
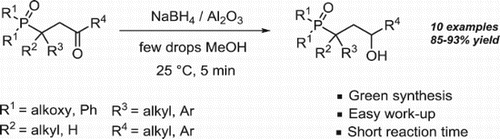
ABSTRACT
A green and versatile method for the preparation of γ-hydroxyphosphonate and phosphine oxide derivatives in 5 min, is accomplished through the reduction of γ-phosphonylketones with sodium borohydride supported on alumina, at room temperature, under solvent free conditions.
GRAPHICAL ABSTRACT
Introduction
An increasing interest has been paid for several years to the synthesis of hydroxyphosphonates due to their promising pharmacological activities. Much of these activities has been attributed to the physical and structural similarity of hydroxyphosphonates and phosphonic acids to the biologically important natural hydroxycarboxylic acids, in which a carboxylic group is replaced by phosphonic or related functionality ( Citation1). Such structural similarity enables hydroxyphosphonates to inhibit, by mimetic effect, enzymes involved in hydroxycarboxylic acid metabolism and thus affects a variety of physiological processes ( Citation2). For instance, α-hydroxyphosphonates, which are structural analogs of α-hydroxyacids, are an important class of compounds in medicinal chemistry with potential biological effects as inhibitors of human renin ( Citation3) and HIV protease ( Citation4). Some are also medicinally important as antitumor ( Citation5), antibacterial ( Citation6), antiviral ( Citation7) and antioxidant ( Citation8) agents.
On the other hand, β-hydroxyphosphonates are known for their interesting biological activities as 5′-nucleotidase ( Citation9), autotaxin ( Citation10) and sphingomyelinase ( Citation2) inhibitors, antioxidant ( Citation11) and antibiotic ( Citation12) agents.
Although α- and β-hydroxyphosphonates are well described in the literature, their γ-hydroxyphosphonate homologues did not receive sufficient attention despite their structural resemblance to γ-hydroxybutyric acid (GHB), wich is a naturally occurring neurotransmitter and a psychoactive drug used in a medical setting as a general anesthetic and as a treatment for cataplexy, narcolepsy and alcoholism ( Citation13–15). It is a precursor to GABA, glutamate, and glycine in certain brain areas, and it is an agonist at the GHB and GABAB receptors ( Citation16, Citation17).
There is only a limited number of synthetic approaches to γ-hydroxyphosphonates. This includes (i) the BF3.OEt2- or TiCl4-catalyzed nucleophilic ring opening reaction of epoxides by methanephosphonates ( Citation18–20) and (ii) the oxazaborolidine-catalyzed catecholborane reduction of γ–ketophosphonates ( Citation21, Citation22). However, in spite of their potential utility, these procedures suffer from one or the other drawbacks such as the use of expensive catalysts and hydride reducing agents, where specialized handling techniques and tedious work-up are necessary, as well as long reaction time, use of solvent and lack of generality. Therefore, the development of a simple, mild, eco-benign and low cost protocol is still desirable to obtain a wider variety of γ-hydroxyphosphonates for biological screening.
In recent years, NaBH4 supported on inorganic solid supports especially alumina, has been reported in literature ( Citation23) as an efficient and eco-benign reagent for the reduction of aldehydes and ketones. With this in mind, and in the continuation of our interest to develop efficient protocols for the synthesis of biologically active phosphonates ( Citation24–27), we report herein an operationally simple, green and fast methodology for the synthesis of γ-hydroxyphosphonates and phosphine oxides, in excellent yields, through the reduction of γ-phosphonylketones with NaBH4 supported on alumina, at room temperature, under solvent-free conditions, within few minutes.
Results and discussion
In order to establish the optimum reaction conditions for the formation of the target compounds, we used γ-ketophosphonate 1a as model substrate. The reduction of 1a was initially carried out with one hydride equivalent of LiAlH4 in anhydrous THF. Unfortunately, the expected γ-hydroxyphosphonate 2a was not obtained (, entry 1). Although the starting materials were completely consumed, the 31P NMR analysis of the reaction mixture showed complete dephosphonylation due to C–P bond cleaving ( Citation24, Citation25, Citation28). When the reaction was conducted with 1 equiv of NaBH4 in EtOH at room temperature, the desired product 2a was isolated in only 30% yield (, entry 2). Further improvement of the yield to 65% was observed when using 3 equiv of NaBH4, under the same reaction conditions (, entry 3). It was gratifying to observe that performing the reduction with NaBH4 (3 equiv) supported on alumina (12 equiv), in the presence of few drops of MeOH (5 drops for 1 mmol 1a), at room temperature, under solvent-free conditions, afforded the desired product 2a in 90% yield, within 5 min (, entry 4).
Table 1. Optimization of the reaction conditions.
With optimized reaction conditions in hand, we next studied the scope of this methodology. A variety of structurally diverse γ-phosphonylketones were investigated and a series of γ-hydroxyphosphonates and phosphine oxides of type 2 were afforded in high to excellent yields (). Pure products can be isolated by simple extraction without the need for chromatography or recrystallization.
Table 2. Substrate scope studies.
Compounds 2a–g, presenting two stereocenters, were obtained as a mixture of two unseparable diastereoisomers, as evidenced by their NMR spectral data. The relative proportions of these diastereoisomers were estimated from the 31P NMR spectra where a singlet for each one was found ().
Conclusion
We have successfully developed an efficient, green and fast methodology for the synthesis of γ-hydroxyphosphonates and phosphine oxides, through the reduction of γ-phosphonylketones with NaBH4 supported on alumina, in the presence of few drops of MeOH, at room temperature, under solvent-free conditions. This synthetic strategy offers significant advantages such as high to excellent yields, short reaction time, versatile substituents, mild reaction conditions, easy work-up and environmental safety, what make this protocol more amenable for high throughput library synthesis. These compounds are potential agonists at the GHB and GABAB receptors. This aspect is currently under study in our laboratory and will be reported in due course.
Experimental
General informations
Commercially available analytical grade reagents were used without further purification. Melting points were measured in an open capillary, using Büchi melting point B-540 apparatus and are uncorrected. 1H, 31P and 13C NMR spectra were recorded with CDCl3 as the solvent, on a Bruker AC-300 spectrometer operating at 300.1 MHz for 1H, 121.5 MHz for 31P and 75.5 MHz for 13C. The chemical shifts are reported in ppm relative to Tetramethylsilane (internal reference) for 1H and 13C NMR and relative to 85% H3PO4 (external reference) for 31P NMR. The coupling constants are reported in Hz. For the 1H NMR, the multiplicities of signals are indicated by the following abbreviations: s: singlet; d: doublet; t: triplet; q: quartet; quint: quintet; m: multiplet and br s: broad singlet. Mass spectra were determined on a Thermo LTQ Orbitrap XL spectrometer, or on a VOYAGER DE STR spectrometer. IR spectra were recorded on a Nicolet IR200 spectrometer. The progress of the reactions was monitored by TLC (Thin Layer Chromatography), using 5 × 20 cm plates with a layer thickness of 0.25 mm (Silica gel 60 F254), and ether as eluent.
Synthesis of γ-phosphonylketones 1
The starting γ-ketophosphonates and phosphine oxides 1 were prepared according to the reported procedures ( Citation29, Citation30) (see Electronic Supplementary Information).
General procedure for the synthesis of γ-hydroxyphosphonates and phosphine oxides 2
A round-bottomed flask equipped with a magnetic stir bar was charged with Al2O3 (12 mmol) and NaBH4 (3 mmol). The mixture was stirred for 30 min at 25°C, under nitrogen atmosphere. The γ-ketophosphonate or phosphine oxide (1 mmol) and five drops of MeOH were added to this mixture and stirring was continued for 5 min (reactions were monitored by TLC). The reaction mixture was poured into ether (15 mL) and then filtred. The filtrate was washed with water (2 × 5 mL). The organic phase was dried over Na2SO4 and concentrated under vacuum to afford the pure products in 85–93% isolated yields.
Characterization data of compounds 2
Diethyl (3-hydroxy-1-phenylbutyl) phosphonate (2a) (Citation31). Transparent oil; 1H NMR (300 MHz, CDCl3): δ = 1.09 (t, 3H, 3JHH = 6.0 Hz, CH3-CH2-O, min); 1.10 (t, 3H, 3JHH = 6.0 Hz, CH3-CH2-O, min); 1.15 (t, 3H, 3JHH = 6.0 Hz, CH3-CH2-O, maj); 1.16 (t, 3H, 3JHH = 6.0 Hz, CH3-CH2-O, maj); 1.24 (d, 3H, 3JHH = 6.0 Hz, CH3-CH, min); 1.26 (d, 3H, 3JHH = 6.0 Hz, CH3-CH, maj); 2.04–2.19 (m, 2H, CH2-CH); 2.68 (br s, 1H, OH); 3.20–3.47 (m, 1H, CH-P=O); 3.65–3.79 (m, 1H, CH3-CH); 3.85–4.10 (m, 4H, 2CH3-CH2-O); 7.24–7.32 (m, 5H, arom-H); 13C NMR (75.5 MHz, CDCl3): δ = 16.2 (d, 3JCP = 4.5 Hz, 2CH3-CH2-O, maj); 16.3 (d, 3JCP = 4.5 Hz, 2CH3-CH2-O, min); 22.7 (s, CH3-CH, min); 24.2 (s, CH3-CH, maj); 39.3 (s, CH2-CH, maj); 39.9 (s, CH2-CH, min); 41.2 (d, 1JCP = 103.4 Hz, CH-P, min); 41.4 (d, 1JCP = 102.6 Hz, CH-P, maj); 62.0 (d, 2JCP = 6.0 Hz, 2CH3-CH2-O, maj); 62.7 (d, 2JCP = 5.3 Hz, 2 CH3-CH2-O, min); 64.7 (d, 3JCP = 10.6 Hz, CH-OH, min); 65.6 (d, 3JCP = 8.3 Hz, CH-OH, maj); phenyl carbons: δ = 127.1; 127.2; 127.2; 127.2; 128.5; 128.6; 128.6; 129.1; 129.2; 129.4; 129.4; 136.0; 136.1; 136.5; 136.6; 31P NMR (CDCl3, 121.5 MHz): δ (ppm) = 29.1 (s, 61%, maj); 29.8 (s, 39%, min); IR (neat): νP = O = 1266 cm−1; νOH = 3427 cm−1; ESI-HRMS: m/z calculated for C14H24O4P [M + H]+: 287.14122; found: 287.14208.
Diethyl (3-hydroxy-1-(thiophen-2-yl)butyl) phosphonate (2b). Yellow oil; 1H NMR (300 MHz, CDCl3) : δ = 1.04 (d, 3H, 3JHH = 6.0 Hz, CH3-CH, min); 1.10 (d, 3H, 3JHH = 9.0 Hz, CH3-CH, maj); 1.13 (t, 3H, 3JHH = 6.0 Hz, CH3-CH2-O, maj); 1.15 (t, 3H, 3JHH = 6.0 Hz, CH3-CH2-O, min); 1.22 (t, 3H, 3JHH = 6.0 Hz, CH3-CH2-O, maj); 1.29 (t, 3H, 3JHH = 6.0 Hz, CH3-CH2-O, min); 1.85–2.12 (m, 2H, CH2-CH); 3.21 (br s, 1H, OH); 3.42–3.64 (m, 1H, CH-P=O); 3.66–3.78 (m, 1H, CH3-CH); 3.64–4.06 (m, 4H, 2CH3-CH2-O); 6.88–7.26 (m, 3H, arom-H); 13C NMR(75.5 MHz, CDCl3): δ = 16,2(d, 3JCP = 6.0 Hz, CH3-CH2-O); 16.3 (d, 3JCP = 6.0 Hz, CH3-CH2-O); 22.6 (s, CH3-CH, min); 24.1 (s, CH3-CH, maj); 36.3 (d, 1JCP = 144,1 Hz, CH-P, maj); 36.4 (d, 1JCP = 141,9 Hz, CH-P, min); 40.6 (s, CH2-CH, maj); 41.1 (s, CH2-CH, min); 62.3 (d, 2JCP = 7.5 Hz, CH3-CH2-O); 62.8 (d, 2JCP = 6.8 Hz, CH3-CH2-O); 64.3 (d, 3JCP = 13.6 Hz, CH-OH, maj); 65.2 (d, 3JCP = 10.6 Hz, CH-OH, min); phenyl carbons: δ = 124.5; 124.5; 125.9; 126.4; 126.5; 126.8; 126.8; 126.9; 127.0; 133.4; 138.1; 138.3; 138.4; 138.6; 138.7; 31P NMR (CDCl3, 121.5 MHz): δ (ppm) = 26.9 (s, 16%, min); 27.6 (s, 84%, maj); IR (neat): νP=O = 1242 cm−1; νOH = 3396 cm−1; MALDI-MS: m/z = 293.2 ([M + H]+).
Diethyl (3-hydroxy-1,3-diphenylpropyl) phosphonate (2c). Transparent oil; 1H NMR (300 MHz, CDCl3): δ = 1.03 (t, 3H, 3JHH = 6.0 Hz, CH3-CH2-O, min); 1.04 (t, 3H, 3JHH = 6.0 Hz, CH3-CH2-O, min); 1.16 (t, 3H, 3JHH = 6.0 Hz, CH3-CH2-O, maj); 1.17 (t, 3H, 3JHH = 6.0 Hz, CH3-CH2-O, maj); 1.80 (br s, 1H, OH); 2.20–2.46 (m, 2H, CH2-CH); 2.89–3.50 (m, 1H, CH-P=O); 3.62–3.99 (m, 4H, 2CH3-CH2-O); 4.32–4.64 (m, 1H, CH-OH); 7.14–7.34 (m, 10H, arom-H); 13C NMR (75.5 MHz, CDCl3): δ = 16.2 (d, 3JCP = 6.0 Hz, 2CH3-CH2-O, min); 16.4 (d, 3JCP = 6.0 Hz, 2CH3-CH2-O, maj); 39.4 (s, CH2-CH, maj); 39.5 (s, CH2-CH, min); 41.1 (d, 1JCP = 104.1 Hz, CH-P, maj); 41.4 (d, 1JCP = 102.6 Hz, CH-P, min); 62.1 (d, 2JCP = 7.1 Hz, 2CH3-CH2-O, min); 62.7 (d, 2JCP = 7.2 Hz, 2CH3-CH2-O, maj); 72.2 (d, 3JCP = 14.4 Hz, CH-OH, maj); 72.3 (d, 3JCP = 13.8 Hz, CH-OH, min); phenyl carbons: δ = 125.6; 126.3; 127.3; 127.3; 127.6; 127.6; 127.9; 127.9; 128.5; 128.5; 128.6; 128.7; 129.2; 129.3; 129.4; 129.5; 135.8; 135.9; 136.1; 136.2; 136.3; 136.3; 143.3; 143.4; 31P NMR (CDCl3, 121.5 MHz): δ (ppm) = 28.6 (s, 65%, maj); 29.3 (s, 35%, min) IR (neat): νP=O = 1236 cm−1; νOH = 3388 cm−1; ESI-HRMS: m/z calculated for C19H26O4P [M + H]+: 349.15687; found: 349.15766.
(1-(Furan-2-yl)-3-hydroxybutyl) diphenylphosphine oxide (2d). Brown solid; mp 146–148°C; 1H NMR (300 MHz, CDCl3) : δ = 1.10 (d, 3H, 3JHH = 6.0 Hz, CH3-CH, maj); 1.13 (d, 3H, 3JHH = 6,0 Hz, CH3-CH, min); 1.85–2.23 (m, 2H, CH2-CH); 2.94 (br s, 1H, OH); 3.85–4.06 (m, 1H, CH-P=O); 3.62–4.29 (m, 1H, CH3-CH); 6.10–7.87 (m, 13H, arom-H); 13C NMR (75.5 MHz, CDCl3): δ = 22.6 (s, CH3-CH, min); 24.2 (s, CH3-CH, maj); 37.3 (s, CH2-CH, maj); 37.6 (s, CH2-CH, min); 38.0 (d, 1JCP = 69.6 Hz, CH-P, min); 38.2 (d, 1JCP = 68,3 Hz, CH-P, maj); 64.9 (d, 3JCP = 8.3 Hz, CH-OH, maj); 65.3 (d, 3JCP = 11.3 Hz, CH-OH, min). Phenyl carbons: δ = 109.1; 109.2; 109.3; 109.3; 110.7; 110.7; 110.8; 110.9; 128.0; 128.2; 128.2; 128.4; 128.6; 128.7; 129.9; 130.5; 130.6; 130.7; 131.2; 131.4; 131.5; 131.6; 131.6; 131.7; 131.8; 131.8; 131.8; 131.9; 132.0; 132.0; 141.6; 141.6; 141.7; 141.8; 149.4; 149.5; 149.6; 149.6; 31P NMR (CDCl3, 121.5 MHz): δ (ppm) = 33.0 (s, 46%, min), 33.8 (s, 54%, maj); IR (neat): νP=O = 1222 cm−1; νOH = 3351 cm−1; ESI-HRMS: m/z calculated for C20H22O3P [M + H]+: 341.13066; found: 341.13105.
(3-Hydroxy-1-(thiophen-2-yl)butyl) diphenylphosphine oxide (2e). White solid; mp 200–202°C; 1H NMR (300 MHz, CDCl3): δ = 1.03 (d, 3H, 3JHH = 6.0 Hz, CH3-CH, maj); 1.04 (d, 3H, 3JHH = 6.0 Hz, CH3-CH, min); 1.77–2.21 (m, 2H, CH2-CH); 3.63 (br s, 1H, OH); 3.76–4.00 (m, 1H, CH-P=O); 3.92–4.29 (m, 1H, CH3-CH); 6.75–7.88 (m, 13H, arom-H); 13C NMR (75.5 MHz, CDCl3): δ = 21.6 (s, CH3-CH, maj); 23.3 (s, CH3-CH, min); 37.7 (d, 1JCP = 70.9 Hz, CH-P, min); 38.4 (d, 1JCP = 68.7 Hz, CH-P, maj); 39.4 (s, CH2-CH); 63.8 (d, 3JCP = 8.3 Hz, CH-OH). Phenyl carbons: δ = 123.7; 123.7; 127.0; 127.1; 127.1; 127.3; 127.3; 127.7; 127.9; 127.9; 130.0; 130.0; 130.1; 130.1; 130.2; 130.2; 130.2; 130.2; 130.3; 130.3; 130.4; 130.4; 130.4; 130.4; 130.5; 136.4; 136.5; 136.6; 31P NMR (CDCl3, 121.5 MHz): δ (ppm) = 33.1 (s, 38%, min), 34.0 (s, 62%, maj); IR (neat): νP=O = 1267 cm−1; νOH = 3395 cm−1; ESI-HRMS: m/z calculated for C20H22O2PS [M + H]+: 357.10781; found: 357.10643.
(3-Hydroxy-1-phenylbutyl) diphenylphosphine oxide (2f). White solid; mp 133–135°C; 1H NMR (300 MHz, CDCl3) : δ = 0.97 (d, 3H, 3JHH = 6.0 Hz, CH3-CH, min); 0.98 (d, 3H, 3JHH = 6.0 Hz, CH3-CH, maj); 1.76–2.35 (m, 2H, CH2-CH); 3.16 (br s, 1H, OH); 3.34–3.59 (m, 1H, CH-P=O); 3.62–3.97 (m, 1H, CH3-CH); 6.85–7.90 (m, 15H, arom-H); 13C NMR (75.5 MHz, CDCl3): δ = 21.4 (s, CH3-CH, maj); 23.3 (s, CH3-CH, min); 37.6 (s, CH2-CH, maj); 37.9 (s, CH2-CH, min); 42.0 (d, 1JCP = 68.7 Hz, CH-P, min); 43.1 (d, 1JCP = 67.2 Hz, CH-P, maj); 63.6 (d, 3JCP = 12.1 Hz, CH-OH, min); 64.1 (d, 3JCP = 10.6 Hz, CH-OH, maj). Phenyl carbons: δ = 125.9; 125.9; 126.0; 126.0; 126.8; 126.9; 126.9; 127.0; 127.0; 127.1; 127.2; 127.2; 127.4; 127.4; 127.5; 127.6; 127.8; 128.7; 128.8; 129.0; 129.1; 129.7;129.8; 129.9; 130.0; 130.0; 130.1; 130.1; 130.3; 130.4; 130.4; 130.4; 130.5; 130.7; 130.7; 130.8; 130.9; 131.0; 131.3; 131.8; 134.7; 134.8; 134.8; 134.9; 31P NMR (CDCl3, 121.5 MHz): δ (ppm) = 34.2 (s, 48%, min); 34.3 (s, 52%, maj); IR (neat): νP=O = 1266 cm−1; νOH = 3427 cm−1; MALDI-MS: m/z = 351.2 ([M + H]+).
(3-Hydroxy-1,3-diphenylpropyl) diphenylphosphine oxide (2g) (Citation32). White solid; mp 204–206°C; 1H NMR (300 MHz, CDCl3): δ = 2.14–2.77 (m, 2H, CH2-CH); 3.08–3.24 (m, 1H, CH-P=O); 3.33–3.73 (m, 1H, CH-OH); 4.45 (br s, 1H, OH); 7.15–7.57 (m, 20H, arom-H). 13C NMR (75.5 MHz, CDCl3): δ = 38.6 (s, CH2-CH); 43.4 (d, 1JCP = 67.4 Hz, CH-P); 72.2 (d, 3JCP = 13.6 Hz, CH-OH); Phenyl carbons: δ = 125.5; 126.2; 126.8; 127.1;127.8; 127.9; 128.0; 128.4; 128.4; 128.6; 129.7; 129.1; 129.8; 129.9; 130.3; 130.7; 130.9; 131.0; 131.3; 131.4; 131.6; 131.8; 132.0; 132.6; 135.1; 135.2; 139.3; 143.1. 31P NMR (CDCl3, 121.5 MHz): δ (ppm) = 34.1 (s, 8%, min), 34.2 (s, 92%, maj). IR (neat): νP=O = 1239 cm−1; νOH = 3251 cm−1; MALDI-MS: m/z = 413.3 ([M + H]+).
Diethyl (4-hydroxy-2-methylpentan-2-yl) phosphonate (2h). Transparent oil; 1H NMR (300 MHz, CDCl3) : δ = 0.92 (d, 3H, 3JHH = 6.0 Hz, CH3-CH); 1.00 (d, 3H, 3JPH = 18.0 Hz, CH3-C); 1.06 (d, 3H, 3JPH = 15.0 Hz, CH3-C); 1.09 (t, 6H, 3JHH = 6.0 Hz, 2 CH3-CH2-O); 1.52–1.92 (m, 2H, CH2-CH), 3.76–3.85 (m, 1H, CH-CH3); 3.86–4.24 (m, 4H, 2 CH3-CH2-O); 4.04 (br s, 1H, OH); 13C NMR (75.5 MHz, CDCl3): δ = 16.2 (d, 3JCP = 6.0 Hz, 2 CH3-CH2-O); 21.6 (s, CH3-CH); 23.5 (s, CH3-C); 24.4 (s, CH3-C); 33.6 (d, 1JCP = 139.5 Hz, C–P); 48.2 (s, CH2-C); 61.9 (d, 2JCP = 7.5 Hz, 2CH3-CH2-O); 72.1 (d, 3JCP = 9.0 Hz, CH-OH). 31P NMR (CDCl3, 121.5 MHz): δ (ppm) = 28.2; IR (neat): νP=O = 1238 cm−1; νOH = 3462 cm−1; ESI-HRMS: m/z calculated for C10H24O4P [M + H]+: 239.14122; found: 239.14215.
Dimethyl (4-hydroxy-2-methylpentan-2-yl) phosphonate (2i) (Citation33). Transparent oil; 1H NMR (300 MHz, CDCl3): δ = 0.72 (d, 3H, 3JHH = 6.0 Hz, CH3-CH); 0.75 (d, 3H, 3JPH = 15.0 Hz, CH3-C); 0.81 (d, 3H, 3JPH = 18.0 Hz, CH3-C); 1.25–1.44 (m, 2H, CH2-CH), 3.25–3.31 (m, 1H, CH-CH3); 3.34 (d, 6H, 3JPH = 6.0 Hz, 2CH3-O); 3.91 (br s, 1H, OH); 13C NMR (75.5 MHz, CDCl3): δ = 21.8 (s, CH3-C); 23.5 (s, CH3-C); 24.5 (s, CH3-CH); 33.9 (d, 1JCP = 139.7 Hz, C–P); 47.2 (s, CH2-C); 52.3 (d, 2JCP = 7.5 Hz, 2CH3-O); 71.6 (d, 3JCP = 8.8 Hz, CH-OH); 31P NMR (CDCl3, 121.5 MHz): δ (ppm) = 39.5; IR (neat): νP=O = 1239 cm−1; νOH = 3368 cm−1; ESI-HRMS: m/z calculated for C8H20O4P [M + H]+: 211.10992; found: 211.11067.
(4-Hydroxy-2-methylpentan-2-yl) diphenylphosphine oxide (2j). White solid; mp 160–162°C; 1H NMR (300 MHz, CDCl3) : δ = 0.99 (d, 3H, 3JHH = 6.0 Hz, CH3-CH); 1.15 (d, 3H, 3JPH = 15.0 Hz, CH3-C); 1.17 (d, 3H, 3JPH = 15.0 Hz, CH3-C); 1.31–1.83 (m, 2H, CH2-CH); 3.92–4.01 (m, 1H, CH-OH); 5.43 (br s, 1H, OH); 7.28–7.88 (m, 10H, arom-H); 13C NMR (75.5 MHz, CDCl3): δ = 22.4 (s, CH3-CH); 24.6 (s, CH3-C); 25,7 (s, CH3-C); 37.0 (d, 1JCP = 68.7 Hz, C–P); 49.1 (s, CH2-CH); 63.0 (d, 3JCP = 3.8 Hz, CH-OH); Phenyl carbons: δ = 128.1; 128.3; 129.3; 130.0; 130.1; 130.6; 131.2; 131.3; 131.5; 131.5; 131.6; 132.1; 132.2; 132.3; 132.4; 31P NMR (CDCl3, 121.5 MHz): δ (ppm) = 41.8; IR (neat): νP = O = 1266 cm−1; νOH = 3289 cm−1; ESI-HRMS: m/z calculated for C18H24O2P [M + H]+: 303.15139; found: 303.15082.
Supplement_Material.doc
Download MS Word (1.7 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Notes on contributors
Aicha Mhamdi is studying as a Ph.D. candidate at the University of Carthage – Faculty of Sciences of Bizerte (Tunisia), under supervision of Prof. Soufiane Touil. Her research has focused on the synthesis and reactivity of γ-ketophosphonates and phosphine oxides.
Soufiane Touil is Professor of Chemistry and Head of the Laboratory of Heteroatom Organic Chemistry, at the University of Carthage – Faculty of Sciences of Bizerte (Tunisia). He received his Ph.D. in organic chemistry, from the University of Tunis El Manar – Faculty of Sciences of Tunis (Tunisia) in 1998, and his Habilitation (HDR) in 2003, from the same university. Professor Touil’s research interests involve the chemistry and physical organic chemistry of organophosphorus, organosulfur and heterocyclic compounds: Development of synthetic methods, the study of mechanisms, reactivity, stereochemistry, investigation of biochemical properties and, taking this as a basis, creation of new approaches for finding biologically active substances of medicinal interest.
Additional information
Funding
Related Research Data
References
- Kolodiazhnyi, O.I. In Advances in Asymmetric Synthesis;Hassner, A., Ed.; JAI Press, Inc. Stamford-London, 1998, Vol. 3, Chap. 5; pp. 273–357; Olszewski, T. K. Synthesis (Stuttg) 2014, 46, 403–429; Olszewski T. K. Tetrahedron: Asymmetry 2015, 26, 393–399.
- Yokomatsu, T.; Murano, T.; Akiyama, T.; Koizumi, J.; Shibuya, S.; Tsuji, Y.; Soeda, S.; Shimeno, H. Bioorg. Med. Chem. Lett. 2003, 13, 229–236; Mauritz, R. P.; Schmelz, F. S.; Meier, C. Nucleosides Nucleotides 1999, 18, 1417–1418; Demmer, C. S.; Krogsgaard-Larsen, N.; Bunch, L. Chem. Rev., 2011, 111, 7981–8006; Proniewicz, E.; Pięta, E.; Zborowski, K.; Kudelski, A.; Boduszek, B.; Olszewski, T. K.; Kim, Y.; Proniewicz, L. M. J. Phys. Chem. A 2014, 118, 5614–5625; Pięta, E.; Proniewicz, E.; Boduszek, B.; Olszewski, T. K.; Nattich-Rak, M.; Kim, Y. Appl. Surf. Sci. 2015, 335, 167–183. doi: 10.1016/S0960-894X(02)00888-0
- Tao, M.; Bihovsky, R.; Wells, G.J.; Mallamo, J.P. J. Med. Chem. 1998, 41, 3912–3916. doi: 10.1021/jm980325e
- Stowasser, B.; Budt, K.-H.; Jian-Qi, L.; Peyman, A.; Ruppert, D. Tetrahedron Lett. 1992, 33, 6625–6628. doi: 10.1016/S0040-4039(00)61002-X
- Eummer, J.T.; Gibbs, B.S.; Zahn, T.J.; Sebolt-Leopold, J.S.; Gibbs, R.A. Bioorg. Med. Chem. 1999, 7, 241–250; Leung, C.-H.; Zhong, H.; Yang, H.; Cheng, Z.; Chan, D. S.; Ma, V. P.; Abagyan, R.; Wong, C.; Ma, D.-L. Angew. Chem. Int. Ed. 2012, 51, 9010–9014. doi: 10.1016/S0968-0896(98)00202-8
- Kategaonkar, A.H.; Pokalwar, R.U.; Sonar, S.S.; Gawali, V.U.; Shingate, B.B.; Shingare, M.S. Eur. J. Med. Chem. 2010, 45, 1128–1132. doi: 10.1016/j.ejmech.2009.12.013
- Snoeck, R.; Holy, A.; Dewolf-Peeters, C.; Van Den Oord, J.; De Clercq, E.; Andrei, G. Agents Chemother. 2002, 46, 3356–3361; Korshin, E. E.; Pozdeev, O.K. Tetrahedron 2013, 69, 11109–11115. doi: 10.1128/AAC.46.11.3356-3361.2002
- Aouani, I.; Lahbib, K.; Touil, S. Med. Chem. 2015, 11, 206–213. doi: 10.2174/1573406410666140706154046
- Hospital, A.; Meurillon, M.; Peyrottes, S.; Périgaud, C. Org. Lett, 2013, 15, 4778–4781. doi: 10.1021/ol402143y
- Cui, P.; Tomsig, J.L.; McCalmont, W.F.; Lee, S.; Becker, C.J.; Lynch, K.R.; Macdonald, T.L. Bioorg. Med. Chem. Lett. 2007, 17, 1634–1640. doi: 10.1016/j.bmcl.2006.12.114
- Gao, Y.; Wu, J.; Xu, J.; Zhang, P.; Tang, G.; Zhao, Y. RSC Adv. 2014, 4, 51776–51779. doi: 10.1039/C4RA10593A
- Takahashi, E.; Kimura, T.; Nakamura, K.; Arahira, M.; Iida, M. J. Antibiot. 1995, 48, 1124–1129. doi: 10.7164/antibiotics.48.1124
- Mayer, G. Expert Rev. Neurother. 2012, 12, 519–529. doi: 10.1586/ern.12.42
- Caputo, F.; Mirijello, A.; Cibin, M.; Mosti, A.; Ceccanti, M.; Domenicali, M.; Bernardi, M.; Maremmani, I.; Addolorato, G. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 1315–1320.
- Keating, G.M. Clin. Drug Investig. 2014, 34, 63–80. doi: 10.1007/s40261-013-0158-x
- Cash, C.; Gobaille, S.; Kemmel, V.; Andriamampandry, C.; Maitre, M. Biochem. Pharmacol. 1999, 58, 1815–1819. doi: 10.1016/S0006-2952(99)00265-8
- Maitre, M.; Humbert, J.P.; Kemmel, V.; Aunis, D.; Andriamampandry, C. Med. Sci. (Paris) 2005, 21, 284–289. doi: 10.1051/medsci/2005213284
- Racha, S.; Li, Z.; El-Subbagh, H.; Abushanab, E. Tetrahedron Lett. 1992, 33, 5491–5494. doi: 10.1016/S0040-4039(00)61125-5
- Li, Z.; Racha, S.; Dan, L.; El-Subbagh, H.; Abushanab, E. J. Org. Chem. 1993, 58, 5779–5783. doi: 10.1021/jo00073a043
- Pajkert, R.; Kolomeitsev, A.A.; Milewska, M.; Roschenthaler, G.V.; Koroniak, H. Tetrahedron Lett. 2008, 49, 6046–6049. doi: 10.1016/j.tetlet.2008.07.146
- Meier, C.; Laux, W.H.G. Tetrahedron: Asymmetry 1995, 6, 1089–1092. doi: 10.1016/0957-4166(95)00132-9
- Meier, C.; Laux, W.H.G. Tetrahedron 1996, 52, 589–598. doi: 10.1016/0040-4020(95)00855-1
- Varma, R.S.; Saini, R.K. Tetrahedron Lett. 1997, 38, 4337–4338; Shalbaf, H. Asian J. Chem. 2010, 22, 6761–6764. doi: 10.1016/S0040-4039(97)00968-4
- Samarat, A.; Yahyaoui, M.; Aouani, I.; Touil, S. Chem. Lett. 2013, 42, 687–688. doi: 10.1246/cl.130160
- Wahbi, A.; Mhamdi, A.; Hassen, Z.; Touil, S. Green Chem. Lett. Rev. 2014, 7, 73–78. doi: 10.1080/17518253.2014.895863
- Ben Gaied, L.; Touil, S.; Zantour, H. Phosphorus, Sulfur Silicon Relat. Elem. 2006, 181, 601–608. doi: 10.1080/10426500500269844
- Essid, I.; Touil, S. Curr. Org. Synth. 2017, 14, 272–278. doi: 10.2174/1570179413666160624092814
- Grimaldi, J.; Amedjkouh, M. Tetrahedron Lett. 2002, 43, 3761–3764. doi: 10.1016/S0040-4039(02)00521-X
- Touil, S.; Kremp, E.; Baccar, B.; Zantour, H. J. Soc. Chim. Tunisie 1997, 4, 85–95; Mironov, V. F.; Buzykin, B. I.; Garaev, R. S.; Tatarinov, D. A.; Kashapov, L. R.; Chestnova, R. V.; Nabiullin, V. N.; Iĺyasov, A. V.; Zobov, V. V. Russ. Chem. Bull. Int. Ed. 2014, 63, 2114–2125.
- Conant, J.B.; Braverman, J.; Hussey, R. J. Am. Chem. Soc. 1923, 45, 165–171; Bell, A.; Davidson, A. H.; Earnshaw, C.; Norrish, H. K.; Torr, R. S.; Trowbridge, D. B.; Warren, S. J. Chem. Soc., Perkin Trans. 1983, 1, 2879–2891. doi: 10.1021/ja01654a024
- Tomioka, H.; Inagaki, T.; Izawa, Y. J. Chem. Soc., Chem. Commun. 1976, 1023–1024. doi: 10.1039/C39760001023
- Miller, J.A.; Stewart, D. J. Chem. Soc., Perkin Trans. 1977, 1, 2416–2422. doi: 10.1039/p19770002416
- Pryazhnikov, E.G.; Skorinkin, A.I.; Garaev, R.S.; Giniatullin, R.A.; Vizel’, A.O.; Shchukina, L.I. Bull. Exp. Biol. Med. 2005, 139, 437–440. doi: 10.1007/s10517-005-0316-0