
ABSTRACT
Luffa aegyptiaca fruit juice was used as a low purity source of enzyme, which contained peroxidase activity of 180 IU/mL. The results of UV/VIS and IR studies suggested that L. aegyptiaca fruit juice works efficiently in the enzymatic conversion of phenolic compounds, namely guaiacol, m-cresol, p-cresol, o-cresol, anisole, resorcinol, catechol, pyrogallol, hydroquinone, veratryl alcohol and phoreguicinol to quinones at 30°C. The p-cresol and pyrogallol were converted to quinones more efficiently as compared to other phenolic compounds. Determination of enzymatic characteristic properties such as Michaelis–Menten constant (Km), temperature optima and pH optima using different phenolic compounds, indicated p-cresol as a potential substrate for the peroxidase enzyme assay at room temperature, whereas guaiacol, which is widely used as a substrate for enzyme assay, has a higher temperature optima at 60°C for its maximum catalytic activity. Enzyme activity is inhibited by sodium azide using different phenolic substrates in the reaction mixture.
GRAPHICAL ABSTRACT
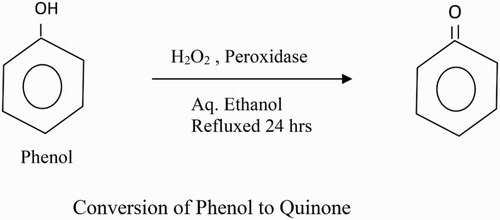
Introduction
Harmful aromatic compounds are generally found in wastewaters discharged from industries such as petroleum refining, coal, plastics, resins, textiles, paper and pulp, iron and steel manufacturing units, and agricultural activities. Phenolic compounds and their derivatives are considered priority pollutants because they are harmful to living organisms even at low concentrations (Citation1). Many of these compounds are toxic and carcinogenic (Citation2) and have been introduced into the environment in the last few decades and have, therefore, become an issue of great environmental concern. Most bioremediation studies have been carried out with bacteria and basidiomycetes which have developed the capability to utilize environmental pollutants (Citation3–6). Conventional processes for the removal of phenolics from industrial wastewaters include extraction, adsorption on activated carbon, steam distillation, bacterial and chemical oxidation, electrochemical techniques and irradiation. All of these methods have high costs, very low efficiency, incomplete purification, discharge hazardous byproducts and only apply to a limited range of concentration (Citation7). Due to these disadvantages, alternative methods must be developed on a large scale in the near future. The treatment through enzymatic catalysis seems to be a potential application to substitute conventional methods (Citation8), providing ecofriendly green methods.
Peroxidase [E.C. 1. 11.1.7] is a heme-containing enzyme, which catalyzes the oxidation of a wide variety of organic and inorganic substrates using hydrogen peroxide as the electron acceptor (Citation9–12). One-electron oxidation of aromatic substrates (AH2) catalyzed by peroxidases is depicted by the Chance–George mechanism (Citation13) is the following:(1)
(2)
(3)
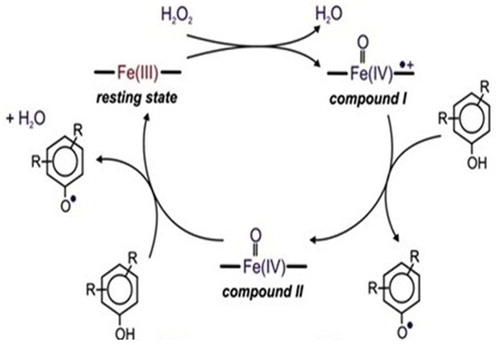
(4) The native enzyme (E) is oxidized by peroxide (H2O2) to an active intermediate enzymatic form called complex compound I (E(I)). Complex compound I oxidizes organic substrate AH2 and as a result a free radical (AH*) is produced in the reaction mixture. Enzyme remains in the complex compound II (E(II)) state. Complex compound II oxidizes the second organic substrate to a free radical product and the enzyme returns to its native state. The overall peroxidase reaction consists of the reactions described by Equations (1), (2) and (3) and a catalytic cycle (Citation14) is shown in . Free radicals formed during the cycle diffuse from the enzyme into the bulk solution where they react to form polyaromatic products. These polyaromatic compounds are insoluble in water and can be removed by solid–liquid operations (Citation13).
Figure 1. Peroxidase-catalytic cycle (Citation14).
Horseradish peroxidase, an enzyme isolated from the roots of horseradishes, acts as a catalyst for the treatment of discharged wastewater from industries (Citation14–16). The main advantages of this method (Citation7, Citation15, Citation17) are: (1) broad substrate specificity; (2) effectiveness over a wide range of operating conditions including pH, temperature, salinity and substrate concentrations; and (3) ability to remove other organic compounds by co-precipitation. Peroxidases have found applications in different industrial processes due to their high stability in solution, broad substrate specificity, tolerance to a wide range of pH and temperature, and easy availability from plant materials (Citation18–20).
Keeping these points in view, we have isolated a peroxidase enzyme from Luffa aegyptiaca fruit juice, which has high peroxidase activity to the order of 180 IU/mL, to study the degradation of phenolics to quinones. The efficiency of enzyme quality was found to be independent of the enzyme purity and therefore, it was feasible to effectively use a crude enzyme preparation instead of a purified enzyme. This characteristic feature of the enzyme preparation leads to a significant reduction in the treatment cost (Citation14, Citation15). The objective of this research work is to develop a technology based on an enzyme treatment to remove the toxic organic carcinogenic compounds from discharged wastewaters from industries and agricultural activities.
Experimental
Enzyme assay
Guaiacol was used as the target phenolic compound because it is a widely known substrate used for assaying peroxidase activity in the extract from different sources. Guaiacol was obtained from Sigma Chemical Company, St. Louis, U.S.A. Phenol and their derivatives, ethanol and H2O2 were purchased from S.D. Fine Chem. Ltd., Mumbai, India, and were used without further purification.
To isolate the enzyme, L. aegyptiaca fruit was cut into small pieces, crushed with a mortar and pestle, and the juice was extracted through four layers of cheese cloth. The juice was centrifuged using Sigma (Germany) model 3K-30 refrigerated centrifuge at 4000 g for 20 min at 4°C to remove the cloudiness of the juice. The clear juice was stored at 4°C. The enzyme stored does not lose activity for 3 months. The activity of peroxidase was determined spectrophotometrically by using guaiacol (Citation21, Citation22) as the substrate and monitoring the absorbance change at λ = 470 nm due to the formation of product tetraguaiacol in the reaction solution using molar extinction coefficient value of 2.66 × 104 M−1 cm−1. 1.8 IU of the enzyme solution was added at 30°C to the reaction solution of 1 mL consisting of 50 mM sodium phosphate buffer (pH 7.0), 0.6 mM H2O2 and using guaiacol 5 mM as the substrate. One enzyme unit transformed 1 μM of the substrate into the product under specified assay condition. A UV/VIS spectrophotometer (SHIMADZU UV 2600) was used for spectrophotometric measurements. The least count of the absorbance measurement was 0.001 absorbance unit.
Enzymatic characterization
Enzymatic characterization such as Km, pH and temperature optima was determined for different phenolic compounds taken in order to explore enzyme specificity. The enzymatic characteristic, Km value of peroxidase was determined using different phenolic compounds namely guaiacol, m-cresol, p-cresol, o-cresol, anisole, resorcinol, catechol, pyrogallol, hydroquinone, veratryl alcohol and phoreguicinol and H2O2 as the substrates. The Km values were determined by measuring steady-state velocities of peroxidase-catalyzed reactions at different concentrations of substrates varied in the range 0.0–2.0 mM and drawing double reciprocal plots. 1.8 IU of the crude enzyme was added to the reaction solution of 1 mL consisting of 250 μM, H2O2 in 100 mM phosphate buffer (pH 7.0) at 30°C, wherein the concentrations of phenolic compounds were varied in the range of 0.0–2.0 mM. The Km value was calculated by the linear regression analysis of the data points of double reciprocal plots (Citation23, Citation24). The pH optimum was determined by measuring the steady-state velocities of peroxidase-catalyzed reaction in the reaction mixture having different pH ranges, from 2.0 to 10.0 and plotting steady-state velocity against the pH values. Similarly, the temperature optimum was determined by measuring the steady-state velocities in reaction solutions at different temperatures ranging from 12°C to 80°C and plotting the steady-state velocities against temperatures of the reaction solutions.
Bioconversion of phenolic compounds
In order to evaluate the catalytic activity of peroxidase, the catalytic oxidation of phenolic compounds using H2O2 as the mild oxidant was studied to prove the efficiency of the peroxidase. The reaction solution under study contained H2O2, phenol, 1 mL of enzyme stock (180 IU/mL) in 50 mM sodium phosphate buffer, pH 7.0 (Citation22, Citation25). The reaction was carried out for a fixed period of time at 30°C and the change in phenolic compounds was monitored using a UV/VIS spectrophotometer (SHIMADZU UV 2600). An FTIR Spectrophotometer (SHIMADZU) IR Affinity-I was also used for the study of product formation in the reaction solution. The reaction mixture was incubated for 24 h and then harvested. The IR spectra were recorded in the region 4000–400 cm−1 for the samples in KBr pellets (Citation26). In order to study the biotransformations, 1 mL of the reaction mixture containing 250 μM, H2O2 in 100 mM phosphate buffer pH 7.0 at 30°C, substrate (phenolic compounds) was varied in the range 0.01–0.5 mM and 1.80 IU of the crude enzyme was added. H2O2 was added to initiate the reaction and the reaction progress was monitored spectrophotometrically at different time intervals. The different phenolic compounds taken are listed in with Km values, pH and temperature optima determined for the maximum activity of the enzyme peroxidase.
Table 1. Enzymatic characterization using different substrates.
Inhibition of enzyme activity
The steady-state velocity of peroxidase-catalyzed reaction has been measured (Citation27, Citation28) at different concentrations of sodium azide and a graph has been plotted in velocity versus azide concentration.
Results and discussion
The analysis of the steady-state kinetics measurements showed the presence of 180 IU/mL of peroxidase activity in fruit juice (Citation25). Thus, L. aegyptiaca fruit juice is a very rich and convenient source of peroxidase enzyme. The Michaelis constant (Km) for the L. aegyptiaca peroxidase enzyme is reported (Citation25) to be 2.0 mM for guaiacol and 0.2 mM for H2O2, which are greater than the Km values for horseradish peroxidase, Turkish black radish and Solanum melongena fruit juice. Thus, this signifies that L. aegyptiaca enzyme is less specific for the substrate guaiacol and may show more specificity or affinity for other phenolic compounds. In order to explore the specificity of this enzyme for the substrate, we studied the enzymatic oxidation of phenol and their derivatives, namely guaiacol, m-cresol, p-cresol, o-cresol, anisole, resorcinol, catechol, pyrogallol, hydroquinone, veratryl alcohol and phoreguicinol in reaction solution at room temperature and found that the enzyme effectively converted phenol and their derivatives to their respective quinones as shown by enzymatic reaction in . The steady-state enzyme kinetics of the peroxidase-catalyzed reaction was studied by measuring absorbance change per minute at λ = 280 nm. The substrate binds to the available empty catalytic site of the active enzyme. If the concentration of the substrate is increased in the reaction mixture, the enzyme gets saturated with the substrate. The rate of reaction is at a maximum when the active site of the enzyme is completely saturated with the substrate. The rate of reaction is at a maximum at 2 mM enzyme saturating concentration of p-cresol and at 0.15 mM for H2O2 in the reaction solution as visible in and . In order to determine the Km value, two sets of experiments were done. In one set of experiments, the steady-state velocity of the enzyme catalyzed reaction was measured at different concentrations of p-cresol in the range of 0.0–2 mM at the fixed 0.15 mM enzyme saturating concentration of H2O2 and 1.8 IU of the enzyme in 50 mM sodium phosphate buffer pH 7 at 30°C. In the second set of experiments, steady-state velocity was measured at a different concentration of H2O2 in the range 0.0–0.2 mM at a fixed 2 mM enzyme saturating concentration of p-cresol. Michaelis–Menten curves for p-cresol and H2O2 were drawn by plotting steady-state velocity versus the concentration of the substrates as shown in and . Each data point was an average of three measurements. From the graphs, the Km values were calculated. Using p-cresol as a substrate, the Km value was 0.8 mM, and using H2O2 as a substrate, the Km value was 0.08 mM. The p-cresol was selected as the best substrate because it showed better catalytic activity with lower Km value as compared to other phenolic compounds as given in . Low Km value signifies that the enzyme has more affinity for the substrate, p-cresol. The variation of the activity of the enzyme with temperature is shown in . The 1 mL of reaction solution contained 1.8 IU of the enzyme, 100 μM H2O2, 200 μM p-cresol in 50 mM sodium phosphate buffer. The temperature of the reaction solution was varied at a fixed optimum pH of 7.0. The optimum temperature determined from the graph was found to be 30°C. The variation of the activity of the enzyme with pH is shown in . The reaction solution 1 mL contained 1.8 IU of the enzyme, 100 μM H2O2, 200 μM p-cresol in 50 mM sodium phosphate buffer at 30°C. The pH of the reaction solution was varied from 2.0 to 10.0. The optimum pH as determined from the graph was found to be 7.0. Km values indicating that p-cresol and pyrogallol are better substrates for the enzyme assay because the enzyme is more specific for these substrates and has a temperature optima of 30°C (i.e. room temperature) with pH nearly neutral 7.0. The catalytic activity of the enzyme with p-cresol as the substrate with time is shown in . (a) shows the spectra of p-cresol substrate in 50 mM sodium phosphate buffer, pH 7.0, which shows a peak at 280 nm. (b) shows the spectra of the enzyme in the same buffer having Soret band at 403 nm which is the characteristic band of the heme active site present in the enzyme. (c) shows the change in absorption spectra of the p-cresol as the reaction proceeds with the addition of H2O2 in the reaction mixture consisting of 1 mM p-cresol, 1.8 IU of the enzyme in 50 mM sodium phosphate buffer pH 7.0 at 30°C. Spectra of the reaction mixture were monitored at zero time and at subsequent 2, 4, 6, 8 min and after 1 h. It is clear from the absorption spectra that the absorption depth at 280 nm is decreased, indicating that the p-cresol is being consumed by the enzyme as the reaction proceeds with time. (d) shows the increased absorption at 403 nm due to the formation of the active heme–substrate complex called activated complex as the reaction proceeds after the addition of H2O2 to initiate the reaction and simultaneous increase in absorption near 600 nm due to formation of quinone from p-cresol, which was being consumed in the reaction. (e) shows the absorption spectra with no peak at 280 nm indicating that p-cresol has been consumed in the reaction and complete formation of quinone at 600 nm after 1 h. Thus, p-cresol acts as a better substrate for peroxidase enzyme assay at room temperature and easily available reagent, whereas guaiacol has a temperature optima of 60°C for its maximum catalytic activity to show. The rest of the substrate was also converted to quinones, but less efficiently as compared to p-cresol and pyrogallol. IR spectra of the product in comparison to those of the respective substrate were analyzed. In both cases, the intensity of absorption appeared in the range 690–900 cm−1 which may be considered due to υ (C–C) and υ (C–H) vibrations of an aromatic ring and intensities at 1500 and 3200 cm−1 are denoted as υ (>C=C<) and υ (−OH) vibrations of an aromatic ring, respectively, in the substrate spectra. An intense absorption band at 1650 cm−1 of the product indicated the formation of a benzoquinone structure.
Figure 2. Bioconversion of phenolic compounds to quinone.
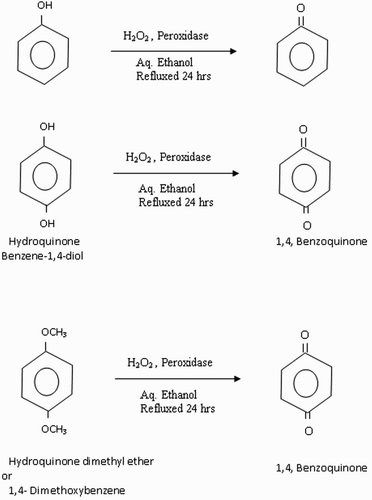
Figure 3. (a) Michaelis–Menten and (b) double reciprocal plots using p-cresol as the variable substrates, respectively. The reaction 1 mL contained 0.1 mM H2O2, 1.80 IU of the enzyme in 50 mM sodium phosphate buffer pH 7 at 30°C.
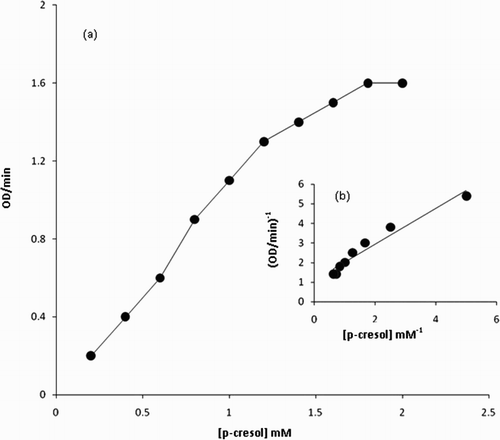
Figure 4. (a) Michaelis–Menten and (b) double reciprocal plots using H2O2 as the variable substrate. The reaction 1 mL contained 2 mM p-cresol, 1.80 IU of the enzyme in 50 mM sodium phosphate buffer pH 7 at 30°C.
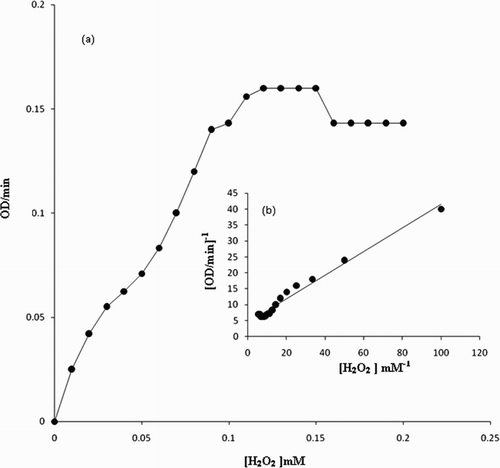
Figure 5. The variation of the activity of the enzyme with temperature. The reaction solution 1 mL contained 1.80 IU of the enzyme, 100 μM H2O2, 200 μM p-cresol in 50 mM sodium phosphate buffer. The temperature of the reaction solution was varied at fixed pH of 7.0.
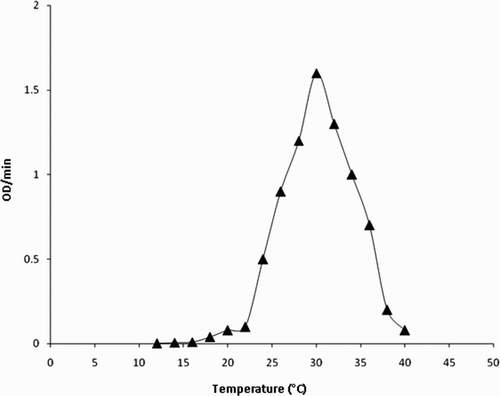
Figure 6. The variation of the activity of the enzyme with pH. The reaction solution 1 mL contained 1.80 IU of the enzyme, 100 μM H2O2, 200 μM p-cresol in 50 mM sodium phosphate buffer at 30°C. The pH of the reaction solution was varied from 2 to 10.
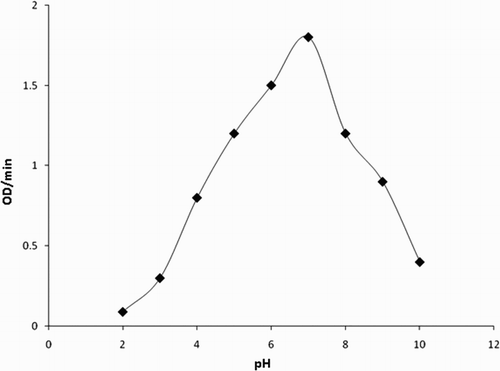
Figure 7. UV–VIS spectra of (a) p-cresol in 50 mM phosphate buffer pH 7.0. (b) Enzyme with p-cresol in 50 mM phosphate buffer pH 7.0. (c) Added H2O2 to initiate the reaction. Change in spectra of p-cresol at zero time, 2 min, 4 min, 6 min and 8 min. (d) Added H2O2 to initiate the reaction. Change in spectra of enzyme at zero time, 2, 4 and 6 min. (e) Change in spectra of enzyme and substrate p-cresol after 1 h.
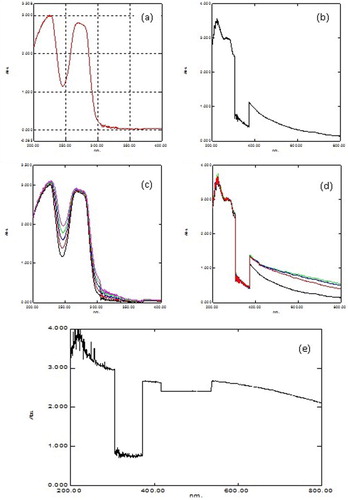
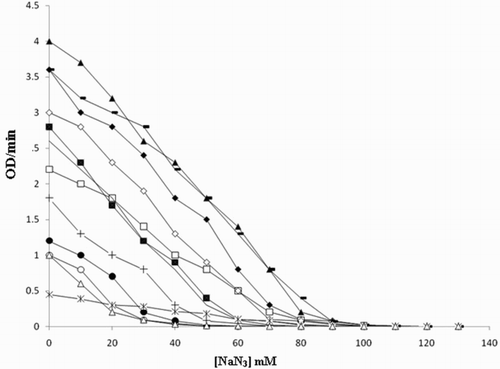
Peroxidase activity was inhibited by sodium azide (Citation27, Citation28) when studied using different phenolic substrates as shown in . Enzyme activity is inhibited using guaiacol as a substrate at 80 mM, m-cresol at 80 mM, p-cresol at 100 mM, o-cresol at 60 mM, anisole at 40 mM, resorcinol at 40 mM, catechol at 50 mM, pyrogallol at 100 mM, hydroquinone at 60 mM, phenol at 80 mM, veratryl alcohol at 80 mM and phoroguicinol at 40 mM in the reaction mixture. It is noted that greater concentrations of inhibitor were required to inhibit the activity of peroxidase, when p-cresol and pyrogallol were used as substrates. Thus, it is convenient to use p-cresol as a substrate for enzyme assay. Inhibitors such as potassium cyanide and ammonium hydroxide were also checked for activity inhibition and found that sodium azide was significantly more effective in reducing the activity of the peroxidase compared to these inhibitors.
Figure 8. Inhibition of peroxidase activity by sodium azide using different substrates: (i) ◆ guaiacol, (ii) □ m-cresol, (iii) ▲ p-cresol, (iv) ■ ocresol, (v) ○ anisole, (vi) ● resorcinol, (vii)+ catechol, (viii) −pyrogallol, (ix)−hydroquinone, (x) ◇phenol, (xi) ∗ veratrylalcohol and (xii) △ phoroguicinol.
Conclusion
In conclusion, this communication reports that the enzyme isolated from L. aegyptiaca efficiently converts phenol and their derivatives to the respective quinones at low cost and within ecofriendly green chemistry domain. This research work demonstrates an enzymatic process to remove toxic phenolic organic compounds from industrial wastewaters and agricultural wastes. Enzyme-based technology is to be explored as a viable treatment process for the conversion of phenol and its derivatives into less harmful quinones, which can be removed using adsorption methods more conveniently.
Acknowledgements
The authors are thankful to Department of Chemistry, North Eastern Regional Institute of Science and Technology, Nirjuli for providing facilities to do this work.
Disclosure statement
No potential conflict of interest was reported by the authors.
Notes on contributors
Dr. Meera Yadav obtained her Ph.D. from D.D.U. Gorakhpur University, Gorakhpur. She is presently working as an Assistant Professor in the Chemistry Department of North Eastern Regional Institute of Science and Technology, Nirjuli, Itanagar. Her current interest is in purification, characterization and applications of Metalloenzymes in organic and Inorganic chemistry.
Nivedita Rai is a research scholar working under the supervision of Dr. Meera Yadav, Department of chemistry, North Eastern Regional Institute of science and Technology, Nirjuli, Itanagar, AP, India.
Hardeo Singh Yadav is Senior Professor in the Department of chemistry, North Eastern Regional Institute of science and Technology, Nirjuli, Itanagar, AP, India. His research area of interest is in synthesis of inorganic & bio-inorganic complexes, coordination chemistry of oxocations, macrocyclic complexes, amavadin chemistry, green chemistry synthesis and catalysis, peroxidase activities in green vegetables / fruits, thallium chemistry in MRI and clean water technology.
Additional information
Funding
References
- Cohen, S.; Paula, A.; Belinky, P.A.; Hadar, Y.; Dosoretz, C.G. Characterization of Catechol Derivative Removal by Lignin Peroxidase in Aqueous Mixture. Bioresour. Technol. 2009, 100, 2247–2253. doi: 10.1016/j.biortech.2008.11.007
- Nicell, J.A.; Bewtra, J.K.; Biswas, N.; St. Pierre, C.C.; Taylor, K.E. Enzyme Catalyzed Polymerization and Precipitation of Aromatic Compounds from Aqueous Solution. Can. J. Civ. Eng. 1993, 20, 725–735. doi: 10.1139/l93-097
- Bogan, B.W.; Lamar, R.T.; Burgos, W.D.; Tien, M. Extent of Humification of Anthracene, Flouranthene and Benzo[a]pyrene by Pleurotus Ostreatus During Growth in PAH-Contaminated Soils. Lett. App. Microbiol. 1999, 28, 250–254. doi: 10.1046/j.1365-2672.1999.00537.x
- Bogan, B.W.; Lamar, R.T. Polycyclic Aromatic Hydrocarbons Degrading Capabilities of Phenerochaete Laevis HHB-1625 and Its Extracellular Ligninolytic Enzymes. Appl. Environ. Microbiol. 1996, 62, 1597–1603.
- Bogan, B.W.; Lamar, R.T.; Hammel, K.E. Fluorene Oxidation In Vivo by Phanerochaete Chrysosporium and In Vitro During Manganese Peroxidase-Dependent Lipid Peroxidation. Appl. Environ. Microbiol. 1996, 62, 1788–1792.
- Bumpus, J.A. Biodegradation of Polycyclic Aromatic Hydrocarbons by Phanerochaete Chrysosporium. Appl. Environ. Microbiol. 1989, 55, 154–158.
- Klibanov, A.M.; Alberti, B.N.; Morris, E.D.; Felshin, L.M. Enzymatic Removal of Toxic Phenols and Anilines from Waste Waters. J. Appl. Biochem. 1980, 2, 414–421.
- Karam, J.; Nicell, J.A. Potential Applications of Enzymes in Waste Treatment. J. Chem. Technol. Biotechnol. 1997, 69, 141–153. doi: 10.1002/(SICI)1097-4660(199706)69:2<141::AID-JCTB694>3.0.CO;2-U
- Banci, L. Structural Properties of Peroxidases. J. Biotechnol. 1997, 53, 253–263. doi: 10.1016/S0168-1656(97)01677-5
- Yemenicioglu, A.; Ozkan, M.; Cemeroglu, B. Partial Purification and Thermal Characterization of Peroxidase from Okra (Hibiscus Esculentum). J. Agri. Food Chem. 1998, 46, 4158–4163. doi: 10.1021/jf980456o
- Gholami-Borujeni, F.; Mahvi, A.H.; Naseri, S.; Faramarzi, M.A.; Nabizadeh, R.; Alimohammadi, M. Application of Immobilized Horseradish Peroxidase for Removal and Detoxification of Azo Dye from Aqueous Solution. Res. J. Chem. Environ. 2011, 15, 217–222.
- Temoçin, Z.; Yiğitoğlu, M. Studies on the Activity and Stability of Immobilized Horseradish Peroxidase on Poly(Ethylene Terephthalate) Grafted Acrylamide Fiber. Bioprocess Biosyst. Eng. 2009, 32, 467–474. doi: 10.1007/s00449-008-0266-9
- Nicell, J.A. Kinetics of Horseradish Peroxidase-Catalyzed Polymerization and Precipitation of Aqueous 4-Clorophenol. J. Chem. Technol. Biotechnol. 1994, 60, 203–215. doi: 10.1002/jctb.280600214
- Guzik, U.; Hupert-Kocurek, K.; Wojcieszyńska, D. Immobilization as a Strategy for Improving Enzyme Properties – Application to Oxidoreductase. Molecules 2014, 19, 8995–9018. doi: 10.3390/molecules19078995
- Alberti, B.N.; Klibanov, A.M. Enzymatic Removal of Dissolved Aromatics from Industrial Aqueous Effluents. Biotechnol. Bioeng. Symp. 1981, 11, 373–379.
- Klibanov, A.M.; Tu, T.; Scott, K.P. Peroxidase-Catalyzed Removal of Phenols from Coal-Conversion Waste Waters. Science 1983, 221, 259–261. doi: 10.1126/science.221.4607.259-a
- Cooper, V.A.; Nicell, J.A. Removal of Phenols from a Foundry Wastewater Using Horseradish Peroxidase. Water Res. 1996, 30, 954–964. doi: 10.1016/0043-1354(95)00237-5
- Bayramoğlu, G.; Arıca, M.Y. Enzymatic Removal of Phenol and p-Chlorophenol in Enzyme Reactor: Horseradish Peroxidase Immobilized on Magnetic Beads. J. Hazard. Mater. 2008, 156, 148–155. doi: 10.1016/j.jhazmat.2007.12.008
- Galarraga, J.C.; dos Santos, A.F.; Bassan, J.C.; Goulart, A.J.; Monti, R. Bromophenol Blue Discoloration Using Peroxidase Immobilized on Highly Activated Corncob Powder. Rev. Ciênc. Farm. Basica. Apl. 2013, 34, 321–326.
- Klibanov, A.M. Massachusetts Institute of Technology, Removal of Combined Organic Substances from Aqueous Solutions, Int. Cl.4 C02F 1/54, U.S. n. 4,623,465, oct. 29 (1985), nov.18 (1986).
- Whitaker, J.R.Principles of Enzymology for Food Sciences;Marcel Decker: New York, 1972.
- Vernwal, S.K.; Yadav, R.S.S.; Yadav, K.D.S. Musa Paradisiaca Stem Juice as a Source of Peroxidase and Lignin Peroxidase. Indian J. Exp. Biol. 2000, 38, 1036–1040.
- Engel, P.C.Enzyme Kinetics: The Steady State Approach;Chapman and Hall: London, 1977.
- Yadav, M., Yadav, K.D.S. Enzymatic Characteristics of Ligninperoxidase by Penicillium Citrinum, Fusarium Oxysporum and Aspergillus Terreus Using n-Propanol as the Substrate. Indian J. Biochem. Biophys. 2006, 43, 48–51.
- Yadav, R.S.S.; Yadav, K.S.; Yadav, H.S. Luffa aegyptiaca (Gourd) Fruit Juice as a Source of Peroxidase. Enzyme Res. 2011, Article ID 319105, 5 Pages.
- El-Hamshary, H.; El-Newehy, M.H.; Al-Deyab, S. Oxidation of Phenol by Hydrogen Peroxide Catalysed by Metal-Containing Poly (Amidoxime) Grafted Starch. Molecules 2011, 16, 9900–9911. doi: 10.3390/molecules16129900
- Yadav, M., Singh, S.K.; Yadav, K.D.S. Purification and Characterization of Lignin Peroxidase from Pleurotus Sajor Caju MTCC-141. J. Wood Chem. Technol. 2009, 29, 59–74. doi: 10.1080/02773810802596489
- Hochman, A.; Shemesh, A. Purification and Characterization of a Catalase-Peroxidase from the Photosynthetic Bacterium Rhodo Pseudomonas Capsulate. J. Biol. Chem. 1987, 262, 6871–6876.