
ABSTRACT
The efficiency of conventional heating energy source compared with Infrared (IR), Ultrasound (US), Microwave and the simultaneous combination US–IR eco-friendly approaches for preparation of new N-(5-R1 -amino-2-nitrophenyl)acetamides and 5-R1-amino-2-nitroaniline by Nucleophilic Aromatic Substitution (SNAr) via addition–elimination reactions on the halogens F, Cl, Br, I, employing amines as nucleophiles were explored. Moreover, phenyldiazenyl derivatives in good yields by an oxidative one-pot SNAr-based amination reaction from an unusual oxidation of 2-phenylhydrazinyl derivatives in DMSO was prepared.
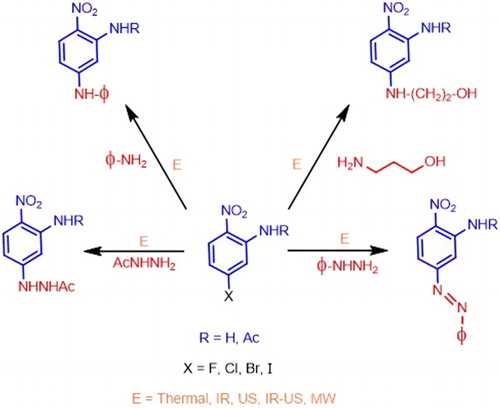
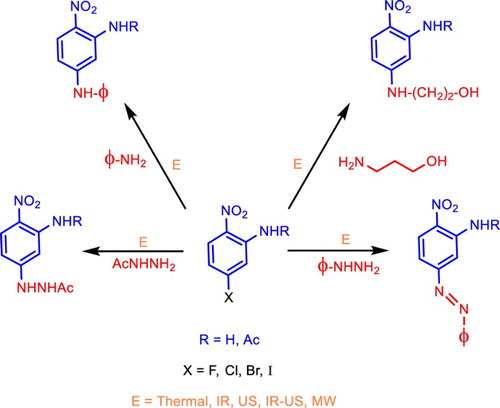
GRAPHICAL ABSTRACT
Introduction
In general, SNAr reactions are chemical transformations in which an anionic or neutral nucleophile displaces a potential leaving group attached to an aromatic ring. The presence of electron withdrawing groups in ortho and/or para positions with respect to the leaving group provides the main driving force to achieve SNAr, which reduce the electronic density of the aromatic ring, favoring the nucleophilic reaction.
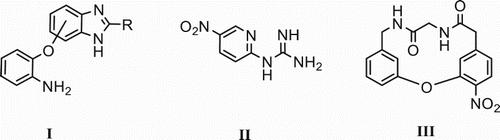
Nitro-halo aromatic derivatives are valuable building blocks to assemble simple and complex molecular structures of pharmaceutical and industrial importance, such as Vancomycin ( Citation1). Due to their high capacity for reactivity under a wide range of conditions, SNAr reactions have found important applications in the preparation of various kinds of biologically relevant intermediates, including azaheterocycle compounds such as benzimidazoles ( Citation2) I, 1-(phenyldi-2-yl) guanidine derivatives ( Citation3) II, and macrocyclic compounds ( Citation1) III ().
Figure 1. Selected compounds ensemble for a SNAr reaction.
The SNAr-based reactions not only as a new avenue of inquiry to synthesize strategic functionalized building blocks and fine chemicals have been explored, but also as an important synthetic pathway to access elaborated natural products as well as their application in the combinatorial chemistry for the preparation of important heterocyclic compounds.
Among many reported SNAr reactions are those made using 5-fluoro-2-nitroaniline ( Citation4) and 5-chloro-2-nitroaniline ( Citation5) as nucleophiles toward methanol ( Citation5) and phenol ( Citation6), sulfur, in the form of thiophenol ( Citation7) and butanethiol ( Citation8) and phenol, in sodium carbonate to form the thiophenoxide and phenoxide ions, are SNAr reactions that use different primary amines, aliphatic and aromatic compounds, but they have been less widely reported ( Citation9,Citation10).
Modifications of organic reactions not only aim to reduce reaction times or the number of synthetic steps in a process, but also to contribute to new trends in chemistry. Then, the implementation of greener technologies in some types of organic process may afford more reaction efficiency, high atom-economy and reaction yields, shorter reaction times and low-environmental charge, among other advantages. In consequence, there is a great tendency to reduce the high dependence on traditional chemical processes, while permitting the development of environmentally friendly chemical protocols that conserve both resources and energy and that best meet the principals of chemical sustainability and green chemistry. However, reinvestigation of novel greener protocols applicable to the preparative chemistry field is still a challenging problem. Processes including energy consumption, operating costs, improvement of overall yield of reaction, industrial, and environmental aspects remain to be fully explored.
In this paper, we present a SNAr reaction under the protocol of Green Chemistry, using various energy sources such as thermal, Infrared (IR), Ultrasound (US), a simultaneous combination of IR–US, and Microwave (MW) for activating the reactions. We have implemented a series of reactions of SNAr under an eco-friendly program toward the preparation of N-(5-R1amino-2-nitrophenyl)acetamides 3a-d, , and 5-R1amino-2-nitroanilines 5a-d from appealing chemical precursors 1a-d and 4a-d, respectively. Conscious that many practical problems remain unsolved in this field, we undertook the use of environmentally friendly technologies such as IR, MW, US, and the simultaneous combination of US–IR to explore SNAr-based amination reactions to prepare new nitro arylamines 5/7, with the look toward the synthesis of appealing building blocks to synthesize many other molecules.
Figure 2. General reactions to obtain target molecules.
Materials and methods
General
All commercial reagents and solvents were of reagent grade and used as received without further purification. Analytical thin-layer and preparative chromatography was performed on precoated Kieselgel 60F254 plates and Gel silice MN-Kieselgel G/UV254 respectively; spots were located using UV (254 nm). All melting points were measured with a Fisher-Johns SEV PF-300 apparatus and remain uncorrected. 1H and 13C NMR spectra were obtained in either DMSO-d6 or CDCl3 using a 300 MHz spectrometer. Chemical shifts (δ) are expressed in ppm relative to the tetramethylsilane peak used as internal standard. The J values are in Hertz, and the splitting patterns are designated as follows: s, singlet; d, doublet; t, triplet; m, multiplet; and bs, broad singlet. IR spectra on a FT–IR Bruker spectrophotometer using Attenuated Total Reflection (ATR) method were recorded. Mass spectra (MS) and high-resolution mass spectra (HRMS) were obtained using a JEOL AccuTOF JMS-T100LC spectrometer, by Direct Analysis in Real Time (DART) method.
Microwave-assisted reactions using a MW reactor ESEVE-MIC-1 with output power 762 W, irradiating at 60 Hz were performed. IR reactions were heating with wavelength of 1100 nm (9.09 cm−1) using a home-made device as reported previously ( Citation11). Ultrasound experiments were carry out in a Cole Parmer (500 W/115 V) equipment with an irradiation frequency of 20 KHz.
General procedure
Syntheses of 3a-d
Into a 25 mL flask equipped with a stirring bar was placed the corresponding aminoderivate 2a-d (7 mmol), N-(5-haloderivate-2-nitrophenyl)acetamide 1a-d (3.2 mmol) and dimethyl sulfoxide (10 mL), except for 5a were dimethyl formamide was used as solvent. The mixture was submitted to react under the conditions indicated in . At the end of the required time, the mixture was poured onto crushed ice and the precipitated solid was filtered off and purified by preparative TLC (hexane/AcOEt, 7:3, v/v) to afford the pure products 3a-d.
N-(5-(2-Acetylhydrazino)-2-nitrophenyl)acetamide (3a). Yield 80%; mp 182°C; 1H NMR (300 MHz; DMSO-d6/TMS): δ 2.16 (s, 3H), 3.07 (s, 3H), 4.41 (t, J = 6.0 Hz, 1H); 6.56 (dd, J = 7.2, 2.4 Hz, 1H), 7.61 (d, J = 2.4 Hz, 1H), 8.02 (d, J = 9.6 Hz, 1H), 10.60 (s, 1H); 13C NMR (75 MHz, DMSO-d6/TMS): δ 22.7, 25.4, 102.4, 107.6, 126.6, 128.4, 136.8, 155.0, 169.5, 169.6; IR (ATR): 3470, 3358 (NH), 1699 (NHC=O), 1614 (CN), 1572, 1366 (NO2), 962, 897, 802 (Ar1,2,4-subst) cm−1 ; MS (DART [+1]) m/z 253; HRMS (DART [+1]) Found: 306.95918, calcd for C8H8IN2O3: 306.95796.
N-(2-Nitro-5-(phenylamino)phenyl)acetamide (3b). Yield 78%; mp 128 °C; 1H NMR (300 MHz; CDCl3/TMS): δ 2.27 (s, 3H), 6.52 (s, 1H), 6.65 (dd, J = 9.3, 2.4 Hz, 1H), 7.06–7.36 (m, 3H), 7.27–753 (m, 2H), 8.15 (d, J = 9.6 Hz, 1H), 8.36 (d, J = 2.4 Hz, 1H), 10.93 (s, 1H); 13C NMR (75 MHz, CDCl3/TMS): δ 25.8, 104.6, 108.4, 122.3, 125.1, 128.8, 129.7, 137.8, 138.8, 151.5, 169.5; IR (ATR): 3269 (NH), 1682 (HNC=O) 1620 (CN), 1544, 1298 (NO2), 813, 752 (Ar1,2,4-subst) cm−1; MS (DART [+1]) m/z 272; HRMS (DART [+1]) Found: 272.10348, calcd for C14H14N3O3: 272.10352.
N-(5-(2-Hydroxyethylamino)-2-nitrophenyl)acetamide (3c). Yield 93%; mp 180°C; 1H NMR (300 MHz; CDCl3/TMS): δ 2.27 (s, 3H), 3.42 (t, J = 5.4 Hz, 2H), 3.88 (t, J = 5.4 Hz, 2H), 5.01 (s, 1H), 6.30 (dd, J = 6.9, 2.7 Hz, 1H), 8.06 (d, J = 2.7 Hz, 1H), 8.13 (d, J = 9 Hz, 1H), 11.06 (s, 1H); 13C NMR (75 MHz, CDCl3/TMS): δ 25.9, 45.2, 60.6, 107.8, 126.3, 126.9, 128.9, 138.1, 149.5, 169.6; IR (ATR): 3181, 3313 (NH), 1674 (NHC=O), 1621 (CN), 1593, 1317 (NO2), 1059 (OH), 817, 852 (Ar1,2,4-subst) cm−1; MS (DART [+1]) m/z 240; HRMS (DART [+1]) Found: 240.09853, calcd for C10H14N3O4: 240.09843.
N-(2-Nitro-5-(phenyldiazenyl)phenyl)acetamide (3d). Yield 70%; mp 155°C; 1H NMR (300 MHz; CDCl3/TMS): δ 2.35 (s, 3H), 7.44–7.66 (m, 3H), 7.64 (dd, J = 9.0, 2.1 Hz, 1H), 7.87–8.10 (m, 2H), 8.36 (d, J = 9.0 Hz, 1H), 9.30 (d, J = 2.1 Hz, 1H), 10.46 (s, 1H); 13C NMR (75 MHz, CDCl3/TMS): δ 25.8, 104.6, 108.2, 122.3, 124.9, 128.8, 129.6, 137.7, 138.9, 151.7,169.6; IR (ATR): 3472, 3358 (NH); 1691 (NHC=O); 1614 (CN); 1540, 1364, 1262 (NO2); 1498, 1383 (N=N); 773, 755 (Aripso), 848, 827, 807 (Ar 1,2,4-subst) cm−1; MS (DART [+1]) m/z 285; HRMS (DART [+1]) Found: 285.09936, calcd for C14H13N4O3: 285.09876.
Syntheses of 4a-d
Method 1 (4a and 4c). A 50 mL round-bottomed flask fitted with a reflux condenser, was charged with a stirring bar, N-(5-halo-2-nitrophenyl)acetamide 1a or 1c (16.736 mmol), 2 ml of aqueous KOH (16.736 mmol) and 20 mL of dimethyl sulfoxide. The mixture was heated at 60°C for 20 min and poured onto crushed ice after cooling at room temperature. The precipitated solid was filtered off and purified by preparative TLC (hexane/AcOEt, 6:4, v/v) to afford either 4a or 4c.
Method 2 (4b/4d). Following the above approach, the corresponding N-(5-halo-2-nitrophenyl)acetamide 1b or 1d (16.736 mmol), 2 ml of aqueous solution of K2CO3 (16.736 mmol) and 20 mL of EtOH was submitted to react at 60°C for 50 min, and the crude product was processed according to Method 1 to afford either 4b or 4d.
5-Fluoro-2-nitroaniline (4a). Yield 99%, mp 86-87°C; 1H NMR (300 MHz; DMSO-d6/TMS): δ 6.44–6.61 (m, 1H), 6.77 (dd, J = 9.0, 2.1 Hz, 1H), 7.62 (s, 2H), 8.07 (dd, J = 6.0, 3.3 Hz, 1H); 13C NMR (75 MHz, DMSO-d6/TMS): δ 103.6 (2 JCF = 25.8 Hz), 104.6 (2 JCF = 25.1 Hz), 128.1, 129.4 (4 JCF = 12.4 Hz), 148.6 (3 JCF = 14.4 Hz), 166.5 (1 JCF = 250.5 Hz); IR (ATR): 3489, 3467 (NH2), 3363, 3344 (NH2), 1636 (CN), 1594, 1331, (NO2), 1244 (C-F), 837, 747 (Ar1,2,4-subst) cm−1; MS (DART[+1]) m/z 157; HRMS (DART [+1]) Found: 157.04104, calcd for C6H6FN2O2: 157.04133.
5-Chloro-2-nitroaniline (4b). Yield 99%; m.p. 126-129°C; 1H NMR (300 MHz; CDCl3/TMS): δ 6.12 (s, 2H), 6.67 (dd, J = 9.0, 2.4 Hz, 1H), 6.83 (d, J = 2.1 Hz, 1H), 8.06 (d, J = 9.0 Hz, 1H); 13C NMR (75 MHz, CDCl3/TMS): δ 117.6, 117.8, 127.7, 139.6, 142.0, 145.1; IR (ATR): 3493, 3381 (NH2), 1633 (CN), 1558, 1493 (NO2), 1245 (C-Cl), 849, 797, 748 (Ar1,2,4-subst) cm−1; MS (DART[+1]) m/z 173; HRMS (DART [+1]) Found: 173.01120, calcd for C6H6ClN2O2: 173.01178.
5-Bromo-2-nitroaniline (4c). Yield 88%; mp 151-153°C; 1H NMR (300 MHz; DMSO-d6/TMS): δ 6.97 (d, J = 9.0 Hz, 1H), 7.48 (dd, J = 9.3, 2.4 Hz, 1H), 7.56 (s, 2H), 8.03 (d, J = 2.1 Hz, 1H); 13C NMR (75 MHz, DMSO-d6/TMS): δ 105.5, 121.8, 127.4, 131.0, 138.5, 145.5; IR (ATR): 3470, 3347 (NH2), 1557 (CN), 1495, 1243 (NO2), 1751 (C–Br), 813, 713 (Ar1,2,4-subst) cm−1; MS (DART[+1]) m/z 216; HRMS (DART [+1]) Found: 216.96168, calcd for C6H6BrN2O2: 216.96127.
5-Iodo-2-nitroaniline (4d). Yield 85%; mp 174°C; 1H NMR (300 MHz; DMSO-d6/TMS): δ 6.59 (dd, J = 2.4 Hz, J = 9.6 Hz, 1H); 7.06 (d, J = 2.1 Hz, 1H); 7.52 (s, 2H, NH2); 7.95 (d, J = 9.3 Hz, 1H); 13C NMR (75 MHz, DMSO-d6/TMS): 115.5, 117.6, 127.5, 129.3, 140.2, 146.7; IR (ATR): 3287,3252 (NH2), 1579 (CN), 1482, 1253 (NO2), 1899 (C-I), 843, 827, 734 (Ar1,2,4-subst) cm−1; MS (DART[+1]) m/z 264; HRMS (DART [+1]) Found: 264.94637, calcd for C6H6IN2O2: 264.94739.
Syntheses of 5a-d
A mixture of the corresponding compound 2a-d (7 mmol) and 5-haloderivative-2-nitroaniline 4a-d (3.2 mmol) in dimethyl sulfoxide (10 mL) (except for 4a were dimethyl formamide as solvent) was submitted to react under the conditions shown in . After the required time, the mixture of reaction was poured onto crushed ice and the precipitated solid was filtered off and purified by preparative TLC (hexane/AcOEt, 6:4, v/v) to afford 5a-d.
3-Amino-4-nitrophenylhydrazine (5a). Yield 48%; mp 177–178°C; 1H NMR (300 MHz; DMSO-d6/TMS): δ 3.07 (s, 3H), 5.96 (s, 1H), 6.21 (d, J = 9.6 Hz, 1H), 7.26 (s, 2H), 7.63 (s, 1H), 7.81 (d, J = 9.6 Hz, 1H); 13C NMR (75 MHz, DMSO-d6/TMS): δ 25.4, 95.2, 104.6, 122.6,127.7,148.7,155.0, 169.5; IR (ATR): 3472, 3358 (NH); 1691 (C=O); 1614 (CN); 1540, 1364, 1262 (NO2); 1498, 1383, 1465, 1437, 1420 (N=N); 848, 827, 807 (sust 1,2,4); MS (DART[+1]) m/z 211; HRMS (DART [+1]) Found: 211.05260, calcd for C8H11N4O3: 211.05339.
2-Nitro-5-anilinoaniline (5b). Yield 72%; mp 143°C; 1H NMR (300 MHz; CDCl3/TMS): δ 6.14 (s, 3H), 6.18 (d, J = 2.4 Hz, 1H), 6.23 (dd, J = 9.6, 2.4 Hz, 1H), 7.03-7.30 (m, 3H), 7.25–7.50 (m, 2H), 8.02 (d, J = 9.3 Hz, 1H); 13C NMR (75 MHz, CDCl3/TMS): δ 98.2, 107.0, 122.4, 124.6, 128.6, 129.6, 136.3, 139.4, 147.3, 150.8; IR (ATR): 3451, 3325 (NH), 1626 (CN), 1579, 1545 (as), 1226 (NO2), 850, 821, 809 (Ar1,2,4-subst) cm−1; MS (DART[+1]) m/z 230; HRMS (DART [+1]) Found: 230.09333, calcd for C12H12N3O2: 230.09295.
5-(2-Hydroxyethylamino)-2-nitroaniline (5c). Yield 62%, mp 177 °C; 1H NMR (300 MHz; DMSO-d6/TMS): δ 3.15 (td, J = 6.0, 5.1 Hz, 2H), 3.58 (td, J = 6.0, 5.4 Hz, 2H), 4.83 (t, J = 5.4 Hz, 1H), 5.87 (d, J = 2.4 Hz, 1H), 6.08 (dd, J = 7.2, 2.4 Hz,1H), 6.96 (t, J = 5.4 Hz, 1H), 7.35 (s, 2H), 7.74 (d, J = 9.3 Hz, 1H); 13C NMR (75 MHz, DMSO-d6/TMS): δ 45.6, 59.6, 93.3, 106.9, 122.3, 127.7, 149.7, 154.9; IR (ATR): 3467, 3351 (NH2), 3324 (NH), 3275; 1051 (OH), 1632 (CN), 1632, 1287 (NO2), 848, 827, 807 (Ar1,2,4-subst) cm−1; MS (DART[+1]) m/z 198; HRMS (DART [+1]) Found: 198.08810, calcd for C8H12N3O3: 198.08787.
2-Nitro-5-(phenyldiazenyl)aniline (5d). Yield 69%; mp 77°C; 1H NMR (300 MHz; CDCl3/TMS): δ 6.23 (s, 2H), 7.10-7.30 (m, 2H), 7.33 (d, J = 2.1, 1H), 7.42–7.60 (m, 2H), 7.82–8.00 (m, 2H), 8.26 (d, J = 9 Hz, 1H); RMN 13C NMR (75 MHz, DMSO-d6/TMS): δ 110.5, 114.1, 123.5, 124.6, 127.8, 129.5, 132.4, 145.3, 151.7, 156.8; IR (ATR): 3475, 3364 (NH2); 1542, 1378, 1249 (NO2), 1499, 1461, 1426 (N=N), 875, 838, 808 (Ar1,2,4-subst) cm−1, 769 (Aripso); MS (DART[+1]) m/z 243; HRMS (DART [+1]) Found: 243.08915, calcd for C12H11N4O2: 243.08820.
Results and discussion
The general overview behind our synthetic design in is depicted, starting with compounds 1a-d, previously prepared in our laboratory by a nitration reaction and subsequent acetylation reaction of the corresponding 3-haloaniline.
Scheme 1. Synthetic Pathway to synthesize 3a-d and 5a-d.
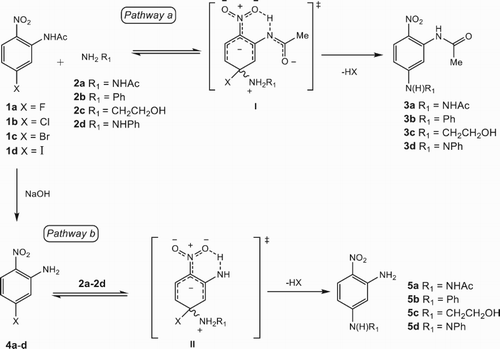
To investigate the generality of the SNAr-based amination, we examined the model reactions of the conversion of N-(5-fluoro-2-nitrophenyl)acetamide 1a into N-(2-nitro-5-(phenylamino)phenyl)acetamide 3b as well as N-(5-(2-hydroxyethylamino)-2-nitrophenyl)acetamide 3c, focusing on the effects of leaving groups, the base as catalyst and solvent used (). When 1a with 1.1 equiv. of base at 150°C in the appropriate solvent reacts with either base 2b or 2c under conventional heating source, the generation of the desired products ranged from moderate to good overall yield (17-88%) within 40 min determined by TLC and confirmed by NMR spectroscopy after their isolation and purification.
Table 1. Screening the SNAr reaction conditions for the preparation of 3b and 3c.
Among the various bases that we examined, excess of aniline in DMSO (entry 5) provided the desired product with the best yield (88%). Conversely, stronger organic bases, such as trimethylamine and pyridine, decreased by almost 50% the overall yield of the target product, regardless of the nature of the solvent used even under solventless condition (entries 2, 4 and 8). Such results were comparable to the yield obtained with uncatalyzed reaction and NaHCO3 in a protic solvent and (entries 1 and 7). In contrast, improved results with K2CO3 and NaOH in DMSO and THF, respectively (entries 3 and 6) were obtained; further, 3c was formed in similar yield, but when NaOH in THF was used, the overall yield decreased substantially (17%, entry 6).
In the same way, when the binucleophile 2-aminoethanol 2c was reacted with 1a in the presence of a strong non-nucleophilic base (entry 2), N-(5-(2-hydroxyethylamino)-2-nitrophenyl)acetamide 3c was isolated as the unique product in good yield without the formation of N-(5-(2-aminoethoxy)-2-nitrophenyl)acetamide. This outcome suggested that the reactivity by orbital control instead of charge control, apart from the easy of stabilization ( Citation12) of δL-complex I might be ruled.
Thus, ready formation of intramolecular N-H••••-O-N hydrogen bonds between the amide moiety and the adjacent nitro group to form the stable δL-complex I at the transition state would contribute significantly to the success of the reaction. So, the combination of a weak base, aprotic solvents (mainly DMSO), and good nucleophiles was proposed as the standard screening condition to be implemented in the preparation of (5-R1amino-2-nitrophenyl)acetamides.
To explore the scope and limitations of these transformations using IR, MW, US, and simultaneous combination US–IR as alternative heating methods, a series of (5-R1amino-2-nitrophenyl)acetamides 3a-3d were prepared () by SNAr reaction of N-(5-halo-2-nitrophenyl)acetamides 1a-d with 2a-d as shown in Scheme 1 (Pathway a).
Table 2. Preparation of 3a-d from 1a-d by thermal and alternative energies.
Among the explored methods, MW heating at 100°C in DMSO afforded the desired products with the best time of reaction and good overall yields (entries 1, 5, 9 and 13, using DMF as solvent). These outcomes were reasonably comparable with the yields obtained with the remaining methods. For example, in the preparation of compound 3a by MW (entry 1), similar results were obtained with IR (50%) or US (52%) methods, although longer reaction times were needed. By the same reason, the US–IR combination gave improved yield (69%) while the thermal approach afforded the lowest yield (32%). Importantly, the overall yields strongly depended upon the nucleofugue capacity of the halogen atoms present in the starting material as well as the nature of nucleophile used in the reaction, 3c vs 3b (entries 3 and 6).
Therefore, the general order of reactivity deduced from all examined reactions, regardless of the approach used, appeared as F > Cl > Br > I. Further, the SNAr reactions proceeded better with an alkylamine, which contrasted with an arylamine (e.g. 3c vs 3b, entries 7 and 6), and even acyl or arylhydrazines (for example 3c vs 3a/3d, entries 15 and 16/13), in spite of the α-effect contribution present in the latter. Therefore, the relative nucleophilicity seemed to decrease in the following order: NH2CH2CH2OH > NH2NHAc > NH2NHAr > NH2Ar. Accordingly, the worst yields and the longer time of reaction were observed under thermal (entry 10) and US (entry 14) conditions. The simultaneous combination of IR–US failed to give 3b and 3d, in spite of prolonged time of reaction (entries 14 and 16). Similar results were generated for the reactions of 4a-d with 2a-d to obtain 5a-d (Scheme 1, Pathway b, ), but in general with either lower reaction yields or no reaction of substrates (entries 26, 28, 30 and 32). We proposed that this behavior can be explained by the formation and stabilization of the δL-complex at TSI vs TSII (Scheme 1, Pathways a and b, respectively), because a weaker intramolecular H••••-O-N hydrogen bonding is formed in TS II, as well as the involvement of the amino group in π-electron delocalization on the aromatic ring, which would lead to the destabilization of TS II.
Table 3. Preparation of 5a-d.
Interestingly, we observed that when either 1a-d or 4a-d reacted with phenylhydrazine 2d, the unexpected N-[2-nitro-5-(phenyldiazenyl)phenyl]acetamide 3d (, entries 4, 8, 12 and 16) and 2-nitro-5-(phenyldiazenyl)aniline 5d (, entries 20, 24, 28 and 32) were isolated as the main products in moderate yields through an oxidative SNAr-based amination reaction. This reaction by an unusual oxidation of the 2-phenylhydrazinyl derivative III through its reaction with DMSO can be explained, which allegedly led to the formation of the species IV, which finally decomposed into the phenyldiazenyl derivative 3d/5d, dimethyl sulfide and water ().
Scheme 2. Proposed mechanism to the preparation of phenyldiazenyl derivatives 3d and 5d.
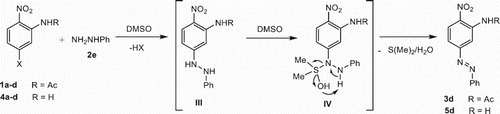
Notably, 3d was obtained in high yield (70%, entry 4) when 1a was used as the feedstock and the reaction was activated by thermal energy. However, the MW approach afforded better results with 1b-d, while the IR–US method gave the poorer result when 1c-d were used as starting materials in spite of prolonged time of reaction ( entries 12 and 16). A similar result was found with 5d, however, no desired product was obtained when 1d was reacted under US, US–IR, and MW conditions. Product 3d was supported by the diagnostic hydrogens of the 2-nitrophenylacetamide moiety at δ 7.64 (H-4) and 9.30 (H-6) in 1H NMR and 151.7 (C-5) in 13C NMR as well as their HRMS and IR data which showed the characteristic diazenyl fingerprint at 1383 cm−1.
Conclusion
Nucleophilic Aromatic Substitution reactions with several amines on 5-halo-2-nitroaniline(acetanilide) using infrared, ultrasound, simultaneous IR–US irradiation and microwave alternative energy sources and conventional thermal energy were successfully realized. In general, according with the results obtained microwave energy represents the best option to achieve target molecules starting from either amine or acetanilide derivatives, where the azo-oxidation product is obtained when phenylhydrazine as nuclephilic agent is used.
Supplementary_Material.pdf
Download PDF (2.7 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Notes on contributors
Ricardo A. Luna-Mora is Ph.D. candidate in Organic Chemistry at National Autonomous University of Mexico (UNAM). His research interests lie in synthesis and reactivity of benzimidazoles and Green Chemistry.
Ángeles Torres-Reyes is student of the Degree in Chemistry at UNAM.
Oscar A. González-Cruz is Student of the Degree in Chemistry at UNAM.
Fernando Ortega-Jiménez is Ph.D. in Organometallic Chemistry. He is currently assistant professor in the Department of Chemistry in FES Cuautitlán-UNAM. His research interests lie in synthesis and catalyst of palladium complexes and Green Chemistry.
Hulme Ríos-Guerra is Ph.D. in Medicine Investigation. He is currently assistant professor in the Department of Chemistry in FES Cuautitlán-UNAM. His research interests lie in synthesis and application in medicine of heterocycles.
Jessica V. González-Carrillo is student of the Degree in Chemical Engineering at UNAM.
Francisco Barrera-Téllez is student of the M.S. Degree in Chemistry at UNAM.
Javier Perez-Flores is Ph.D. in Organic Chemistry. He is currently academic technician in the Department of Mass Spectrometry in Chemistry Institute – UNAM. His research interests lie in the study of mass spectrometry of organic compounds.
José G. Penieres-Carrillo is Ph.D. in Organic Chemistry. He is currently titular professor of university in the Department of Chemistry in FES Cuautitlán – UNAM. His research interests lie in Green Chemistry and synthesis and reactivity of heterocyclic compounds.
ORCID
José G. Penieres-Carrillo http://orcid.org/0000-0003-2085-7813
Additional information
Funding
References
- Ono, N. The Nitro Group in Organic Synthesis, Wiley-VCH: New York, 2001; pp 302–309.
- Dolzhenko, A.V.; Chui, W.K.; Dolzhenko, A.V. Synthesis of ethyl 6‐aryl‐4‐oxo‐4,6‐dihydro‐1(12)(13)H‐pyrimido‐[2′,1′:4,5][1,3,5]triazino[1,2‐a]benzimidazole‐3‐carboxylates. J. Heterocycl. Chem. 2006, 43, 1513–1521. doi: 10.1002/jhet.5570430614
- Ishimoto, K.; Nagata, T.; Murabayashi, M.; Ikemoto, T. Oxidative Cyclization of 1-(pyridin-2-yl)Guanidine Derivatives: A Synthesis of [1,2,4]triazolo[1,5-a]Pyridin-2Amines and an Unexpected Synthesis of [1,2,4]Triazolo [4,3-a]Pyridin-3-Amines. Tetrahedron. 2015, 71, 407–418. doi: 10.1016/j.tet.2014.12.015
- Loewe, H.; Urbanietz, J.; Kirsch, R.; Duewel, D. 2-carbalkoxy-amino-benzimidazol-5(6)phenylaether, ihre herstellung und verwendung in mitteln gegen helminthen. Ger. Often. 1975, 90, 2–486 .
- Marquet, J.; Casado, F.; Cervera, M.; Espin, M.; Gallardo, I.; Mir, M. Reductively Activated ‘Polar’ Nucleophilic Aromatic Substitution. A New Mechanism in Aromatic Chemistry? Niat. M. Pure & Appl. Chem. 1995, 6 (5), 703–710 . doi: 10.1351/pac199567050703
- Begunov, R.S.; Valyaeva, A.N.; Belyaev, V.V.; Dobretsova, N.O. Ultrasound Synthesis of Diaryl Ethers. Russ. Chem. Bull. 2015, 64, 1971–1974. doi: 10.1007/s11172-015-1102-4
- Herlekar, O.P. Lasa Laboratory Pvt Ltd files patent “An Improved Process for the Preparation of Methyl 5-(phenylthio)-1H-benzo[d]Imidazole-2-Ylcarbamate”. Indian Pat. Appl. 2014, Application No. 1953/MUM/2014 A.
- Hay, M.P.; Gamage, S.A.; Kovacs, M.S.; Pruijn, F.B.; Anderson, R.F.; Patterson, A.V.; Wilson, W.R.; Brown, J.M.; Denny, W.A.Structure−Activity Relationships of 1,2,4-Benzotriazine 1,4-Dioxides as Hypoxia-Selective Analogues of Tirapazamine. J. Med. Chem. 2003, 46, 169–182. doi: 10.1021/jm020367+
- Chen, D.; Yang, K.; Xiang, H.; Jiang, S. New Ligands for Copper-Catalyzed C–N Coupling Reactions with Aryl Halides. Tetrahedron Lett. 2012, 53, 7121–7124. doi: 10.1016/j.tetlet.2012.10.083
- Scholz, U.; Schlummer, B. Arylamines. In Science of Synthesis; Georg Thieme Verlag: Stuttgart, 2007; 31b, pp 1565–1678.
- Ortega-Jiménez, F.; Domínguez-Villa, F.X.; Rosas-Sánchez, A.; Penieres-Carrillo, J.G.; López-Cortés, J.G.; Ortega-Alfaro, M.C. An Expedient Approach to Enhance Mizoroki-Heck Coupling Reaction by Infrared Irradiation using Palladacycle Compounds. Appl. Organometal. Chem. 2015, 29, 556–560. doi: 10.1002/aoc.3331
- Vlasov, V.M. Nucleophilic Substitution of the Nitro Group, Fluorine and Chlorine in Aromatic Compounds. Russ. Chem. Rev. 2003, 72, 681–703. doi: 10.1070/RC2003v072n08ABEH000809