
ABSTRACT
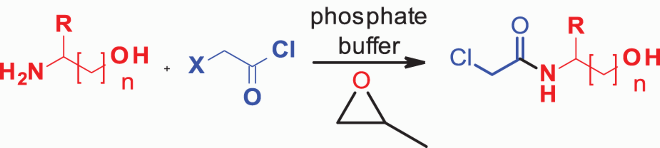
We are reporting for the first time, efficient, highly chemoselective N-chloroacetylation of amino compounds (amino alcohols, amino acids) by chloroacetyl chloride, without compromising its high reactivity, to give chloroacetamides in phosphate buffer within 20 min. We have systematically studied the effects of buffers, metal salts and HCl scavengers to optimize the reaction conditions. We have carried out intermolecular competitive reactions and shown that anilines and aliphatic amines can be selectively N-chloroacylated in the presence of alcohols and phenols. The acylated products are obtained in high yields and can be easily isolated without chromatographic separation. This reaction represents the first example of a metal-free bio-compatible synthesis under neutral conditions. This method is eco-friendly, easily scalable and robust. We have further studied the intramolecular competitive reactions of aminoalcohols and prepared various N-chloroacetamides in very good yields. Finally, this protocol was conveniently extended to ceramides synthesis as well.
GRAPHICAL ABSTRACT
1. Introduction
Amide bond ( Citation1, Citation2) is a fundamentally important linkage that is present in natural (proteins) and synthetic compounds (polymers). The difficulty in the controlled synthesis of amides ( Citation3–5) and their applications in different fields like polymers, engineering materials, detergents and lubricants led to the development of many synthetic methods ( Citation4, Citation6). The physio-chemical properties of amides, their stability, 3-dimensional conformational structure and conversion to other active groups has been extensively exploited by researchers in various fields.
With simplicity and accuracy, nature carries out myriad selective N-acylation reactions as post-translational modifications without affecting other functional groups (alcohols, phenols, thiols, etc.) in histones, p53, tubulins and other biomolecules. Many natural products and bioactive compounds have N-acylated units. What nature does with innate ease is difficult to apply to synthesis, owing to the limited reaction repertoire at our disposal. Modification at a single amino acid or site, without affecting other reactive sites, is a significant and exciting challenge in terms of both chemo- and regioselectivity. To replicate what nature does, we need to design reaction conditions similar to biologically ambient conditions (that is, <37°C, pH 6–8, an aqueous solvent), so as not to disrupt protein architecture and/or function. Ideally, this process should proceed with near total conversion to produce a single product ( Citation7).
Sphingolipids ( Citation8, Citation9) belong to an important class of lipids that are present in all membranes, especially in the myelin sheath. Structurally, these compounds consist of two major units, sphingoid bases ( Citation10) (sphingosine, an aliphatic amino alcohols unit) and a fatty acid. Sphingolipids play vital roles in many cellular functions ( Citation9, Citation11, Citation12) including, signal transmission and cell recognition. A sphingolipid with an R group consisting of a hydrogen atom only is a ceramide. Ceramides are of commercial and therapeutic utility ( Citation13). Currently, ceramides, are primarily obtained via extraction and isolation from animal sources. The presence of other similar compounds makes isolation of ceramides very difficult. It would thus be desirable to find an alternative, cost-effective, high-yield method for obtaining these valuable products. Thus, chemical synthesis methods may provide a suitable alternative ( Citation14–18).
Many bioactive molecules contain functional groups that differ very little in their size or electronic nature. Therefore, it is difficult to modify one functional group over the other ( Citation19, Citation20). In order to achieve protection free synthesis ( Citation21), we wanted to develop selective functionalization of amines in the presence of other groups, especially alcohols ( Citation22, Citation23), which can be extended to ceramides synthesis.
In the case of amino alcohols, N- versus O-acylation can be controlled effectively by running the reaction under either basic or acidic conditions, respectively ( Citation24, Citation25). It has been shown that chemoselective N-acylation of amino alcohols can be carried out using catalytic amounts of dibutyltin oxide ( Citation26). For hydroxy substituted aromatic amines, it is possible to obtain selective O-acylation ( Citation27). There is also a recent report mentioning selective amidation of acids and unprotected amino alcohols, using EDC-HOBt-surfactant mediated coupling in water ( Citation28). Some of the limitations of the reported procedures include use of toxic materials (dibutyltin oxide), the need to run the reaction under either acidic or basic conditions or the reactions taking considerably longer time (more than 15 h) for completion.
Chloroacetyl chloride (CAC) ( Citation29) plays multiple roles in synthetic and biological chemistry as a bifunctional linker ( Citation30), protecting group ( Citation31) for alcohols and amines, in dendrimer synthesis ( Citation32–36), and as a glycosyl donor in oligosaccharide synthesis ( Citation33, Citation35, Citation37, Citation38).
In our efforts to develop synthetic methodologies focusing on bioconjugation reactions ( Citation39), we have employed amide bond formation reactions ( Citation40, Citation41). Although acylation using acetyl chloride or chloroacetyl chloride seems to produce chemically similar reactions, the synthetic utility and stability of the products obtained differ vastly. Moreover, the reactivity and selectivity profiles of chloroacetyl chloride are less explored compared to acetyl chloride. We wanted to expand the scope and utility of chloroacetyl chloride. The major challenge posed was to develop a method having high selectivity without reducing the high reactivity of acid chlorides.
To alleviate environmental pollution caused by using toxic and volatile organic solvents, particularly chlorinated hydrocarbons, many synthetic processes are turning to greener alternatives. The Innovative Medicines Initiative (IMI)-CHEM21 solvent guide had identified water as the most preferred green solvent, with a safety, health and environmental score of 1. To develop a method in bio-compatible medium (aqueous media) to ceramide derivatives, we were attracted by two principles ( Citation42) of the green chemistry approaches, namely (i) amide formation avoiding poor atom economy reagents, and (ii) replacements for dipolar aprotic solvents. Furthermore, reactions conducted in aqueous media minimize protection–deprotection sequences ( Citation21) that are commonly associated with conventional methods.
We were surprised to see that there are only very few reports utilizing near neutral conditions with water as the solvent for chloroacetylation ( Citation43, Citation44) even though CAC is used for functionalization of amines ( Citation45). One of the methods utilizes a large excess of CAC (4 eq.), and the reaction requires longer times (12–16 h) to complete ( Citation43). An aqueous method developed recently for ranolazine ( Citation44) synthesis uses a stoichiometric amount of water (5 eq.) for chloroacetylation using chloroacetic anhydride. There are problems associated with acetylation reactions in water ( Citation46). It is reported that CAC undergoes very slow hydrolysis in water to the corresponding acid ( Citation47–49). We presumed, if the chloroacetylation can be effected faster than the hydrolysis, then it will be possible to carry out the reaction in water.
2. Results and discussion
2.1. Optimization of chemoselective N-chloroacetylation of aminobenzyl alcohol (ABA) with chloroacetyl chloride (CAC)
In our effort to prepare various chloroacetamides using CAC for use in bioconjugation reactions, we tried many known procedures with different solvents (CH2Cl2, MeCN, H2O, toluene, DMF, HOAc) and with different bases (TEA, DIPEA, K2CO3, Na2CO3, NaHCO3). Maximum yield and minimal workup of the corresponding chloroacetamide derivatives varied depending on the amine. Aniline required no base (AcOH/NaOAc), benzylamine required an inorganic base (K2CO3/MeCN), whereas butylamine, cyclohexylamine and dicyclohexylamine required reverse addition (no base, 2 eq. amine, MeCN). Most of these reactions required longer times (>3 h or overnight) for completion as well.
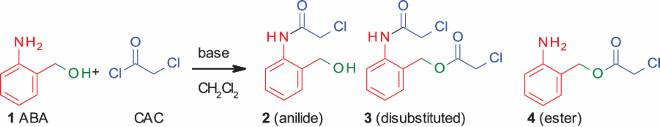
To optimize chloroacetylation using CAC, we chose 2-aminobenzyl alcohol (ABA) as a model substrate. Since ABA has both aniline and alcohol moieties in a single substrate, we envisioned that it can also help us to study selectivity (N- vs. O-chloroactylation).
In this chloroacetylation, HCl is the only by-product. Keeping this in mind, as the first variation we focused on screening different bases with CH2Cl2 as the solvent under argon. Strong bases like DBU, DABCO (, entries 1, 2), DMAP in DMF (, entry 3) or TEA in MeCN (, entry 8) gave roughly a 1:1 mixture of the anilide:disubstituted (both N- and O-chloroacetylated) products. Lutidine (, entry 5) gave the maximum amount of the disubstituted product (3). Interestingly, when no base was used, the dichloroacylated product was obtained in maximum yield (85% by NMR). On the contrary, TEA (Table1, entry 7) was the only base which gave the maximum yield of the anilide (2) (83% by NMR), followed by DBN (, entry 6). Although TEA in CH2Cl2 gave the anilide as the major product, changing the solvent to MeCN (, entry 8) gave 17% of the ester (4) (O-chloroacylated product). Even a slow addition of the base to the reaction medium did not suppress the disubstituted product formation.
Table 1. Reaction of ABA with CAC: effect of bases in CH2Cl2.
2.1.1. To study the solvent effect for the chemoselective N-chloroacetylation of ABA with CAC
Because the organic solvent and base combination did not give the anilide as the sole product, we planned to change the solvent system to the aqueous medium. We started studying the reactivity in aqueous buffers, expecting that if successful, this method could be directly applied to bioconjugation reactions under physiological conditions. When the reaction was carried out in water (, entry 1), the anilide was formed in only 40% yield, along with 8% of the ester.
Table 2. Reaction of ABA with CAC: effect of buffers.
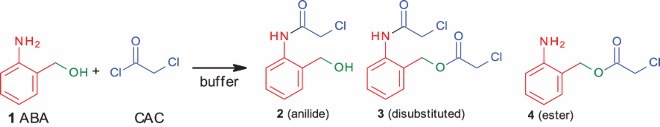
It is known that slow hydrolysis of chloroacetyl chloride produces chloroacetic acid. However, under the reaction conditions employed, we did not see any appreciable hydrolysis. Therefore, we started exploring the effect of buffers. Citrate, oxalate and carbonate buffer (, entries 2–4) gave 2 as the major product, along with 7–12% of the ester (4). These reactions were completed within 20 min. The use of phosphate buffer (, entry 5) gave 77% of the anilide. It is important to mention the two distinct advantages with phosphate buffer: (i) the reaction was over within 20 min, and (ii) there was no ester formation. Although the yield of anilide was still not high compared to carbonate buffer, there is room for improvement. Finally, Tris and MES buffers (, entries 6, 7) gave around 80–83% of anilide, with 10–15% of esters.Footnote1
Although all these reactions were analyzed by NMR for product distribution, the yield of the crude products ranged from 55% to 65% only. Since no separate base is used for neutralizing the HCl formed, theoretically 50% of the aniline will form an HCl salt, thereby limiting the crude product to 50% isolated yield. To probe how we were getting higher than 50% yield, we treated ABA with 1 eq. of HCl to form the ABA•HCl salt. The salt was then treated with CAC. To our surprise, we observed that 10–15% of anilide was formed from the HCl salt. It is reported that the HCl salt of aminoalcohols can be acylated to give exclusively esters ( Citation50). However under the current protocol with phosphate buffer, only the N-acylation took place, and we did not see any appreciable O-acylation. Computational studies are currently underway to ascertain the role of phosphate ions in the current chemoselective chloroacetylation reactions.
2.1.2. To study the effect of metal salts on the chemoselective N-chloroacetylation of ABA with CAC
To suppress the reactivity of alcohols completely, metal coordination based on the HSAB principle was invoked. The hydroxyl is a hard base, and aniline is a borderline base. Hence attempts were made to use various hard acids for selective coordination. As expected Fe+3 (, entry 1) gave the maximum amount of anilide. However, use of Mg+2 (, entry 2), another hard acid, was not effective. Hence it was decided to try other metal salts. The use of soft or borderline acids Cu+2, Ag+1, Cd+2, Zn+2 (, entries 3–6) gave 75–85% of anilide. Mn+2 and Co+2 gave around 65% (, entries 7, 8) of anilide. The only exception was K+1, which gave exclusively the disubstituted product in MeCN (, entry10).
Table 3. Reaction of ABA with CAC: effect of metal salts.
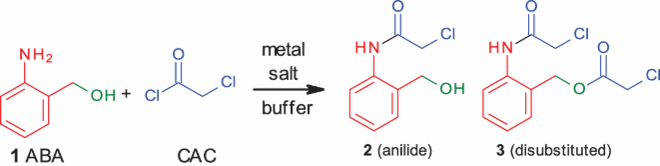
From these results, it became evident that metal-hydroxyl coordination based on the HSAB principle cannot be exploited effectively for specificity in the present scenario.
2.1.3. To study the effect of HCl scavengers for the chemoselective N-chloroacetylation of ABA with CAC
We then wanted to study the effect of various phosphate salts. When NaH2PO4 or Na2HPO4 was used, large amounts of ABA remained unreacted (, entries 1, 2), 52% and 20%, respectively. Water with K3PO4 produced the anilide (88%) (, entry 3). The use of milder bases like NH4OH, NaOAc and TEA gave approximately 75% of anilides (, entries 4–6). However, the isolated yield was still only around 60–70%. Next, we wanted to find out the effect of neutral HCl scavengers like limonene (, entry 7) and propylene oxide (, entry 9). Only propylene oxide gave a higher yield (86%) of isolated anilide. Use of Amberlyst21 (, entry 8) gave 84% anilide (80% crude). Finally, use of pyridine (, entry 10) as the HCl scavenger gave comparable results with propylene oxide. Thus, we have developed a mild and clean procedure for chloroacetylation of amines which can be used in neutral conditions (with propylene oxide) or can also be used with 1 equiv. of the base (pyridine).
Table 4. Reaction of ABA with CAC: effect of scavengers.
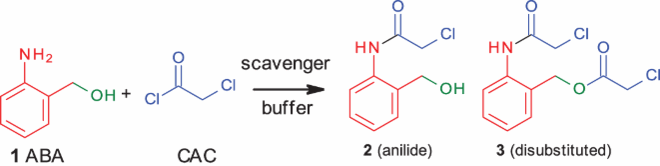
2.2. Study of chemoselective intermolecular N-chloroacetylation of anilines in the presence of alcohols/phenols
Further, we investigated the selectivity between anilines and alcohols/phenols. The results are reported in . We found out earlier that FeCl3 decreased the formation of disubstituted product (, entry 1). Moreover, FeCl3 completely suppressed the reactivity of benzylamine. Hence, we decided to check its effect on benzyl alcohol as well. Organic solvent or phosphate buffer with or without FeCl3 was not selective towards N-chloroacetylation (, entries 1–3). However, we observed complete selectivity (, entry 4) when phosphate buffer was used for the reaction. The use of FeCl3 has little effect for selectivity in case of cinnamyl alcohol (, entry 6). Once again complete selectivity (100:0, , entry 7) was observed when phosphate buffer was used. In the case of phenols (, entries 8–11), complete selectivity was observed towards N-chloroacetylation. The use of FeCl3 had little or no effect on selectivity.
Table 5. Competitive reaction of aniline and alcohols/phenols.
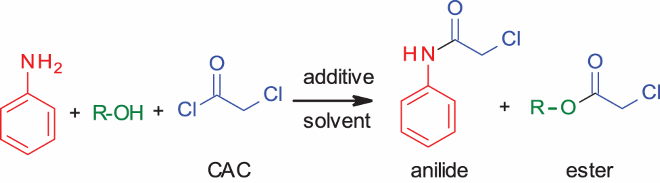
2.3. Study of chemoselective intermolecular N-chloroacetylation of benzylamine in the presence of alcohols/phenols
We then checked the selectivity between benzylamine with alcohols/phenols. describes the results. Use of organic solvent or phosphate buffer with or without FeCl3gavemixed results (, entries 1–3). Use of FeCl3 reduced the reactivity of benzylamine. The use of phosphate buffer with no additive gave very good selectivity (, entry 4), which was increased further when pyridine was used as the additive (, entry 5). In contrast, the use of CH2Cl2 with pyridine gave 1:1 mixture with no selectivity (, entry 6). In the case of cinnamyl alcohol, phosphate buffer gave the best results, compared to the use of water or phosphate buffer with FeCl3 (, entries 7–9). Similarly, excellent selectivity was observed for reactions with phenols (, entries 10–12) or with furfuryl alcohol (, entry 13).
Table 6. Competitive reaction of amines and alcohols/phenols.
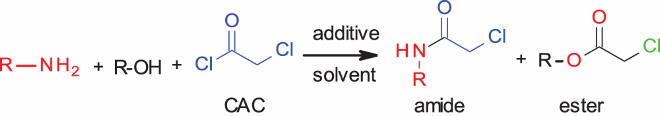
Thus, this protocol can be used for selective N-chloroacetylation of benzylamines in the presence of alcohols or phenols. When butylamine was used in the competitive reaction with benzyl alcohol in different solvents, different results were obtained (, entries 14–16). For the butylamine reaction, the addition of TEA (1 eq.) increased the amide formation from 38% to 70%. However, the most important result was the suppression of alcohol reactivity in phosphate buffer. Large amount of benzyl alcohol remained unreacted (84%, entry 15) in phosphate buffer, versus 55% unreacted alcohol in CH2Cl2 ( entry 16). Even with hindered cyclic amines (, entries 17, 18), good selectivity in amide formation was observed. In these cases as well, a substantial amount (72–90%) of alcohol did not react. Although the cause for this selectivity is currently under investigation, we presume that this observation can be attributed to the basic nature of the aliphatic amines, which can abstract a proton from benzyl alcohol to give the ester. Bulky dicyclohexylamine, being a hindered base, cannot efficiently abstract the hydroxyl proton and gave the least amount of ester (10%) compared to small and linear butylamine (50%). Finally, a substrate having both amine and phenol on the same aromatic ring (, entry 19) was reacted to give the amide substituted product selectively, with no trace of the ester.
2.4. Study of chemoselective N-chloroacetylation various amino derivatives
In this respect, we then attempted selective chloroacetylation of amino alcohols (). All of the amino alcohols (, entries 1, 3, 5–11) gave the corresponding N-chloroacylated product exclusively. Use of K3PO4 was not effective (, entries 2, 4). However, one of the key observations came during the purification of prolinol, 4-aminobutanol, and 2-amino butanol products (, entries 7, 8 and 11). It was observed that for the monosubstituted product, the CH2 protons attached to the OH have chemical shifts around 3.5–3.7 ppm (–CH2OH), whereas the same CH2 protons (–CH2OCOCH2Cl) in O-chloroacyl ester have chemical shifts around 4.2. Although the crude NMR of aminobutanol and prolinol products showed no chloroacetyl ester peak, these products rearranged to the disubstituted product during column chromatography (silica gel) ().
Figure 1. Chloroacetyl products from aminoalcohols and aminoacids.
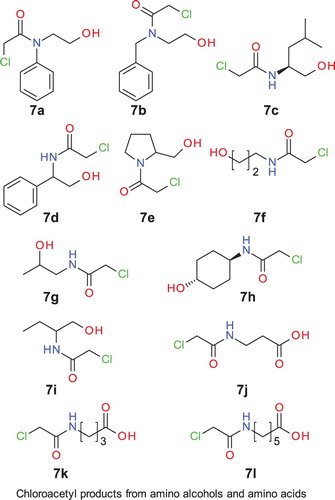
Table 7. Reaction of amino alcohols and amino acids with CAC.
It is known that, under acidic conditions, N- to O-acyl migration is possible for an amino alcohol ( Citation25). In order to extend our methodology further, a few amino acids (, entries 12–14) were subjected to the same reaction conditions. All these amino acids were converted to the corresponding chloroacetamides, albeit in good to moderate yields. It was also observed that for α-amino acids the N-chloroacylation reaction was very sluggish (data not shown).
2.5. Study of chemoselective acylation of aminoalcohols using fatty acid chlorides
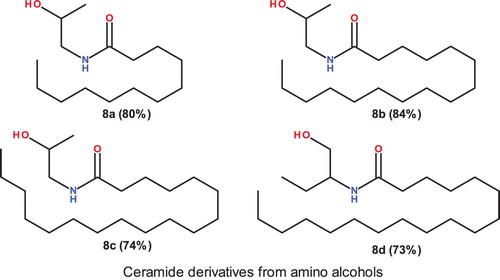
When aminoalcohols were reacted with acyl chlorides from lipophilic long chain fatty acids (Lauroyl, Plamitoyl and Stearoyl) they were efficiently converted (isolated yield 73–84%) to the corresponding amides. depicts the compounds synthesized. 8a, 8b and 8c were formed by the reaction of 1-amino-2-propanol with Lauroyl, Palmitoyl and Stearoyl chlorides respectively. 8d was formed by the reaction of 2-amino-1-butanol with Stearoyl chloride ().
Figure 2. Ceramide derivatives from aminoalcohols.
In conclusion, we have shown for the first time that chloroacetyl chloride can be effectively and efficiently used for chloroacetylation of aminoalcohols and amino acids under neutral, metal-free and green chemical conditions. The generality of the reaction has been studied. It was found that the reaction occurs within 20 min, and the isolated yields are high in the presence of an HCl scavenger. Anilines and amines react selectively in the presence of alcohols and phenols using the current protocol, in contrast to poor selectivity in organic solvents. The selective N-chloroacetylation over O-chloroacetylation has been exploited for the formation of N-chloroacetamides using amino alcohols. This method was extended to the formation of ceramides with various fatty acid chlorides.
Supplemental_Material.pdf
Download PDF (621.8 KB)Acknowledgements
We are thankful to AIRF, JNU for NMR and Mass analysis of the samples.
Disclosure statement
No potential conflict of interest was reported by the authors.
Notes on contributors
B. S. Balaji is Ph.D. in Organic Chemistry and MBA in Technology Management. He is currently Associate professor at School of Biotechnology, Jawaharlal Nehru University. His research interests lie in the interface of chemistry and biology focusing on Green Chemistry, anti-cancer research, drug development, bio-orthogonal reactions and synthetic methodologies.
Neha Dalal was a Master student at Jawaharlal Nehru University. She is currently pursuing Ph.D from Indian Institute of Technology, Bombay.
ORCID
B. S. Balaji http://orcid.org/0000-0002-5522-7726
Notes
1 Since the NMR experiments were done on 0.4 mmol scale and due to its extremely low yield, the ester could not be isolated in pure form. Various attempts to run the reactions on large scale (10–15 mmol) consistently produced the anilide and the disubstituted product as the major component.
Related Research Data
References
- Pitzer, J.; Steiner, K. Amides in Nature and Biocatalysis. J. Biotechnol. 2016, 235, 32–46. doi:10.1016/j.jbiotec.2016.03.023.
- Pattabiraman, V.R.; Bode, J.W. Rethinking Amide Bond Synthesis. Nature 2011, 480, 471–479. doi:10.1038/nature10702.
- de Figueiredo, R.M.; Suppo, J.-S.; Campagne, J.-M. Nonclassical Routes for Amide Bond Formation. Chem. Rev. 2016, 116, 12029–12122. doi:10.1021/acs.chemrev.6b00237.
- Dunetz, J.R.; Magano, J.; Weisenburger, G.A. Large-Scale Applications of Amide Coupling Reagents for the Synthesis of Pharmaceuticals. Org. Process Res. Dev. 2016, 20, 140–177. doi:10.1021/op500305s.
- Lundberg, H.; Tinnis, F.; Selander, N.; Adolfsson, H. Catalytic Amide Formation From Non-Activated Carboxylic Acids and Amines. Chem. Soc. Rev. 2014, 43, 2714–2742. doi:10.1039/C3CS60345H.
- Montalbetti, C.A.G.N.; Falque, V. Amide Bond Formation and Peptide Coupling. Tetrahedron 2005, 61, 10827–10852. http://www.ncbi.nlm.nih.gov/pubmed/12399580. doi: 10.1016/j.tet.2005.08.031
- Spicer, C.D.; Davis, B.G. Selective Chemical Protein Modification. Nat. Commun. 2014, 5, 4740. doi:10.1038/ncomms5740.
- Brown, D.A.; London, E. Structure and Function of Sphingolipid- and Cholesterol-Rich Membrane Rafts. J. Biol. Chem. 2000, 275, 17221–17224. doi:10.1074/jbc.R000005200.
- Hannun, Y.A.; Obeid, L.M. Principles of Bioactive Lipid Signalling: Lessons From Sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. doi:10.1038/nrm2329.
- Pruett, S.T.; Bushnev, A.; Hagedorn, K.; Adiga, M.; Haynes, C.A.; Sullards, M.C.; Liotta, D.C.; Merrill, A.H. Thematic Review Series: Sphingolipids. Biodiversity of Sphingoid Bases (“Sphingosines”) and Related Amino Alcohols. J. Lipid Res. 2008, 49, 1621–1639. doi:10.1194/jlr.R800012-JLR200.
- Ohanian, J.; Ohanian, V. Sphingolipids in Mammalian Cell Signalling. Cell. Mol. Life Sci. 2001, 58, 2053–2068. doi:10.1007/PL00000836.
- Simons, K.; Gerl, M.J. Revitalizing Membrane Rafts: New Tools and Insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 688. doi: 10.1038/nrm2977
- Brodowicz, J.; Przegaliński, E.; Müller, C.P.; Filip, M. Ceramide and Its Related Neurochemical Networks as Targets for Some Brain Disorder Therapies. Neurotox. Res. 2018, 33, 474–484. doi:10.1007/s12640-017-9798-6.
- Click Reactions with Functional Sphingolipids Biol. Chem. 2018, 399, 1157. doi:10.1515/hsz-2018-0169.
- Kim, S.; Cho, M.; Lee, T.; Lee, S.; Min, H.-Y.; Lee, S.K. Design, Synthesis, and Preliminary Biological Evaluation of a Novel Triazole Analogue of Ceramide. Bioorg. Med. Chem. Lett. 2007, 17, 4584–4587. doi:10.1016/j.bmcl.2007.05.086.
- Sinkó, B.; Pálfi, M.; Béni, S.; Kökösi, J.; Takács-Novák, K. Synthesis and Characterization of Long-Chain Tartaric Acid Diamides as Novel Ceramide-Like Compounds. Molecules 2010, 15, 824–833. doi:10.3390/molecules15020824.
- Reynolds, C.P.; Maurer, B.J.; Kolesnick, R.N. Ceramide Synthesis and Metabolism as a Target for Cancer Therapy. Cancer Lett. 2004, 206, 169–180. doi:10.1016/j.canlet.2003.08.034.
- Mormeneo, D.; Casas, J.; Llebaria, A.; Delgado, A. Synthesis and Preliminary Antifungal Evaluation of a Library of Phytosphingolipid Analogues. Org. Biomol. Chem. 2007, 5, 3769–3777. doi:10.1039/B709421C.
- Tadross, P.M.; Jacobsen, E.N. Site-selective Reactions: Remodelling by Diversity and Design. Nat. Chem. 2012, 4, 963–965. doi:10.1038/nchem.1509.
- Afagh, N.A.; Yudin, A.K. Chemoselectivity and the Curious Reactivity Preferences of Functional Groups. Angew. Chem. Int. Ed. Engl. 2010, 49, 262–310. doi:10.1002/anie.200901317.
- Young, I.S.; Baran, P.S. Protecting-group-free Synthesis as an Opportunity for Invention. Nat. Chem. 2009, 1, 193–205. doi:10.1038/nchem.216.
- Vibhute, A.M.; Vidyasagar, A.; Sarala, S.; Sureshan, K.M. Regioselectivity Among Six Secondary Hydroxyl Groups: Selective Acylation of the Least Reactive Hydroxyl Groups of Inositol. Chem. Commun. 2012, 48, 2448–2450. doi:10.1039/C2CC17241K.
- Vibhute, A.M.; Sureshan, K.M. H2SO4-silica: An Eco-Friendly Heterogeneous Catalyst for the Differential Protection of Myo-Inositol Hydroxyl Groups. RSC Adv. 2013, 3, 7321–7329. doi:10.1039/C3RA40506K.
- Kristensen, T.E. Chemoselective O-Acylation of Hydroxyamino Acids and Amino Alcohols Under Acidic Reaction Conditions: History, Scope and Applications. Beilstein J. Org. Chem. 2015, 11, 446–468. doi:10.3762/bjoc.11.51.
- Markey, S.P.; Dudding, T.; Wang, T.C. Base- and Acid-Catalyzed Interconversions of O-Acyl- and N-Acyl-Ethanolamines: A Cautionary Note for Lipid Analyses. J. Lipid Res. 2000, 41, 657–662. http://www.ncbi.nlm.nih.gov/pubmed/10744787.
- Morcuende, A.; Ors, M.; Valverde, S.; Herradón, B. Microwave-Promoted Transformations: Fast and Chemoselective N-Acylation of Amino Alcohols Using Catalytic Amounts of Dibutyltin Oxide. Influence of the Power Output and the Nature of the Acylating Agent on the Selectivity. J. Org. Chem. 1996, 61, 5264–5270. doi:10.1021/jo9605511.
- Mukaiyama, T.; Pai, F.-C.; Onaka, M.; Narasaka, K. A Useful Method For Selective Acylation Of Alcohols Using 2,2′-Bipyridyl-6-Yl Carboxylate And Cesium Fluoride. Chem. Lett. 1980, 9, 563–566. doi:10.1246/cl.1980.563.
- Parmentier, M.; Wagner, M.K.; Magra, K.; Gallou, F. Selective Amidation of Unprotected Amino Alcohols Using Surfactant-in-Water Technology: A Highly Desirable Alternative to Reprotoxic Polar Aprotic Solvents. Org. Process Res. Dev. 2016, 20, 1104–1107. doi:10.1021/acs.oprd.6b00133.
- Kirk-Othmer. In Kirk-Othmer Encyclopedia of Chemical Technology; Othmer, K., Ed., 5th ed; Wiley: New York, 2004; Vol. 1.
- Olszewska, A.; Pohl, R.; Brázdová, M.; Fojta, M.; Hocek, M. Chloroacetamide-Linked Nucleotides and DNA for Cross-Linking with Peptides and Proteins. Bioconjug. Chem. 2016, 27, 2089–2094. doi:10.1021/acs.bioconjchem.6b00342.
- Wuts, P.G.M.; Greene, T.W. Greene’s Protective Groups in Organic Synthesis Fourth Edition, Wiley-Interscience: New Jersey, 2007.
- Kottari, N.; Chabre, Y.M.; Shiao, T.C.; Rej, R.; Roy, R. Efficient and Accelerated Growth of Multifunctional Dendrimers Using Orthogonal Thiol-ene and SN2 Reactions. Chem. Commun. (Cambridge, UK) 2014, 50, 1983–1985. doi:10.1039/c3cc46633g.
- Sharma, R.; Kottari, N.; Chabre, Y.M.; Abbassi, L.; Shiao, T.C.; Roy, R. A Highly Versatile Convergent/Divergent “Onion Peel” Synthetic Strategy Toward Potent Multivalent Glycodendrimers. Chem. Commun. (Camb). 2014, 50, 13300–13303. doi:10.1039/c4cc06191h.
- Vijayalakshmi, N.; Maitra, U. Hydroxyl-terminated Dendritic Oligomers From Bile Acids: Synthesis and Properties. J. Org. Chem. 2006, 71, 768–774. doi:10.1021/jo052173i.
- Vinogradov, S.A. Arylamide Dendrimers with Flexible Linkers via Haloacyl Halide Method. Org. Lett. 2005, 7, 1761–1764. doi:10.1021/ol050341n.
- Bergmann, M.; Michaud, G.; Visini, R.; Jin, X.; Gillon, E.; Stocker, A.; Imberty, A.; Darbre, T.; Reymond, J.-L. Multivalency Effects on Pseudomonas aeruginosa Biofilm Inhibition and Dispersal by Glycopeptide Dendrimers Targeting Lectin LecA. Org. Biomol. Chem. 2016, 14, 138–148. doi:10.1039/C5OB01682G.
- Kiso, M.; Anderson, L. Protected Glycosides and Disaccharides of 2-Amino-2-Deoxy-d-Glucopyranose by Ferric Chloride-Catalyzed Coupling. Carbohydr. Res. 1985, 136, 309–323. doi:10.1016/0008-6215(85)85205-8.
- Bongat, A.F.G.; Demchenko, A.V. Recent Trends in the Synthesis of O-Glycosides of 2-Amino-2-Deoxysugars. Carbohydr. Res. 2007, 342, 374–406. doi:10.1016/j.carres.2006.10.021.
- Subramanian, N.; Sreemanthula, J.B.; Balaji, B.; Kanwar, J.R.; Biswas, J.; Krishnakumar, S. A Strain-Promoted Alkyne–Azide Cycloaddition (SPAAC) Reaction of a Novel EpCAM Aptamer–Fluorescent Conjugate for Imaging of Cancer Cells. Chem. Commun. 2014, 50, 11810–11813. doi:10.1039/C4CC02996H.
- Balaji, B.S.; Lewis, M.R. Double Exponential Growth of Aliphatic Polyamide Dendrimers via AB2 Hypermonomer Strategy. Chem. Commun. (Cambridge, UK) 2009. doi:10.1039/b903948a.
- Balaji, B.S.; Gallazzi, F.; Jia, F.; Lewis, M.R. An Efficient, Convenient Solid-Phase Synthesis of Amino Acid-Modified Peptide Nucleic Acid Monomers and Oligomers. Bioconjug. Chem. 2006, 17, 551–558. doi:10.1021/bc0502208.
- Constable, D.J.C.; Dunn, P.J.; Hayler, J.D.; Humphrey, G.R.; Leazer, Jr, J.L.; Linderman, R.J.; Lorenz, K.; Manley, J.; Pearlman, B.a., Wells, A., et al. Key Green Chemistry Research Areas: A Perspective From Pharmaceutical Manufacturers. Green Chem. 2007, 9, 411–420. doi:10.1039/b703488c.
- Harte, A.J.; Gunnlaugsson, T. Synthesis of Alpha-Chloroamides in Water. Tetrahedron Lett. 2006, 47, 6321–6324. doi:10.1016/j.tetlet.2006.06.090.
- Kommi, D.N.; Kumar, D.; Chakraborti, A.K. “All Water Chemistry” for a Concise Total Synthesis of the Novel Class Anti-Anginal Drug (RS), (R), and (S)-Ranolazine. Green Chem. 2013, 15, 756. doi:10.1039/c3gc36997h.
- Loeser, E.; Prasad, K.; Repic, O. Selective N-Alkylation Of Primary Amines With Chloroacetamides Under Ph-Controlled Aqueous Conditions. Synth. Commun. 2002, 32, 403–409. doi:10.1081/SCC-120002124.
- El-Faham, A.; Albericio, F. Peptide Coupling Reagents, More Than a Letter Soup. Chem. Rev. 2011, 111, 6557–6602. doi:10.1021/cr100048w.
- Ruff, F.; Farkas, O. Concerted SN2 Mechanism for the Hydrolysis of Acid Chlorides: Comparisons of Reactivities Calculated by the Density Functional Theory with Experimental Data. J. Phys. Org. Chem. 2011, 24, 480–491. doi:10.1002/poc.1790.
- Carlson, D.L.; Than, K.D.; Roberts, A.L. Acid- and Base-Catalyzed Hydrolysis of Chloroacetamide Herbicides. J. Agric. Food Chem. 2006, 54, 4740–4750. doi:10.1021/jf0530704.
- Bentley, T.W.; Harris, H.C.; Ryu, Z.H.; Lim, G.T.; Sung, D.D.; Szajda, S.R. Mechanisms of Solvolyses of Acid Chlorides and Chloroformates. Chloroacetyl and Phenylacetyl Chloride as Similarity Models. J. Org. Chem. 2005, 70, 8963–8970. doi:10.1021/jo0514366.
- Cope, A.C.; Hancock, E.M. Benzoates, p-Aminobenzoates and Phenylurethans of 2-Alkylaminoethanols. J. Am. Chem. Soc. 1944, 66, 1448–1453. doi:10.1021/ja01237a009.