
Abstract
The present work describes the efficient ultrasound-assisted synthesis of saturated aliphatic esters from synthetic aliphatic acids in methanol or in ethanol, using p-toluenesulfonic acid as a catalyst. The esters were isolated in good yields after short reaction times under mild conditions. The compounds were analyzed by high resolution mass spectrometry (HRMS), which give a fragmentations pathway common for these molecules.
1. Introduction
Esters and derivatives are widespread in nature and are widely used in industry. The rapid increasing demand for esters in food (Citation1), cosmetics (Citation2), pharmaceutical (Citation3), and lubricants (Citation4) industries makes it necessary to find alternative ways instead of reactions which are too scarce or too expensive for commercial applications. Also, the long-chain esters of acids are being exploited industrially as biodiesel and as waxes (Citation5). The industrial process for ester syntheses are based on direct chemical esterification of fatty acids with alcohol in the presence of inorganic catalysts at high-temperatures (Citation6). However, these chemical procedures are tedious, nonselective, and consume a large amount of energy. In connection, the current situation of environmental degradation requires the development of efficient and cleaner methodologies with regard to energy consumption and reduction of waste.
Ultrasound has increasingly been used in organic chemistry in the last years. Compared with the traditional methods, this technique is more convenient and easily controlled. A large number of organic reactions can be carried out in high yields, shorter reaction time, and milder conditions under ultrasound irradiation. Recently, we have reported a convenient and inexpensive ultrasound-assisted preparation of functionalized arylacetylenes (Citation7), pyrazoles (Citation8, Citation9), thiazoles (Citation10, Citation11), isoxazoles (Citation12), and dihydropyrimidinones (Citation13).
The favorable effects of ultrasound irradiation are playing an increasing role in process chemistry, especially in cases where classical methods require drastic conditions or prolonged reactions times. Specially, the origin of sonochemical effects in liquids is the phenomenon of acoustic cavitation (Citation14, Citation15). Ultrasound is transmitted through a medium via pressure waves by inducing vibrational motion of the molecules which alternately compress and stretch the molecular structure of the medium due to a time-varying pressure. Therefore, the distance among the molecules varies as the molecules oscillate around their mean position. If the intensity of ultrasound in a liquid is increased, a point is reached at which the intramolecular forces are not able to hold the molecular structure intact. In consequence, it breaks down, and a cavity is formed. This cavity is a denominated cavitation bubble and this process is called cavitation, and the point is where it starts the cavitation threshold. A bubble responds to the sound field in the liquid by expanding and contracting (Citation16, Citation17).
Cavitation is a process in which mechanical activation destroys the attractive forces of molecules in the liquid phase. Applying ultrasound, compression of the liquid is followed by rarefaction (expansion), in which a sudden pressure drop forms small, oscillating bubbles of gaseous substances. These bubbles expand with each cycle of the applied ultrasonic energy until they reach an unstable size; they can then collide and/or violently collapse. These cavitation-induced effects can cause physical and chemical effects. Thus, ultrasound has found applications in organic chemistry, materials, and biodiesel synthesis (Citation18).
Currently, the development of simple, efficient, and environmentally friendly methods has been important. In order, acid-catalyzed reactions, under different conditions, has played as key in commons industrial process (Citation19). In recent years, p-toluenesulfonic acid (p-TSA) has been considered as an efficient, cheap, and readily available catalyst for several organic transformations (Citation20, Citation21). Moreover, the first ester synthesis ultrasound-assisted was indicated by Kalva and co-authors (Citation22). Recently, the synthesis of isopropyl esters from palm fatty acids and isopropyl alcohol also via sonochemistry was reported (Citation23). This process had required long reaction time (six hours) in acidic medium. In comparison with base catalyzed, the acid methodology typically leads to a less amount of side reactions. More recently, in our studies, we showed the application of sonochemistry in glycerolysis reaction and enzymatic catalysis (Citation24).
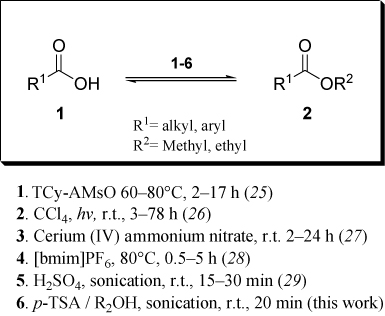
In this respect, eco-friendly methods are shown in the literature on esters synthesis, with or without catalyst (Citation25–Citation29; ). In agreement with this demand, we have prepared several molecules by nonconventional methods such as reaction in aqueous media (Citation30) and sonocatalysis (Citation31).
Eco-friendly methods are also shown in the literature on biodiesel and esters syntheses (Citation18, Citation29, Citation32). We report here that the efficient methodology to synthesize alkyl esters with p-TSA as catalyst and sonication leads to process improvement.
For the identification of compounds for the first time is employed high resolution mass spectrometry (HRMS) in electrospray ionization-quarupole time of flight (ESI-QTOF), providing a fragmentation pathway which can be used for identifying general esters.
2. Method
2.1. Apparatus and analysis
The compounds were dissolved in a solution of 50% (v/v) chromatographic grade acetonitrile (Tedia), 50% (v/v) deionized water, and 0.1% formic acid. The solutions were infused directly and individually into the electrospray ionization (ESI) source by means of a syringe pump (Harvard Apparatus) at a flow rate of 10 µL min−1. ESI(+)-MS and tandem ESI(+)-MS/MS were acquired using a hybrid high-resolution and high-accuracy (5 µL/L) MicrOTOF-Q II mass spectrometer (Bruker Daltonics) under the following conditions: capillary and cone voltages were set to +3500 V and +40 V, respectively, with a desolvation temperature of 200°C. For ESI(+)-MS/MS, the energy for the collision-induced dissociations (CID) was optimized for each component. Diagnostic ions were identified on the comparison of their ESI(+)-MS/MS dissociation patterns with theoretical mass dates. For data acquisition and processing, Micro-TOF software (Bruker Daltonics) was used. The data were collected in the m/z range of 50–400 at the speed of two scans per second, providing the resolution of 50,000 FWHM (full width at half maximum) at m/z 200. The infrared (IR) spectra were obtained on a Shimadzu IR Prestige-21 spectrometer in KBr pellets. The reactions were carried out with a microtip probe connected to a 500W Sonics Vibracell ultrasonic processor operating at 20 kHz at 25% of the maximum power output. The progress of the reactions was monitored on a Shimadzu 2010 Gas Chromatograph equipped with a Rtx-Wax polyethylene glycol capillary column (0.32 mm× 30 m).
2.2. Synthesis of esters by ultrasound irradiation
In a flask, the fatty acid (4.0 mmol) and p-TSA (2.0 mmol) were mixed with ethanol (27.6 mL) or methanol (16.8 mL) and sonicated for 20 minutes at room temperature (25°C). After the time indicated, the alcohol was evaporated under reduced pressure. The solid residue was dissolved into deionized water (35 mL), the product was extracted into ethyl ether (3 × 15 mL), and the combined organic fractions were dried (Na2SO4). The solvent was evaporated under vacuum to give the pure esters.
2.3. Compound characterization by HRMS and IR data
Methyl octanoate: m/z 159.1397 [C9H19O2]+, 117.0926 [C6H13O2]+, 103.0772 [C5H11O2]+. IR: ν (cm−1) 1720–1760, 1160–1200, 2800–2960.
Ethyl octanoate: m/z 173.1554 [C10H21O2]+, 145.1240 [C8H17O2]+, 103.0774 [C5H11O2]+, 89.0612 [C4H9O2]+. IR: ν (cm−1) 1720–1760, 1160–1200, 2800–2960.
Methyl decanoate: m/z 187.1699 [C11H23O2]+, 117.0929 [C6H13O2]+, 103.0769 [C5H11O2]+. IR: ν (cm−1) 1720–1760, 1120–1200, 2800–2960.
Ethyl decanoate: m/z 201.1862 [C12H25O2]+, 173.1552 [C10H21O2]+, 117.0928 [C6H13O2]+, 103.0771 [C5H11O2]+. IR: ν (cm−1) 1720–1760, 1160–1200, 2800–2960.
Methyl dodecanoate: m/z 215.2009 [C13H27O2]+, 201.1849 [C12H25O2]+, 187.1689 [C11H23O2]+, 173.1543 [C10H21O2]+. IR: ν (cm−1) 1720–1760, 1160–1200, 2800–2960.
Ethyl dodecanoate: m/z 251.1972 [C14H28O2 + Na]+, 229.2166 [C14H29O2]+, 201.1854 [C12H25O2]+, 149.0240 [C8H5O3]+. IR: ν (cm−1) 1720–1760, 1160–1200, 2800–2960.
Methyl tetradecanoate: m/z 243.2322 [C15H31O2]+, 229.2162 [C14H29O2]+, 201.1846 [C12H25O2]+, 103.0764 [C5H11O2]+. IR: ν (cm−1) 1720–1760, 1160–1200, 2800–2960.
Ethyl tetradecanoate: m/z 257.2480 [C16H33O2]+, 229.2161 [C14H29O2]+, 201.1845 [C12H25O2]+, 173.1530 [C10H21O2]+. IR: ν (cm−1) 1720–1760, 1120–1200, 2800–2960.
Methyl hexadecanoate: m/z 271.2635 [C17H35O2]+, 257.2481 [C16H33O2]+, 229.2174 [C14H29O2]+, 201.1852 [C12H25O2]+, 103.0733 [C5H11O2]+. IR: ν (cm−1) 1720–1760, 1160–1200, 2800–2960.
Ethyl hexadecanoate: m/z 307.2635 [C18H37O2+Na]+, 285.2807 [C18H37O2]+, 257.2431 [C16H33O2]+, 229.2163 [C14H29O2]+, 201.1852 [C12H25O2]+, 149.0240 [C8H5O3]+. IR: ν (cm−1) 1720–1760, 1160–1200, 2800–2960.
3. Results and discussion
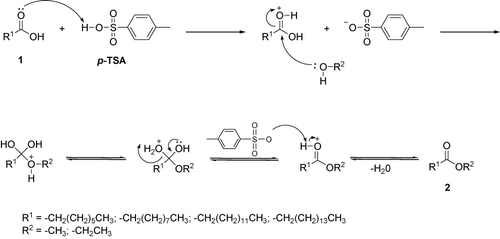
In this work, we have combined acid catalyzed with ultrasound to prepare some esters by the eco-friendly and rapid process. The experimental procedure for these reactions is remarkably simple and does not require the use of expensive catalysts. The mixture of alcohol and p-TSA was reacted with a variety of fatty acid, leading to esters. In particular, by monitoring gas chromatography-flame ionization detector (GC-FID), it was shown that only 20 minutes (ultrasound – 20KHz) are sufficient to convert the fatty acids into short-chain esters. It was not possible to correlate the results with groups of chemical reagents; however, the yields of the products were satisfactory in comparison with the literature (73–98%, , ). The reaction mechanism for the preparation of esters 2 is similar with the Fischer synthesis (Citation33). A possible explanation is that the product was obtained from acid carboxylic 1 due to the attack of the alcohol in acid media (p-TSA) and subsequent water elimination ().
Table 1. Ultrasound-assisted ester synthesis by p-TSA catalysis.
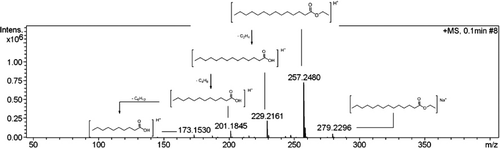
Relating with this work, to analyze the products 2 ESI-MS and ESI-MS/MS were used, which have been important tools to characterize and identify the synthetic compounds. The measurements were made by a direct infusion (DI) in HRMS equipment Micro-QTOF II.
To respond in positive mode, 0.1% of formic acid was introduced into the sample solution. The instrument's accurate mass measurement gives the elemental composition of parent and fragment ions. As is summarized in , mass accuracy is also easily obtained for all the characteristic fragment ions, thus providing two sets of important information for unequivocal identification. Since the widely accepted accuracy threshold for the confirmation of elemental compositions was established as 10 ppm, this usually provides highly reliable identification of the target compounds (Citation34).
Table 2. Synthetic esters analyzed by HRMS ESI-TOF-MS (+).
The QTOF-MS/MS provided HRMS and MS/MS data, which allowed the confirmation of chemical formulas based on its ions beyond the elucidation of the fragments m/z. The [M+Na]+ ions for the investigated esters were hardly observed. The [M+H]+ ions were selected as the precursor ions for CID fragmentation to produce MS/MS spectra. The fragmentation behavior follows an order losing alky groups (). This is a characteristic fragmentation which was observed for all compounds obtained here. In accordance with the chain ester size is formed ions with different stability giving different spectra. Therefore, the combination uses of information provided by the two instruments were helpful to elucidate the structures of the studied compounds.
4. Conclusion
In conclusion, the procedure described here is an economical and environmental methodology for esterification reactions with pharmaceutical and industrial importance. Significant advantages of the method include (1) the catalyst is inexpensive; (2) the reaction is easy to execute; (3) the workup is very simple; (4) the required reaction times are shorter (20 minutes); (5) the reaction can be conducted at ambient temperature; and (6) the products are isolated in good yields (73–98%) and high purities. The analyses based on HRMS are easy and give a fragmentation pathway for the direct identification of similar compounds.
Acknowledgments
The authors are grateful to CNPq, FAPERGS, CAPES, INCT de Estudos do Meio Ambiente (573.667/2008-0) and Secretaria da Ciência e Tecnologia do Estado do Rio Grande do Sul for financial and fellowship support.
References
- Piccicuto, S.; Blecker, C.; Brohee, J.C.; Mbampara, A.; Lognay, G.; Deroanne, C.; Paquot, M.; Marlier, M. Biotechnol. Agron. Soc. Environ. 2001, 5, 209–219.
- Vaze, S. Chem. Eng. World. 1996, 31, 115–116.
- Hopkins, H.; Small, L.V.D. J. Pharm. Sci. 1960, 49, 220–224. 10.1002/jps.3030490410
- Lauer, D.A. In Synthetics, Mineral Oils, and Bio-Based Lubricants; Rudnick, L.R., Ed.; CRC: Boca Raton, FL, 2006; pp 441–458.
- Larios, A.; García, H.S.; Oliart, R.M.; Valerio-Alfaro, G. Appl. Microbiol. Biotechnol. 2004, 65, 373–376. 10.1007/s00253-004-1602-x
- Farris, R.D. J. Am. Oil Chem. Soc. 1979, 56, 770A–773A. 10.1007/BF02667441
- Stefani, H.A.; Cella, R.; Dörr, F.A.; Pereira, C.M.P.; Gomes, F.P.; Zeni, G. Tetrahedron Lett. 2005, 46, 2001–2003. 10.1016/j.tetlet.2005.01.161
- Pizzuti, L.; Piovesan, L.A.; Flores, A.F.C.; Quina, F.H.; Pereira, C.M.P. Ultrason. Sonochem. 2009, 16, 728–731. 10.1016/j.ultsonch.2009.02.005
- Pizzuti, L.; Martins, P.L.; Ribeiro, B.A.; Quina, F.H.; Pinto, E.; Flores, A.F.C.; Venzke, D.; Pereira, C.M.P. Ultrason. Sonochem. 2010, 17, 34–37. 10.1016/j.ultsonch.2009.06.013
- Venzke, D.; Flores, A.F.C.; Quina, F.H.; Pizzuti, L.; Pereira, C.M.P. Ultrason. Sonochem. 2010, 18, 370–374. 10.1016/j.ultsonch.2010.07.002
- Neuenfeldt, P.D.; Duval, A.R.; Drawanz, B.B.; Rosales, P.F.; Gomes, C.R.B.; Pereira, C.M.P.; Cunico, W. Ultrason. Sonochem. 2011, 18, 65–67. 10.1016/j.ultsonch.2010.07.008
- Nascimento, F.; Baltazar, M.; Pizzuti, L.; Gressler, V.; Rivelli, D.; Barros, S.B.M.; Pereira, C.M.P. Lett. Drug Des. Discov. 2009, 6, 323–326.
- Stefani, H.A.; Oliveira, C.B.; Almeida, R.B.; Pereira, C.M.P.; Braga, R.C.; Cella, R.; Borges, V.C.; Savegnago, L.; Nogueira, C.W. Eur. J. Med. Chem. 2006, 41, 513–518. 10.1016/j.ejmech.2006.01.007
- Parkar, P.A.; Choudhary, H.A.; Moholkar, V.S. Chem. Eng. J. 2012, 187, 248–260. 10.1016/j.cej.2012.01.074
- Choudhury, H.A.; Goswami, P.P.; Malani, R.S.; Moholkar, V.S. Ultrason. Sonochem. 2014, 21, 1050–1064. 10.1016/j.ultsonch.2013.10.023
- Choudhury, H.A.; Chakma, S.; Moholkar, V.S. Ultrason. Sonochem. 2014, 21, 169–181. 10.1016/j.ultsonch.2013.04.010
- Choudhury, H.A.; Malani, R.S.; Moholkar, V.S. Chem. Eng. J. 2013, 231, 262–272. 10.1016/j.cej.2013.06.107
- Tuchtenhagen, C.P.; Dias, D.; Muller, B.V.; Ritter, M.; Santos, M.A.Z.; Oliveira, D.; Crizel, M.G.; Mesko, M.F.; Santos, V.; Pereira, C.M.P. Rev. Virtual Quím. 2014, 6, 884–897.
- Karemi-Jaberi, Z.; Pooladian, B. Green Chem. Lett. Rev. 2012, 5, 187–193. 10.1080/17518253.2011.611478
- Thanusu, J.; Kanagarajan, V.; Gopalakrishnan, M. Green Chem. Lett. Rev. 2012, 5, 65–72. 10.1080/17518253.2011.581699
- Das, S.; Thakur, A.J. Green Chem. Lett. Rev. 2011, 4, 131–135. 10.1080/17518253.2010.521775
- Kalva, A.; Sivasankar, T.; Moholkar, V.S. Ind. Eng. Chem. Res. 2009, 48, 534–544. 10.1021/ie800269g
- Deshmane, V.G.; Gogate, P.R.; Pandit, A.B. Ultrason. Sonochem. 2009, 16, 345. 10.1016/j.ultsonch.2008.09.004
- Fiametti, K.G.; Sychoski, M.M.; Cesaro, A.; Furigo, A.; Bretanha, L.C.; Pereira, C.M.P.; Treichel, H.; Oliveira, D.; Oliveira, J.V. Ultrason. Sonochem. 2011, 18, 981–987. 10.1016/j.ultsonch.2010.11.010
- Santi, V.; Cardellini, F.; Brinchi, L.; Germani, R. Tetrahedron Lett. 2012, 53, 5151–5155. 10.1016/j.tetlet.2012.07.063
- Hwu, J.R.; Hsu, C.; Jain, M.L.; Tetrahedron Lett. 2004, 45, 5151–5154. 10.1016/j.tetlet.2004.04.155
- Pan, W.; Chang, F.; Wei, L.; Wu, M.; Wu, Y. Tetrahedron Lett. 2003, 44, 331–334. 10.1016/S0040-4039(02)02578-9
- Yoshino, T.; Imori, S.; Togo, H. Tetrahedron 2006, 62, 1309–1317. 10.1016/j.tet.2005.09.147
- Hobuss, C.B.; Venzke, D.; Pacheco, B.S.; Souza, A.O.; Santos, M.A.Z.; Moura, S.; Quina, F.H.; Fiametti, K.G.; Oliveira, J.V.; Pereira, C.M.P. Ultrason. Sonochem. 2012, 19, 387–389. 10.1016/j.ultsonch.2011.06.020
- Pereira, C.M.P.; Martins, M.A.P.; Moura, S.; Fiss, G.F.; Frizzo, C.P.; Emmerich, D.; Zanatta, N.; Bonacorso, H.G. Arkivoc 2006, xiii, 187–194.
- Venzke, D.; Flores, A.F.C.; Quina, F.H.; Pizzuti, L.; Pereira, C.M.P. Ultrason. Sonochem. 2011, 18, 370–374. 10.1016/j.ultsonch.2010.07.002
- Souza, P.O.; Ferreira, L.F.; Pires, N.R.X.; Sanches, P.J.; Duarte, P.A.; Pereira, C.M.P.; Mesko, M.F. Rev. Bras. Farmacogn. 2012, 22, 825–837. 10.1590/S0102-695X2012005000076
- Carey, F.A. Organic Chemistry; McGraw-Hill Higher Education: Jefferson, USA, 2000.
- Lacorte, S.; Fernandez-Alba, A.R. Mass Spectrom. Rev. 2006, 25, 866–880. 10.1002/mas.20094
- Mori, N.; Togo, H. Tetrahedron 2005, 61, 5915–5925. 10.1016/j.tet.2005.03.097
- Li, X.; Eli, W. J. Mol. Catal. A: Chem. 2008, 279, 159–164. 10.1016/j.molcata.2007.08.032
- Gulati, R.; Arya, P.; Malhotra, B.; Prasad, A.K.; Saxena, R.K.; Kumar, J.; Watterson, A.C.; Parmar, V.S. Arkivoc 2003, iii, 159–170. 10.3998/ark.5550190.0004.316
- Qiao, K.; Hagiwara, H.; Yokoyama, C. J. Mol. Catal. A: Chem. 2006, 246, 65–69. 10.1016/j.molcata.2005.07.031
- Li, C.; Yang, J.; Wang, P.; Liu, J.; Yang, Q. Micropor. Mesopor. Mat. 2009, 123, 228–233. 10.1016/j.micromeso.2009.04.005
- Sun, S.Y.; Xu, Y.; Wang, D. Bioresour. Technol. 2009, 100, 2607–2612. 10.1016/j.biortech.2008.11.006