n
- Simmons, E.M.; Sarpong, R. Org. Lett. 2006, 8, (13), 2883–2886. 10.1021/ol061037c
- Tang, S.; Monot, J.; El-Hellani, A.; Michelet, B.; Guillot, R.; Bour, C.; Gandon, V. Chem. Eur. J. 2012, 18, (33), 10239–10243. 10.1002/chem.201201202
- Petre, A.L.; Bonnetot, B.; Gervasini, A.; Auroux, A. Stud. Surf. Sci. Catal. 2002, 143, 747–755.
- Haneda, M.; Joubert, E.; Menezo, J.C.; Duprez, D.; Barbier, J.; Bion, N.; Daturi, M.; Saussey, J.; Lavalley, J.C.; Hamada, H. J. Mol. Catal. A: Chem. 2001, 175, (1–2), 179–188. 10.1016/S1381-1169(01)00215-1
- Shimizu, K.-I.; Takamatsu, M.; Nishi, K.; Yoshida, H.; Satsuma, A.; Tanaka, T.; Yoshida, S.; Hattori, T. J. Phys. Chem. B. 1999, 103, (9), 1542–1549. 10.1021/jp983790w
- Wang, J.; Zhang, F.; Hua, W.; Yue, Y.; Gao, Z. Catal. Commun. 2012, 18, 63–67. 10.1016/j.catcom.2011.11.023
- Shen, Z.; Liu, J.; Xu, H.; Yue, Y.; Hua, W.; Shen, W. Appl. Catal. A. 2009, 356, (2), 148–153. 10.1016/j.apcata.2008.12.038
- Xu, B.J.; Zheng, B.; Hua, W.M.; Yue, Y.H.; Gao, Z. Stud. Surf. Sci. Catal. 2007, 170B, ( From Zeolites to Porous MOF Materials), 1072–1079.
- Xu, B.; Li, T.; Zheng, B.; Hua, W.; Yue, Y.; Gao, Z. Catal. Lett. 2007, 119, (3–4), 283–288. 10.1007/s10562-007-9232-4
- Xu, B.; Zheng, B.; Hua, W.; Yue, Y.; Gao, Z. J. Catal. 2006, 239, (2), 470–477. 10.1016/j.jcat.2006.02.017
- El, B.Z.; Louis, B.; Tessonnier, J.P.; Ersen, O.; Cherif, L.; Ledoux, M.J.; Pham-Huu, C. Appl. Catal., A. 2007, 316, (2), 219–225. 10.1016/j.apcata.2006.09.033
- Rahmatpour, A. C. R. Chim. 2012, 15, (11–12), 1048–1054. 10.1016/j.crci.2012.08.005
- Rahmatpour, A. J. Organomet. Chem. 2012, 712, 15–19. 10.1016/j.jorganchem.2012.03.025
- Wiles, C.; Watts, P. ChemSusChem. 2012, 5, (2), 332–338. 10.1002/cssc.201100370
- Nur, H.; Ramli, Z.; Efendi, J.; Rahman, A.N.A.; Chandren, S.; Yuan, L. S. Catal. Commun. 2011, 12, (9), 822–825. 10.1016/j.catcom.2011.01.015
- Gryparis, C.; Efe, C.; Raptis, C.; Lykakis, I.N.; Stratakis, M. Org. Lett. 2012, 14, (12), 2956–2959. 10.1021/ol301212j
- Kim, S. M.; Lee, S. I.; Chung, Y. K. Org. Lett. 2006, 8, (24), 5425–5427. 10.1021/ol061801v
- Simmons, E.M.; Sarpong, R. Org. Lett. 2006, 8, (13), 2883–2886. 10.1021/ol061037c
- Hamlin, A.M.; de Jesus Cortez, F.; Lapointe, D.; Sarpong, R. Angew. Chem. Int. Ed. 2013, 52, (18), 4854–4857. 10.1002/anie.201209030
- Chatani, N.; Inoue, H.; Kotsuma, T.; Murai, S. J. Am. Chem. Soc. 2002, 124, (35), 10294–10295. 10.1021/ja0274554
- Godeau, J.; Fontaine-Vive, F.; Antoniotti, S.; Duñach, E. Chem. Eur. J. 2012, 18, (52), 16815–16822. 10.1002/chem.201202263
- Godeau, J.; Olivero, S.; Antoniotti, S.; Duñach, E. Org. Lett. 2011, 13, (13), 3320–3323. 10.1021/ol201028f
- Dia, R.-M.; Belaqziz, R.; Romane, A.; Antoniotti, S.; Duñach, E. Tetrahedron Lett. 2010, 51, (16), 2164–2167. 10.1016/j.tetlet.2010.02.081
- Ben Othman, R.; Affani, R.; Tranchant, M.-J.; Antoniotti, S.; Dalla, V.; Duñach, E. Angew. Chem. Int. Ed. 2010, 49, (4), 776–780. 10.1002/anie.200906036
- Lemechko, P.; Grau, F.; Antoniotti, S.; Duñach, E. Tetrahedron Lett. 2007, 48, 5731–5734. 10.1016/j.tetlet.2007.06.102
- Williams, D.B.G.; Lawton, M. J. Org. Chem. 2010, 75, (24), 8351–8354. 10.1021/jo101589h
- Nishizawa, M.; Yadav, V. K.; Skwarczynski, M.; Takao, H.; Imagawa, H.; Sugihara, T. Org. Lett. 2003, 5, (10), 1609–1611. 10.1021/ol034201u
- Munoz, M.P.; Lloyd-Jones, G.C. Eur. J. Org. Chem. 2009, 2009, (4), 516–524. 10.1002/ejoc.200800970
- Antoniotti, S.; Duñach, E. Tetrahedron Lett. 2009, 50, 2536–2539. 10.1016/j.tetlet.2009.03.055
- Godeau, J.; Antoniotti, S.; Olivero, S.; Duñach, E. CR Chim. 2009, 12, (8), 911–915. 10.1016/j.crci.2008.09.021
- Frenke, M., Clay. Clay Miner. 1974, 22, 435–441.