
ABSTRACT
A series of arylidene thiosemicarbazides have been prepared by a new method, which was titled as transalkylidation. This method is an effective, fast, green and clean method. The mechanism of this reaction has been discussed. Moreover, we clarified the divergences of the structural assignments reported in the literature for the reaction of thiosemicarbazide and aldehydes or ketones in the presence of different catalysts. Where 1,2,4-triazolidine-3-thiones were incorrectly reported as sole product of such reaction, based on occurance of intramolecular cycloaddition of thiosemicarbazones formed in situ. Our findings proved that the reaction stops at earlier stage of thiosemicarbazone and neither cyclization to 1,2,4-triazolidine-3-thiones nor 2-amino-1,3,4-thiadiazoline take place. We have removed the confusion of the NMR interpretation of thiosemicarbazone and their cycloaddition product by carrying out 1H-NMR, 13C-NMR, 15N-NMR and 1H-15N HSQC experiments with temperature gradient. Furthermore, DFT-NMR calculations have been done to make structural distinguish between the three possible structural isomers for this reaction, namely, 1,2,4-triazolidene-3-thione, 2-amino-1,3,4-thiadiazoline and thiosemicarbazone.
GRAPHICAL ABSTRACT
1. Introduction
Thiosemicarbazide and thiosemicarbazone derivatives are especially important due to their diverse biological activity (Citation1–9), including antiviral (Citation10) antibacterial (Citation11), antimalarial (Citation12), antifungal (Citation13), anticancer (Citation14), antitumor (Citation15,Citation16), anti-inflammatory and antiamoebic (Citation17–19) owing to their ease of synthesis and abundance in plants, these compounds have generated great interest for possible therapeutic uses. Furthermore, Metal complexes of thiosemicarbazone (Citation20,Citation21) have gained special attention due to their activity against cancer, small poxvirus, influenza, protozoa, and fungi. Thiosemicarbazones play a significant role in the regulation of plant growth because of their capability for diffusion through the semipermeable membrane of cell lines (Citation22–24). As well as other industrially important activities, including antifouling and anticorrosion effects (Citation25,Citation26) have also been reported for these compounds. Moreover, The imine bond (C = N) in these compounds are useful in the organic synthesis, in particular for the synthesis of many of pharmaceutical and bioactive heterocycles such as, thiazoles (Citation27–31), 1,3,4-thiadiazoles (Citation32), thiazolidinones (Citation33,Citation34), quinolines (Citation35), naphthyridines (Citation36), oxadiazoles (Citation37,Citation38), pyrimidines (Citation39). Water plays an important role in our life. Although, the fact that water as a solvent is the cheapest and most non-toxic solvent, there are many limitations of water use as a solvent in organic synthesis. Additionally, its use as a solvent in organic synthesis avoided sophisticated drying of solvents and substrates before use in many reactions Therefore, one of the recent challenges, due to the environmental and health concerns, is to use green synthesis in aqueous medium (Citation40–44).
Cyclocondensation of thiosemicarbazide with aldehydes could afford either 1,2,4-triazolidine-3-thiones or 2-amino-1,3,4-thiadiazoline using different reaction conditions (Citation45–56). We observed that there is a structural elucidation confusion between the open structure of thiosemicarbazone and their cycloaddition product with aldehyde. That was obvious in several published articles which reported identical spectral data for both thiosemicarbazones and their cycloaddition product (1,2,4-triazolidine-3-thiones). Thus, many authors incorrectly interpreted the NMR spectral data of alkylidene thiosemicarbazones and reported them as triazolidine-3-thiones (Citation45–48,Citation54,Citation55,Citation57), this confusion arises from the presence of three exchangeable N-H signals in both structures. Herein, we would like to report a green and simple method for synthesis of alkylidene thiosemicarbazide and clarify the structural elucidation confusion between alkylidene thiosemicarbazide, and their cyclized forms.
2. Results and discussion
Conventional and unconventional methods for the synthesis of alkylidene thiosemicarbazide had been extensively reported (Citation58–63). Most of these methods involve condensation reaction of carbonyl compounds and thiosemicarbazide in alcohol with catalytic amounts of acetic acid or mineral acids for about 3–72 h at room temperature or under reflux (Citation59–64). Moreover, recently a more facile synthesis under ultrasound or microwave (MW)-assisted methods were reported (Citation58). Herein, we would like to report a catalyst-free, simpler and cleaner (water as solvent) synthesis of alkylidene thiosemicarbazide in only few minutes reflux.
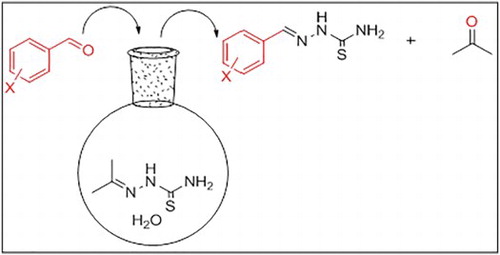
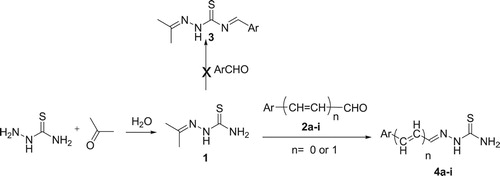
A reaction of acetone with thiosemicarbazide in water as a solvent proceeded effectively to afford isopropylidene thiosemicarbazide (1) in 95% yield. Subsequent reaction of (1) with different aldehydes 2a-i in aqueous medium gave arylidene thiosemicarbazide (4) instead of the expected arylidene isopropylidene thiosemicarbazide 3. We explained this reaction in the term of transalkylidation in which the arylidene group replaces the isopropylidene in good to excellent yields (72–95%) (Scheme 1). To the best of our knowledge, there are no reports published dealing with transalkylidation reaction either for semicarbazones or thiosemicarbazones. Aromatic aldehydes were added to a hot aqueous solution of acetone thiosemicarbazone 1 to afford the corresponding arylidene which filtered while hot as a pure precipitate. The insolubility of the arylidene thiosemicarbazide in hot water makes its isolation as pure product is so easy and gives this method an advantage over the previously reported direct condensation methods of the aromatic aldehydes with thiosemicarbazide that need an extra step for purification of the products.
Scheme 1. Transalkylidation reaction.
To evaluate the solvent effect, the reaction between benzaldehyde and acetone thiosemicarbazone 1 was selected as a model reaction and was conducted using various type of solvents (non-polar, polar protic and polar aprotic solvents) such as toluene, H2O, EtOH, DMF and CH3CN.
When the reaction was carried out in aprotic solvent either polar or non-polar solvents, DMF, CH3CN and toluene, no product was detected (, entries 3–5). Whereas in ethanol, the reaction occurred with good yield (80%) but it requires longer reaction time (4 h) to go to completion (, entry 2). Interestingly, best results obtained in case of employing water as a solvent (, entry 1). based on the criteria such as reaction time (5 min), green nature, cost-effective and excellent yield (95%), water as solvent proved to be supreme solvent for the present procedure. Given this excellent result, we decided not to try additional optimization for the experimental conditions and used these to investigate the substrate scope of the reaction ().
Table 1. Solvent optimization for the synthesis of 4a.
Table 2. Substrate scope for the transalkylidation reactiona.
2.1. Substrate scope
To examine the scope of the transalkylidation reaction in an aqueous medium for the preparation of thiosemicarbazones, a series of aromatic aldehydes or ketones with structurally divergent functional groups were examined ().
It was found that transalkylidation method is tolerant for both electron-rich (Me, OH, Me2N) and electron-deficient (NO2, Cl) aromatic aldehydes. But less tolerant for aromatic ketone such as acetophenone. This could be attributed to lower reactivity of ketones compared to aldehydes towards nucleophilic attack. Moreover, the reaction proceeded effectively with unsaturated aldehyde (cinnamaldehyde) to afford the corresponding thiosemicarbazone (allylidenethiosemicarbazide). Interestingly, different positions (o, m, p) on the phenyl group did not show any significant effect on the reaction time, purity and yield percent. Unfortunately, when aliphatic aldehydes were employed the reaction gives the corresponding thiosemicarbazones in poor yield. (, entries 12,13)
2.2. Reaction mechanism
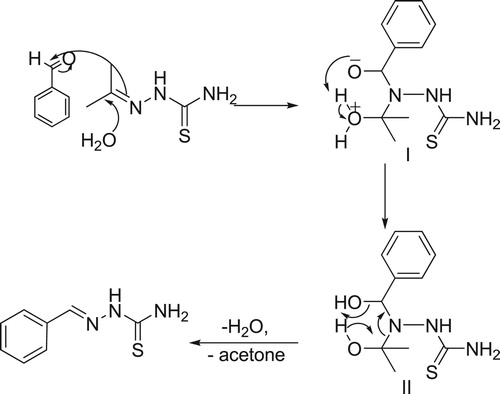
The findings that the transalkylidation reaction only takes place in polar protic solvent and doesn’t proceed in aprotic polar or non-polar solvents, suggests an important role for the solvent. So, we proposed a mechanism of the transalkylidation reaction as shown in Scheme 2. Nucleophilic attack of water molecule on the carbon atom of imine group (C = N) of acetone thiosemicarbazone induces the nucleophilic attack of the π-electrons of the imine double bond on the carbonyl group (C = O) of the aromatic aldehyde to afford the intermediate I. then Proton transfer occur in the second step to give the intermediate II, subsequent loss of water and acetone molecules afford the final product. The nucleophilicity of the solvent (water or ethanol) plays a vital part in this mechanism as it initiates the reaction. Furthermore, its polar character stabilizes the ionic intermediates I and II which was formed during the reaction pathway. That was confirmed when, no product detected either in polar aprotic (DMF, CH3CN) or nonpolar aprotic solvent (toluene) as they are considered to be non-nucleophilic solvents.
Scheme 2: Transalkylidation reaction mechanism.
2.3. NMR argument for thiosemicarbazones and their cycloaddition products
All prepared products were fully identified and characterized by analytical, spectroscopic techniques (see supplementary information).
For all of the prepared thiosemicarbazones, the NH2 group appears as two peaks in proton NMR with one proton for each. This is due to the partial double bond character induced by the thioamide moiety , which restricts the free rotation, making the two protons of the NH2 group diastereotopic protons or electronically nonequivalent. One is trans to the NH and the other is cis. This is a very common characteristic of many amide or thioamide-containing molecules.
Figure 1. Resonance of thioamode group in thiosemicarbazones.
To confirm this assumption, the temperature effect on proton NMR have been investigated for compound 4c. At low temperature, the energy barrier for rotation is high to prevent averaging (fast rotation), the two NH2 protons are non-equivalent and showed two peaks. Rising up the temperature, signal averaging takes place, and the two protons begin to exchange on the NMR timescale. As this rate of exchange becomes faster, the signal coalescing and finally complete signal averaging to the point where the two protons are equivalent .
Figure 2. 1H-NMR spectrum of 4c at different tempertures with the bottom spectrum being at low temperature and the top spectrum being at high temperature.
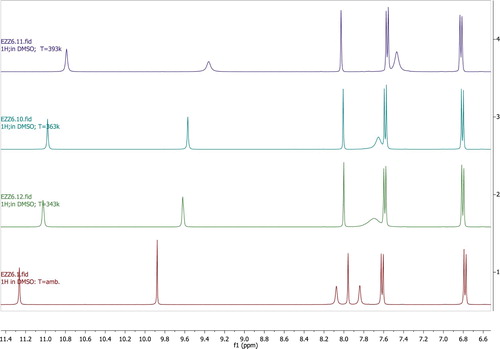
Figure 3. (i) 15N-NMR in DMSO-d6, (ii) 1H-15N (HSQC) 2D-NMR experiments at ambient temperture (iii) and at elevated temperture (343 k) for compound 4c.
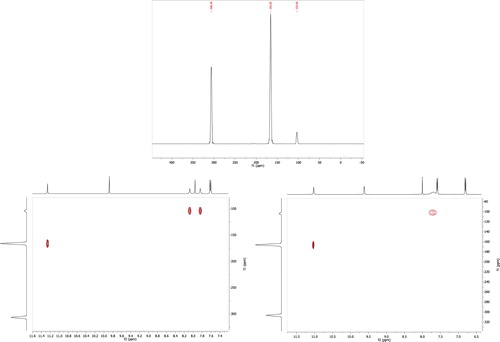
Additionally, 15N-NMR and 1H-15N Heteronuclear Single Quantum Coherence (HSQC) experiments were carried out for compounds 4c and 4d. As shown in and . Three and four 15N-NMR peaks were observed for 4c and 4d respectively. The four nitrogen peaks of compound 4d were assigned to the three nitrogens of the thiosemicarbazone and the fourth nitrogen corresponds to the nitro group. The cross-peaks due to correlating the 15N chemical shifts with H chemical shifts which are spin-coupled showed that the nitrogen appeared at highest field correlates to two protons in 1H-NMR, which clearly proves its chemical nature as NH2 group. Moreover, cross-peak has been observed between the next nitrogen and the most deshielded proton indicating NH group. While no cross peak relates the third nitrogen to any proton which corresponds to imine nitrogen (C = N). For compound 4d the most downfield nitrogen appeared is due to the nitro group of the arylidene moiety.
Figure 4. (i) 15N-NMR in DMSO-d6, (ii) 1H-15N (HSQC) 2D-NMR experiment at ambient temperture (iii) and its expansion for compound 4d.
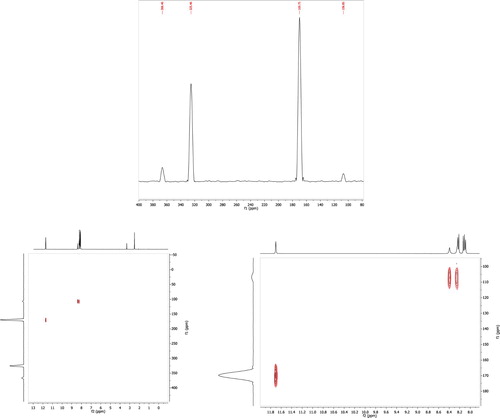
Figure 5. Structure of thiosemicarbazones A and 1,2,4-triazolidene-3-thiones B.
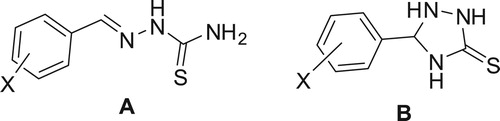
Figure 6. Atom labelling for x-substituted benzylidene thiosemicarbazide and its cycloaddtion products.
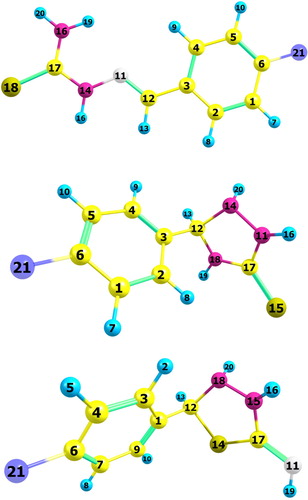
As mentioned before, the reaction of aldehydes or ketones with thiosemicarbazide could afford either condensated product (thiosemicarbazone) or continued further to afford 1,2,4-triazolidene-3-thione or 2-amnio-1,3,4-thiadiazoline as cycloaddition product of thiosemicarbazone intermediate.Herein, we present examples in which incorrect interpretation of NMR was reported. In all of these examples, the products were incorrectly assigned as 1,2,4-triazolidene-3-thione (B) instead of thiosemicarbazones (A) . This confusion is due to the appearance of three exchangeable peaks in 1H NMR, which wrongly interpreted to three NH peaks correspond to 1,2,4-triazolidene-3-thione instead of NH and NH2 peaks corresponding to thiosemicarbazones. Furthermore, in order to make structural distinguish between the three structural isomers namely: triazolidenethione, thiadiazole and thiosemicarbazone. DFT-NMR calculations were done for the previously optimized possible structures using gauge-including atomic orbital GIAO/B3LYP density functional method with 6-31G basis set by Gaussian 09 software. The atom labelling for the three isomers are shown in , and the calculated chemical shifts are listed in .
Table 3. Experimental and calculated NMR chemical shifts for thiosemicarbazones 4a, 4d and 4j and their cyclized forms.
Inspection of showed that besides the presence of the thiocarbonyl group (C = S) which appears at ≈170–180 ppm, thiosemicarbazone is characterized by its azomethine proton and carbon at chemical shift ≈7–8 and 140 ppm in 1H NMR and 13C-NMR respectively. While cycloaddition of thiosemicarbazone to afford triazolidene-3-thione or thiadiazoline led to disappearance of the azomethine group and presence of saturated carbon involved in the ring formation. The saturated carbon and its proton should appear at chemical shift ≈70–80 and 4–5 ppm in 13C-NMR and 1H-NMR respectively. Furthermore, the thiocarbonyl moiety still exists in triazolidine but absent in case of thiadiazoline. To confirm the above data, acetylation of 1 with acetic hydride in pyridine to afford thiadiazoline 5 has been done (Scheme 3).
Scheme 3. Acetylation of acetone thiosemicarbazone.
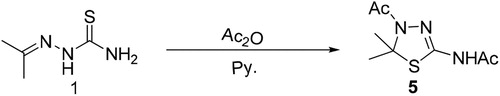
Thiadiazoline 5 showed a peak at 80 ppm in 13C-NMR corresponding to the saturated carbon of the thiadiazoline ring and disappearance of peak at 179.17 ppm corresponding to thiocarbonyl moiety, which agrees to the theoretical calculation.
Based on these findings, cyclocondensation of thiosemicarbazide and aldehydes or ketones affording 1,2,4-triazolidine-3-thiones has been incorrectly reported several times in the literature, either using uncatalyzed reaction (Citation56,Citation65) or using catalyst such as DMAP (Citation46), malononitrile (Citation66), PEG-400 (Citation48), [C16MPy]AlCl3Br (Citation47), Nanostructured Samarium Doped Fluorapatites (Citation67,Citation68), iron-doped fluorapatite (Citation65) and Sm2O3/Fluoroapatite (Citation45). In all these reports’ authors based their structural assignment on the presence of three exchangeable protons for three NH groups but both peaks at ≈80 and 4 ppm were missing in 13C-NMR and 1H-NMR respectively. Instead, peaks at 140 and 8 ppm were present, which are characteristic for the imine functionality; Moreover, the three exchangeable protons could also correspond to the NH and NH2 of the acyclic isomers as proven before. This indicates that the reaction stopped at the earlier stage of condensation affording thiosemicarbazone and no cyclization to triazolidine had been taking place. The presence of three exchangeable protons accounts for the confusion between the acyclic and the cyclized form in these reports.
To support our idea, the spectral data (1H and 13C) for previously reported 5-phenyl-1,2,4-triazolidine-3-thione or 5-(4-methoxyphenyl)-1,2,4-triazolidine-3-thione (Citation45–48,Citation56,Citation69) together with the spectral data of their acyclic forms (compounds 4a and 4j respectively) obtained from this work are listed in and respectively. Inspection of or showed that the reported spectral data of the cyclic forms are identical to that of the acyclic forms. This shows the misleading in the assignment of the structure in these previous studies.
Table 4. Comparison between NMR data for (E)-2-benzylidenehydrazine carbothioamide A prepared in this work and for 5-phenyl-1,2,4-triazolidine-3-thione B previously reported in the literature.
Table 5. Comparison between NMR data for (E)-2-(4-methoxybenzylidene)hydrazine carbothioamide A prepared in this work and for 5-(4-methoxyphenyl)-1,2,4-triazolidine-3-thione B previously reported.
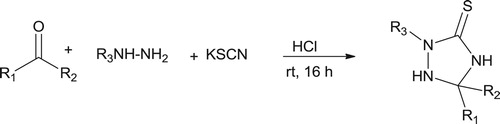
To the best of our knowledge, successful reported synthesis for triazolidine-3-thione neither done by cycloaddition of thiosemicarbazone nor cyclocondensation of thiosemicarbazide with aldehyde or ketones. Alternatively, 1,2,4-triazolidine-3-thione scaffold is readily accessible through a three-component reaction between hydrazines, aldehydes or ketones and potassium thiocyanate in hydrochloricAcid (Citation70) (Scheme 4).
Scheme 4. Synthesis of 1,2,4-triazolidine-3-thione (Citation70).
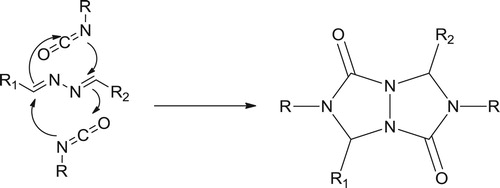
Furthermore, beside this method, there is another reaction titled Criss-Cross Cycloadditions of ketazines led to formation of 3,7-diphenyltetrahydro-[1,2,4]triazolo[1,2-a][1,2,4]triazole-1,5-dithione (Citation71) (Scheme 5).
Scheme 5. Criss-cross cycloadditions of ketazines (Citation71).
3. Materials and methods
Unless otherwise stated, CH3CN, DMF and toluene were purified and dried by distillation over 4A molecular sieves. Thiosemicarbazide, aldehydes and ketones were purchased from Sigma-Aldrich and used without further purification. 1H NMR, 13C NMR, and 15N NMR spectra were recorded at room temperature on a Bruker AC 300 FT, an Avance Bruker DPX 300, or on a Bruker Avance DRX 400 spectrometers.
3.1. Synthesis of 2-(propan-2-ylidene)hydrazinecarbothioamide
A hot solution of thiosemicarbazide (0.91 g, 10 mmol) in 20 mL of water was added to Acetone (10 mmol), Then the mixture was heated under reflux for 2 h. After cooling, the reaction mixture was filtered, and the obtained white solid was washed with water. The crude product was purified by crystallization from hot water.
3.2. General procedure for synthesis of alkylidene thiosemicarbazide
Aldehyde or ketones (2 mmol) were added to hot solution of Acetone Thiosemicarbazone 1 (2 mmol) in 20 ml water then the suspension was heated and stirred for few minutes until starting material 1 is consumed as detected from TLC. The precipitate formed was collected by filtration while hot.
3.3. Synthesis of N-(4-Acetyl-5,5-dimethyl-4,5-dihydro-1,3,4-thiadiazol-2-yl) acetamide (5)
The thiosemicarbazone 1 (1.0 eq.) was added to a stirring solution of acetic anhydride (5.0 eq.) and 3 drops of pyridine. the reaction mixture was heated for 5 h at 100°C. The mixture was poured on ice and the resulting suspension was filtered and washed with water and diethyl ether.
3.4. Computational details
All calculations were performed using the GAUSSIAN 09W. (Citation72) initially a conformational search for obtaining the most stable conformer was done using the semi-empirical PM3 method, the most stable conformer was subjected to full geometrical optimizations using the DFT and Becke’s three-parameter hybrid exchange functional in combination with the gradient-corrected correlation functional of Lee, Yang and Parr B3LYP/6–31++G(d,p) method (Citation73–75). For all optimizations, the vibrational frequencies were checked for imaginary frequencies to ensure all final geometries corresponded to a true minimum on the electronic potential energy surface. 1 H-NMR chemical shifts of the previously optimized compound have been calculated using gauge-including atomic orbital GIAO/B3LYP density functional method with 6-31G basis set. To evaluate the relative chemical shifts, the tetramethylsilane (TMS) shielding constants calculated at B3LYP/6-31G method. The inclusion of solvent in the calculations was done via the inclusion of the polarizable continuum model (PCM) (Citation76).
2-(propan-2-ylidene)hydrazinecarbothioamide (1)
1H NMR (300 MHz, DMSO) δ 9.86 (s, 1H, NH), 7.96, 749 (2s, 2H, NH2), 1.90 (s, 3H, CH3), 1.88 (s, 3H, CH3).13C NMR (75 MHz, CDCl3) δ 179.17 (C = S), 152.62(C), 25.87(CH3), 18.41(CH3).
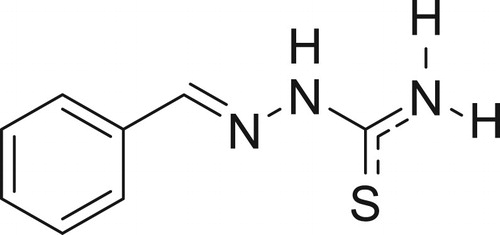
2-benzylidenehydrazinecarbothioamide (4a) (Citation77)
1H NMR (300 MHz, DMSO-d6) δ 11.44 (s, 1H, NH), 8.21, 7.99(2s, 2H, NH2), 8.06 (s, 1H, CH = N), 7.78 (dd, J = 6.4, 2.8 Hz, 2H, Phenyl H), 7.42–7.35 (m, 3H, Phenyl H).13C NMR (75 MHz, CDCl3) δ 178.83 (C = S), 143.20 (CH = N), 134.98(C), 130.70(CH), 129.51(CH), 128.13(CH).
(E)-2-((E)-3-phenylallylidene)hydrazinecarbothioamide (4b)
1H NMR (400 MHz, DMSO-d6) δ 11.40 (s, 1H, NH), 8.18 (s, 1H, aromatic H), 7.90 (d, J = 7.7 Hz, 1H, allylidene H), 7.61, 7.33 (2s, 2H, NH2), 7.56 (m, 2H, aromatic H), 7.40 (m, 2H, aromatic H), 7.03 (d, J = 19.0 Hz, 1H, allylidene H), 6.86 (dd, J = 18.3, 6.6 Hz, 1H, allylidene H).13C NMR (101 MHz, DMSO-d6) δ 178.15(C = S), 145.14, 139.31, 136.35, 129.36, 129.31, 127.39, 125.53.
(E)-2-(4-hydroxybenzylidene)hydrazinecarbothioamide (4c) (Citation77)
1H NMR (400 MHz, DMSO-d6) δ 11.26 (s, 1H, NH), 9.88 (s, 1H, OH), 8.09, 7.84 (2s, 2H, NH2), 7.96(s, 1H, CH = N),7.61 (d, J = 8.5 Hz, 2H, aromatic H), 7.78 (d, J = 8.5 Hz, 2H, aromatic H).13C NMR (101 MHz, DMSO-d6) δ 177.86(C = S), 159.70(C), 143.15(CH), 129.52(CH), 125.60(C), 116.00(CH). 15N NMR (41 MHz, DMSO) δ 306.31(N=), 165.64(NH), 103.35(NH2).
(E)-2-(4-nitrobenzylidene)hydrazinecarbothioamide (4d) (Citation77)
1H NMR (400 MHz, DMSO-d6) δ 11.70 (s, 1H, NH),8.39, 8.25 (2s, 2H, NH2), 8.23 (d, J = 8.0 Hz, 2H, aromatic H), 8.12 (d, J = 8.0 Hz, 2H, aromatic H), 8.09(s, 1H, CH = N).13C NMR (101 MHz, DMSO-d6) δ 178.99(C = S), 148.06(C), 141.22(C), 140.00(CH), 128.64(CH), 124.24(CH). 15N NMR (40.5 MHz, DMSO) δ 366.31(NO2), 325.15(N=), 169.45(NH), 106.50(NH2).
2-(4-chlorobenzylidene)hydrazinecarbothioamide (4e) (Citation77)
1H NMR (400 MHz, DMSO-d6) δ 11.48 (s, 1H, NH), 8.24, 8.08 (2s, 2H, NH2), 8.02 (s, 1H, CH = N), 7.84 (d, J = 8.2 Hz, 1H, aromatic H), 7.46 (d, J = 8.1 Hz, 1H, aromatic H).13C NMR (101 MHz, DMSO-d6) δ 178.54(C = S), 141.28(CH = N), 134.68(C), 133.67(C), 129.42(CH), 129.16(CH).
2-(4-(dimethylamino)benzylidene)hydrazinecarbothioamide (4f) (Citation77)
1H NMR (400 MHz, DMSO-d6) δ 11.17 (s, 1H, NH), 7.99, 7.76 (2s, 2H, NH2), 7.93 (s, 1H, CH = N), 7.58 (d, J = 8.4 Hz, 2H, aromatic H), 6.70 (d, J = 8.5 Hz, 2H, aromatic H), 2.96 (s, 6H, 2CH3). 13C NMR (101 MHz, DMSO-d6) δ 177.47(C = S), 151.85, 143.78, 129.08, 122.00, 112.15, 40.25(CH3).
2-(3-nitrobenzylidene)hydrazinecarbothioamide (4 g) (Citation77)
1H NMR (400 MHz, DMSO-d6) δ 11.62 (s, 1H, NH), 8.66 (s, 1H, CH = N), 8.32 (s, 2H, NH2), 8.24 (d, J = 6.5 Hz, 1H, aromatic H), 8.20 (d, J = 6.3 Hz, 1H, aromatic H), 8.14 (s, 1H, aromatic H), 7.69 (m, 1H, aromatic H).13C NMR (101 MHz, DMSO-d6) δ 178.80(C = S), 148.87, 140.36, 136.66, 134.01, 130.59, 124.40, 121.85.
2-(3-methylbenzylidene)hydrazinecarbothioamide (4 h)
1H NMR (400 MHz, DMSO-d6) δ 11.41 (s, 1H, NH), 8.20, 7.98 (2s, 2H, NH2), 8.01 (s, 1H, CH = N), 7.66 (s, 1H, aromatic H), 7.55 (d, J = 7.7 Hz, 1H, aromatic H), 7.29 (t, J = 7.6 Hz, 1H, aromatic H), 7.21 (d, J = 7.4 Hz, 1H, aromatic H), 2.33 (s, 3H, CH3).13C NMR (101 MHz, DMSO-d6) δ 178.38(C = S), 142.84, 138.39, 134.57, 130.99, 128.99, 128.04, 125.17, 21.31(CH3).
2-(2-chlorobenzylidene)hydrazinecarbothioamide (4i)
1H NMR (400 MHz, DMSO-d6) δ 11.62 (s, 1H, NH), 8.47 (s, 1H, aromatic H), 8.31 (s, 1H, CH = N), 8.12, 8.29 (2s, 2H, NH2), 7.48 (d, J = 7.5 Hz, 1H, aromatic H), 7.44–7.32 (m, 2H, aromatic H). 13C NMR (101 MHz, DMSO-d6) δ 178.69 (C = S), 138.60, 133.57, 131.95, 131.63, 130.20, 127.92, 127.79.
(E)-2-(4-methoxybenzylidene)hydrazinecarbothioamide (4j) (Citation77)
1H NMR (400 MHz, CDCl3) δ 11.32 (s, 1H, NH), 8.12, 7.92 (2s, 1H, NH2), 8.00 (s, 1H, CH = N), 7.74 (d, J = 8.7 Hz, 2H, aromatic H), 6.96 (d, J = 8.7 Hz, 2H, aromatic H), 3.79 (s, 3H, OCH3). 13C NMR (101 MHz, DMSO-d6) δ 178.05(C = S), 161.14(C), 142.67(CH), 129.37(CH), 127.20(C), 114.60(CH), 55.73 (OCH3).
(E)-2-(1-phenylethylidene)hydrazinecarbothioamide (4k)
1H NMR (400 MHz, DMSO-d6) δ 10.22 (s, 1H, NH), 8.28 (s, 1H, NH2), 7.96–7.90 (m, 3H, aromatic 2H, NH2), 7.54–7.15 (m, 3H, aromatic H), 2.30 (s, 3H, CH3).13C NMR (101 MHz, DMSO-d6) δ 179.38(C = S), 148.30(C = N), 138.05(C), 129.67(CH), 128.70(CH), 127.05(CH), 14.46(CH3).
N-(4-acetyl-5,5-dimethyl-4,5-dihydro-1,3,4-thiadiazol-2-yl)acetamide (5) (Citation60)
1H NMR (400 MHz, DMSO-d6) δ 11.47 (s, 1H, NH), 2.11 (s, 3H, CH3), 2.03 (s, 3H, CH3), 1.86(s, 6H, 2CH3).13C NMR (101 MHz, DMSO-d6) δ 169.66(C = O), 168.74(C = O), 143.18(C = N), 76.37(C), 28.82(CH3), 24.27(CH3), 23.01(CH3). 15N NMR (40.5 MHz, DMSO) δ 265.23(N=), 182.79(NAc), 130.09(NHAc).
Conclusions
In conclusion, we introduced a new, green and a more facile reaction titled transalkylidation which can be used for replacing isopropylidene moiety by arylidene group affording a green synthesis of a variety of arylidene thiosemicarbazide. Based on the presence of three exchangeable protons for thiosemicarbazones and their cycloaddition products, it was not so easy to distinguish between them. This confusion promotes us to make full identification for each isomer using spectral data and DFT calculations. Furthermore, simple distinguish between the three isomers could be done using 13C-NMR. Where the appearance of peaks at ≈80 and 180 ppm in 13C-NMR indicates the existence of triazolidine-3-thione ring while lacking a peak at ≈80 ppm and presence of peak at 180 ppm only proves the presence of the acyclic form. Furthermore, appearance of a peak at 80 ppm and absence of peak at 180 ppm verifies the presence of thiadiazoline ring .
Table 6. Summary for the most characterstic peaks 13C-NMR for the three structural isomers.
Supplemental Material
Download MS Word (1.6 MB)Acknowledgements
The authors thank Mr Pasquale Illiano (UNIMI) for doing some NMR spectrum.
Disclosure statement
No potential conflict of interest was reported by the authors.
Notes on contributors
Mohamed A. El-Atawy, an Assistant Professor of Organic Chemistry, his research interest is focused on synthestic methods and structure elucidation of heterocyclic compounds. Moreover, experimental and theoretical studies of the reaction mechanism of certain reactions. Among his recent research “new synthetic methodolgy for pyrroles from nitrodienes”.
Alaa Z. Omar, an Assistant Professor of Organic Chemistry. He currently is the principal investigator for a research group that has ten postgraduate students. His current research interests include the synthesis of unique polymers for solar cell applications, synthesis of novel dyes for industrial applications, design and specified synthesis for antiviral agents, and the synthesis of anticorrosive agents.
Mohamed Hagar, an Associate Professor of Organic Chemistry. His current research interests include the synthesis and structure elucidation of heterocyclic compounds, synthesis of variety of materials used for liquid crystal applications.
Essam M. Shashira, a Professor of Organic Chemistry. His research interest is focused on both photochemistry and synthesis of heterocyclic compounds with evaluation of their biological activities.
ORCID
Mohamed A. El-Atawy http://orcid.org/0000-0002-5042-5221
Mohamed Hagar http://orcid.org/0000-0003-0169-7738
Related Research Data
References
- Hall, I.H.; Chen, S.; Barnes, B.J.; West, D.X. Met.-Based Drugs 1999, 6, 143–147.
- Vengurlekar, S.; Sharma, R.; Trivedi, P. Int J Drug Deliv 2014, 6, 99–112.
- Pérez, J.M.; Matesanz, A.I.; Martín-Ambite, A.; Navarro, P.; Alonso, C.; Souza, P. J. Inorg. Biochem. 1999, 75, 255–261.
- Reddy, K.H.; Reddy, P.S.; Babu, P.R. J. Inorg. Biochem. 1999, 77, 169–176.
- Kelly, S.L.; Lamb, D.C.; Baldwin, B.C.; Corran, A.J.; Kelly, D.E. J. Biol. Chem. 1997, 272, 9986–9988.
- Souza, M.R.; Coelho, N.P.; Baldin, V.P.; Scodro, R.B.; Cardoso, R.F.; da Silva, C.C.; Vandresen, F. Nat. Prod. Res. 2018, 1–6.
- Khan, A.; Jasinski, J.P.; Smolenski, V.A.; Hotchkiss, E.P.; Kelley, P.T.; Shalit, Z.A.; Kaur, M.; Paul, K.; Sharma, R. Bioorg. Chem. 2018.
- Kalaiarasi, G.; Rajkumar, S.R.J.; Dharani, S.; Lynch, V.M.; Prabhakaran, R. Inorg. Chim. Acta 2018, 471, 759–776.
- Dong, H.; Liu, J.; Liu, X.; Yu, Y.; Cao, S. J. Mol. Struct. 2018, 1151, 353–365.
- Shipman, C. Jr; Smith, S.H.; Drach, J.C.; Klayman, D.L. Antimicrob. Agents Chemother 1981, 19, 682.
- Some, A.A.O.; Thiosemicarbazides, N.-S. Indian J. Pharm. Sci. 2003, 65, 423.
- Biot, C.; Pradines, B.; Sergeant, M.-H.; Gut, J.; Rosenthal, P.J.; Chibale, K. Bioorg. Med. Chem. Lett 2007, 17, 6434–6438.
- Siwek, A.; Stefańska, J.; Dzitko, K.; Ruszczak, A. J. Mol. Model. 2012, 18, 4159–4170.
- Banerjee, D.; Yogeeswari, P.; Bhat, P.; Thomas, A.; Srividya, M.; Sriram, D. Eur. J. Med. Chem 2011, 46, 106–121.
- Afrasiabi, Z.; Sinn, E.; Chen, J.; Ma, Y.; Rheingold, A.L.; Zakharov, L.N.; Rath, N.; Padhye, S. Inorg. Chim. Acta 2004, 357, 271–278.
- Singh, N.K.; Srivastava, A.; Sodhi, A.; Ranjan, P. Transit Met. Chem. (London) 2000, 25, 133–140.
- Singh, S.; Bharti, N.; Naqvi, F.; Azam, A. Eur. J. Med. Chem 2004, 39, 459–465.
- Sharma, S.; Athar, F.; Maurya, M.R.; Naqvi, F.; Azam, A. Eur. J. Med. Chem 2005, 40, 557–562.
- Bharti, N.; Athar, F.; Maurya, M.R.; Azam, A. Bioorg. Med. Chem 2004, 12, 4679–4684.
- Pelosi, G.; Bisceglie, F.; Bignami, F.; Ronzi, P.; Schiavone, P.; Re, M.C.; Casoli, C.; Pilotti, E. J. Med. Chem 2010, 53, 8765–8769.
- Netalkar, P.P.; Netalkar, S.P.; Revankar, V.K. Polyhedron 2015, 100, 215–222.
- Thomas, A.M.; Naik, A.D.; Nethaji, M.; Chakravarty, A.R. Inorg. Chim. Acta 2004, 357, 2315–2323.
- Ferrari, M.B.; Bisceglie, F.; Fava, G.G.; Pelosi, G.; Tarasconi, P.; Albertini, R.; Pinelli, S. J. Inorg. Biochem 2002, 89, 36–44.
- Seleem, H.; El-Shetary, B.; Khalil, S.; Mostafa, M.; Shebl, M. J. Coord. Chem 2005, 58, 479–493.
- Reis, C.M.d.; Pereira, D.S.; Paiva, R.d.O.; Kneipp, L.F.; Echevarria, A. Molecules 2011, 16, 10668–10684.
- Shahabi, S.; Norouzi, P. Int. J. Electrochem. Sci 2017, 12, 2628–2646.
- Lino, C.I.; de Souza, I.G.; Borelli, B.M.; Matos, T.T.S.; Teixeira, I.N.S.; Ramos, J.P.; de Souza Fagundes, E.M.; de Oliveira Fernandes, P.; Maltarollo, V.G.; Johann, S. Eur. J. Med. Chem. 2018, 151, 248–260.
- Haroon, M.; Akhtar, T.; Yousuf, M.; Baig, M.W.; Tahir, M.N.; Rasheed, L. J. Mol. Struct. 2018, 1167, 154–160.
- Baba, N.K.; Ashok, D.; Rao, B.A.; Sarasija, M.; Murthy, N. Russ. J. Gen. Chem. 2018, 88, 580–586.
- de Santana, T.I.; de Oliveira Barbosa, M.; de Moraes Gomes, P.A.T.; da Cruz, A.C.N.; da Silva, T.G.; Leite, A.C.L. Eur. J. Med. Chem. 2018, 144, 874–886.
- Gao, W.-W.; Gopala, L.; Bheemanaboina, R.R.Y.; Zhang, G.-B.; Li, S.; Zhou, C.-H. Eur. J. Med. Chem. 2018, 146, 15–37.
- El-Sadek, M.M.; Hassan, S.Y.; Abdelwahab, H.E.; Yacout, G.A. Molecules 2012, 17, 8378–8396.
- Abdel Hafez, N.A.; Elsayed, M.A.; El-Shahawi, M.M.; Awad, G.E.; Ali, K.A. J. Heterocycl. Chem. 2018, 55, 685–691.
- Abdel-Galil, E.; Moawad, E.B.; El-Mekabaty, A.; Said, G.E. Synth. Commun. 2018, 48, 2083–2092.
- Pyrih, A.; Berninger, M.; Gzella, A.; Lesyk, R.; Holzgrabe, U. Synth. Commun. 2018, 48 (14), 1883–1891.
- Omar, F.A.; Abelrasoul, M.; Sheha, M.M.; Hassan, H.Y.; Ibrahiem, Y.M. ChemistrySelect 2018, 3, 2604–2612.
- Chaaban, I.; El Sayeda, M.; El Razik, H.A.A.; El Salamouni, N.S.; Ghareeb, D.A.; Wahab, A.E.A. Monatshefte für Chemie-Chemical Monthly 2018, 149, 127–139.
- Golmohammadi, F.; Balalaie, S.; Hamdan, F.; Maghari, S. New J. Chem. 2018, 42, 4344–4351.
- Gomha, S.M.; Farghaly, T.A.; Abbas, E.M.; Alqurashi, N.T. J. Heterocycl. Chem. 2018, 55, 750–755.
- Lindström, U.M. Chem. Rev. 2002, 102, 2751–2772.
- Shaabani, A.; Hooshmand, S.E. Ultrason. Sonochem. 2018, 40, 84–90.
- Alderson, J.M.; Corbin, J.R.; Schomaker, J.M. Bioorg. Med. Chem. 2018, 26 (19), 5270–5273.
- Xiong, L.; Zhang, H.; He, Z.; Wang, T.; Xu, Y.; Zhou, M.; Huang, K. New J. Chem. 2018, 42, 1368–1372.
- Athanasekou, C.P.; Likodimos, V.; Falaras, P. J. Environ. Chem. Eng. 2018, 6 (6), 7386–7394.
- Maddila, S.N.; Maddila, S.; Gangu, K.K.; van Zyl, W.E.; Jonnalagadda, S.B. J. Fluorine Chem 2017, 195, 79–84.
- Mali, D.A.; Telvekar, V.N. Synth. Commun 2017, 47, 324–329.
- Patil, J.D.; Pore, D.M. RSC Adv. 2014, 4, 14314–14319.
- Ramesh, R.; Lalitha, A. RSC Adv. 2015, 5, 51188–51192.
- Mane, M.M.; Pore, D.M. Tetrahedron Lett. 2014, 55, 6601–6604.
- Meher, S.S.; Naik, S.; Behera, R.K.; Nayak, A. J. Indian Chem. Soc. 1981, 58 (3), 274–276.
- Ayyash1, A.N.; Jaffer, H.J.; Tomma, J.H. Am. J. Org. Chem. 2014, 4, 52–62.
- Hasanen, J. Egypt. J. Chem. 2006, 49 (6), 709–720.
- Yusuf, M.; Jain, P. Arab. J. Chem. 2014, 7, 525–552.
- Kumar, H.; Goyal, R.; Kaur, S.; Anand, R.D.; Parmar, A.; Kumar, B. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 2002, 41B, 2182–2184.
- Gangu, K.; Maddila, S.; Maddila, S.; Jonnalagadda, S. Molecules 2016, 21, 1281.
- Ramesh, R.; Lalitha, A. ChemistrySelect 2016, 1, 2085–2089.
- Elsayed Abdou, S.; Mohamed El-Qusy, S.; Selim Ghabrial, S.; Haggag, M. Mod. Appl. Sci. 2011, 5, 140–149.
- Carradori, S.; Secci, D.; D'Ascenzio, M.; Chimenti, P.; Bolasco, A. J. Heterocycl. Chem. 2014, 51, 1856–1861.
- Aly, M.; El-Mageed, A.; El Kafafy, A.; Nawwar, G. J. Plant Prot. Res. 2011, 51, 114–120.
- De Monte, C.; Carradori, S.; Secci, D.; D'Ascenzio, M.; Guglielmi, P.; Mollica, A.; Morrone, S.; Scarpa, S.; Agliano, A.M.; Giantulli, S. Eur. J. Med. Chem 2015, 105, 245–262.
- Ghaemy, M.; Mighani, H.; Alizadeh, R. Chin. J. Polym. Sci. 2011, 29, 149–155.
- Singh, S.; Jaggi, S. Acta Pharmaceutica Sciencia 2009, 51, 47–55.
- Rodriguez-Argüelles, M.C.; Ferrari, M.B.; Fava, G.G.; Pelizzi, C.; Pelosi, G.; Albertini, R.; Bonati, A.; Dall'Aglio, P.P.; Lunghi, P.; Pinelli, S. J. Inorg. Biochem. 1997, 66, 7–17.
- El-Asmy, A.A.; Al-Gammal, O.A.; Saad, D.A.; Ghazy, S.E. J. Mol. Struct. 2009, 934, 9–22.
- Gangu, K.K.; Maddila, S.; Maddila, S.N.; Jonnalagadda, S.B. Res. Chem. Intermed 2017, 43, 1793–1811.
- Lalitha, A.; Ramesh, R. 11th International Conference on Science Engineering & Technology 2015, 25–28.
- Gangu, K.K.; Maddila, S.; Maddila, S.N.; Jonnalagadda, S.B. Molecules 2016, 21, 1281.
- Gangu, K.K.; Maddila, S.; Maddila, S.N.; Jonnalagadda, S.B. RSC Adv. 2016, 6, 58226–58235.
- Mane, M.; Pore, D. Tetrahedron Lett. 2014, 55, 6601–6604.
- Huggins, W.M.; Minrovic, B.M.; Corey, B.W.; Jacobs, A.C.; Melander, R.J.; Sommer, R.D.; Zurawski, D.V.; Melander, C. ACS. Med. Chem. Lett. 2016, 8, 27–31.
- Safin, D.A.; Babashkina, M.G.; Bolte, M.; Garcia, Y. Polyhedron 2013, 62, 133–137.
- Gaussian09, R.A. Inc., Wallingford, CT 2009, 121, 150–166.
- Becke, A. J. Chem. Phys 1993, 98, 5648.
- Lee, C.; Yang, W.; Parr, R.G. Phys Rev B 1988, 37, 785.
- Hariharan, P.; Pople, J.A. Chem. Phys. Lett 1972, 16, 217–219.
- Scalmani, G.; Frisch, M.J. J. Chem. Phys. 2010, 132, 114110.
- Alam, M.S.; Liu, L.; Lee, D.U. Chem. Pharm. Bull 2011, 59, 1413–1416.