
ABSTRACT
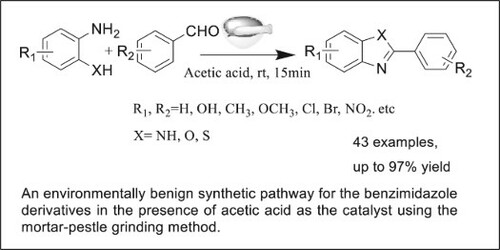
An environmentally friendly and efficient protocol for the synthesis of benzimidazoles by mortar–pestle grinding technique in the presence of acetic acid as a catalyst is reported. This mechanochemical method proceeded with the condensation of aldehyde and o-phenylenediamine followed by cyclization reaction, leading to the formation of the corresponding benzimidazoles in yields up to 97%. This protocol could also be applied for the synthesis of benzothiazoles and benzoxazoles.
GRAPHICAL ABSTRACT
Introduction
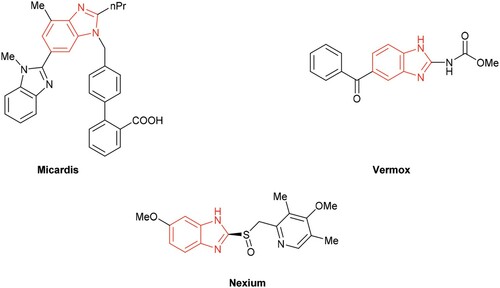
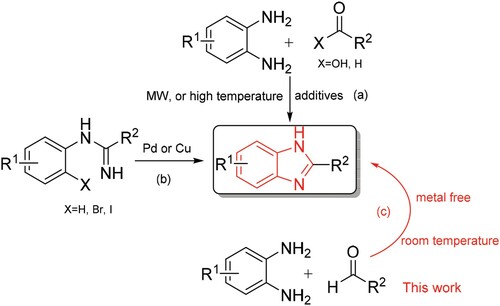
Among the various N-containing heterocyclic compounds, benzimidazole and its derivatives are common building blocks usually found in biologically active and therapeutically useful products. They are also acclaimed for their widespread applications as enzyme inhibitors, drugs, dyes and polymers.Citation1 For example, there exist benzimidazole core structures in drug compounds such as Micardis, Vermox and Nexium (). Therefore, the methods to prepare benzimidazoles have gained considerable attention in recent years. Numerous approaches are available for the construction of benzimidazoles based on condensation reaction between o-phenylenediamine and carboxylic acidsCitation2 or its derivatives (nitriles, amidates and orthoesters),Citation3 or aldehydesCitation4 in the presence of different catalysts such as 4-OMe-TEMPO, polymer-supported hypervalent iodine, iodobenzene diacetate, NaHSO4-SiO2, HCl/H2O2, ionic liquids, the graphene sulfonic acid (Scheme 1a).Citation5 Although several alternative approaches have been developed including transition metal-catalyzed tandem carbonylation–cyclization reaction of o-phenylenediamine or hydroformylation reaction of N-alkenyl phenylenediamines, etc. (Scheme 1b).Citation6 Most of these methods have one or more shortcomings including the usage of strong acidic conditions, high temperature, stoichiometric number of oxidants, environmentally unfriendly solvents, prolonged reaction time or expensive metal catalysts, etc. On the other hand, mechanochemical technique has attracted more and more interest due to environmental concerns as well as low cost.Citation7 One of the common techniques employed in solvent-free reactions is mechanical grinding,Citation8 which became a powerful tool in the paradigm of synthetic organic chemistry because of the simplicity of operation and clean reaction profile as well as its potential application at a larger scale by employing adequate experimental set-up.Citation9
Figure 1. Biologically active benzimidazole derivatives.
Scheme 1. Strategies for the synthesis of benzimidazoles.
Indeed, there has been an upsurge in the use of mechanochemical grinding methods for organocatalytic reactions including Aldol,Citation10 Baylis–HillmanCitation11 and cross-coupling reactions.Citation12 Very recently, Sharma et al. synthesized benzothiazole, benzimidazole and benzoxazole derivatives by using ZnO-NPs via a ball-milling strategy.Citation13a However, the ZnO nanoparticles are difficult to prepare. In 2008, Das et al. reported a one-pot efficient synthesis of benzimidazole derivatives under solvent-free green conditions,Citation13b but this method requires high temperature (140°C). Azarifar et al. reported the synthesis of 2-aryl-1-(arylmethyl)-1H-benzimidazoles by acetic acid-promoted condensation of o-phenylenediamine with aldehydes.Citation13c However, the yields were not very satisfactory and the reaction time was too long. In 2017, Daw et al. discovered a method that used dehydrogenative coupling of aromatic diamines and alcohols to form benzimidazoles catalyzed by pincer complexes of Earth-abundant cobalt under base-free conditions, but this method requires high-temperature conditions and long reaction time.Citation13d
As part of our endeavours to develop ‘green’ catalytic protocols,Citation14 herein is reported a mortar–pestle grinding method for the synthesis of benzimidazoles. This protocol features the following items: simple experimental operation by grinding of a mixture of o-phenylenediamine and aldehydes in an agate mortar–pestle; mild reaction conditions at room temperature without heating and inert atmosphere; short reaction time of 15 min; good functional group tolerance with good to excellent yields; acetic acid as catalyst, avoiding the usage of any metal salts, indicating its potential practical use.
Results and discussion
Our initial efforts focused on searching reaction conditions based on 1,2-diaminobenzene and benzaldehyde as model substrates. In general, the reactions were performed in an agate mortar by simply mixing the two compounds, and then grounded gently with a pestle at ambient temperature. As shown in , control experiments showed that the reaction occurred to form the desired product in a yield around 20% without the addition of any other compounds (, Entry 1). Different acids were then tested, and organic acids bearing carboxyl group resulted in better results than inorganic acids (, Entries 2–11). Among different carboxyl acids used, acetic acid exhibited the best catalytic activity, affording 80% yield in 10 min (, Entry 11). Different amounts of acetic acid and reaction time were also examined, and 0.5 mL acetic acid together with 15 min reaction time seemed to be the best choice, affording a yield 96% (, Entries 12–17).
Table 1. Optimization of the reaction condition for benzomidazole.Table Footnotea
Next, after achieving the optimized reaction conditions, the substrate scope of this reaction was studied. As summarized in , electronic or steric effects seemed to have few influence on the results, and different substrates could proceed smoothly to give the corresponding products in good to excellent yields ranging from 88% to 97% without the formation of side products.
Table 2. Synthesis of benzimidazoles.Table Footnotea
In addition, the catalytic system could well tolerate a variety of functional groups, including nitro, cyano, methoxyl and hydroxyl groups. Meanwhile, heterocyclic compounds, such as 2-pyridinaldehyde and 3-pyridinaldehyde, could also be applied as substrates to form the corresponding products in yields 89% and 91% (, entries 12 and 18).
More importantly, this protocol could also be applied for the synthesis of benzothiazole and benzoxazole by using 2-mercaptonoaniline and 2-aminophenol as starting materials in high yields above 90% (, entries 23–26).
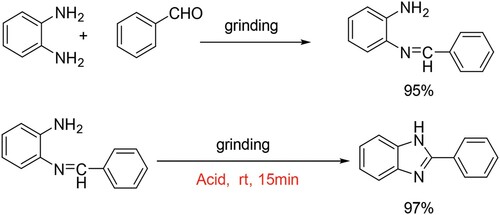
To study the reaction pathway, some control experiments were then carried out. As shown in Scheme 2, 2-(benzylideneamino)aniline could be easily formed in high yield by grinding the mixture of the two substrates in the absence of acid, which could be transformed easily to be the final product after adding acetic acid as the catalyst at room temperature.
Scheme 2. Explore the reaction pathway.
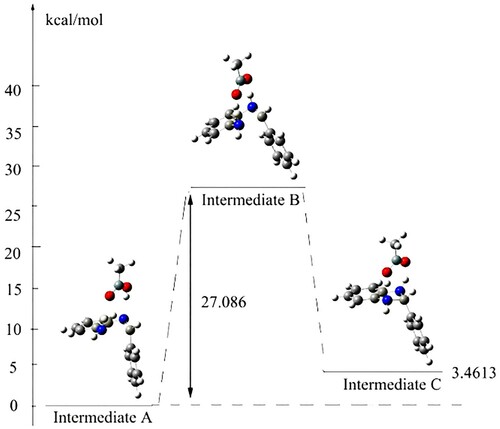
At last, preliminary computational studies using DFT methods implemented in Gaussian 09 (see supporting information) were also carried out, which indicated that the reaction might involve a coordinated cyclic transition state with energy of 27.086 kcal/mol ().
Figure 2. Optimized geometry of transition state B.
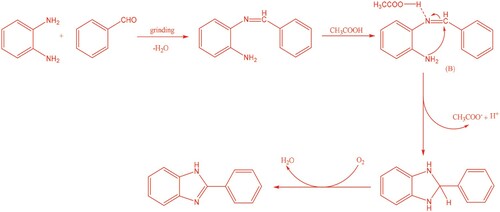
Based on our studies as well as literature,Citation15 a possible reaction pathway is proposed as shown in Scheme 3. The reaction is initiated by the formation of a Schiff’s base when an aldehyde molecule comes in contact with 1,2-diaminobenzene. Next, H+ ion of acetic acid attacks the nitrogen atom of imine, providing the intermediate B. The followed amino group attack of the imine carbon would result in the formation of 2-phenyl-2,3-dihydro-1H-benzo[d]imidazole. Finally, oxidation of 2-phenyl-2,3-dihydro-1H-benzo[d]imidazole by air affords the product.
Scheme 3. Proposed reaction mechanism.
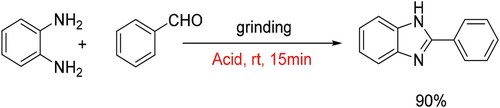
Furthermore, the scaled-up experiment was also carried out by using 1,2-diaminobenzene (0.54 g, 5 mmol) and benzaldehyde (0.53 g, 5 mmol) as substrates. After reaction, the mixture was diluted in 20 mL water, and the crude product was filtered off as yellow precipitate, which could be purified by recrystallization from ethanol in a yield around 90% (Scheme 4).
Scheme 4. Reaction conditions: 1,2-diaminobenzene (5 mmol), benzaldehyde (5 mmol), acetic acid (2.5 mL), grinding 15 min.
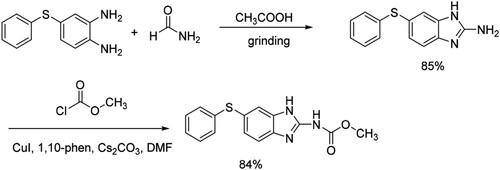
Finally, the possibility of application of this protocol was taken as an example by the synthesis of fenbendazole. As shown in Scheme 5, which is an efficient and low-toxic broad-spectrum anthelmintic,Citation16 it could be obtained by this method without toxic by-products and harsh conditions in a two-step procedure in a total yield of 71%.
Scheme 5. Synthesis of fenbendazole.
Conclusion
In conclusion, we have developed an environmentally benign synthetic pathway for the synthesis of benzimidazoles in the presence of acetic acid as the catalyst using the mortar–pestle grinding technique. 2-phenylbenzothiazoles and 2-phenylbenzoxazoles could also be prepared by this method in good yields. The operational simplicity, good functional group tolerance as well as short reaction time would make this protocol attractive in the practical purpose.
Experimental
General information
All reagents were purchased from commercial suppliers and used without further purification. All experiments were carried out in Agate mortar. Column chromatography was carried out with silica gel (200–300 mesh). Thin-layer chromatography was carried out using Merck silica gel GF254 plates. All products were characterized by NMR. 1H NMR spectra were recorded at 400, 500 MHz and 13C NMR spectra were recorded at 100, 126 MHz (Bruker DPX) with Acetonitrile-d3, Acetone-d6, CDCl3 and DMSO-d6 as solvent. Chemical shifts are reported in ppm using TMS as an internal standard. Gas chromatography (GC) was recorded on an Agilent Technologies 6890 N instrument and an HP5-MS 30 m * 0.25 mm capillary apolar column (Stationary phase: 5% diphenyldimethylpolysiloxane film, 0.25 μm). GC method: Initial temperature: 150°C; Initial time: 1 min; Ramp: about 15°C/min until 250°C then 20 min.
General procedure for compounds 2a–2z
Aldehydes (1 mmol), o-phenylenediamines (1 mmol) were taken in an agate mortar and 0.5 mL of acetic acid was added and the solution was gently ground by a pestle. The reaction mixture turned to a pasty mass after 15 min of grinding. The reaction mixture was quenched with water (3 mL) and diluted with ethyl acetate (5 mL). The layers were separated and the aqueous layer was extracted with (3 × 5 mL) ethyl acetate. The crude reaction mixture was directly subjected to column chromatography (silica gel, 200–300 mesh) and eluted out in pure form by using variable percentage of ethyl acetate in petroleum ether.
White Solid; mp 294–295oC; 1H NMR (400 MHz, Acetonitrile-d3): δ 7.23–7.26 (m, 2H), 7.56 (dd, J = 21.2, 13.6 Hz, 5H), 8.10 (d, J = 4.0 Hz, 2H), 10.92 (s, 1H); 13C NMR (100 MHz, DMSO-d6): δ 151.18, 143.78, 134.96, 130.14, 129.79, 128.90, 126.40, 122.49, 121.62, 118.84, 111.28; ESI-MS (m/z): 195.1 [M + H]+.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- a) Yadav, G.; Ganguly, S. Eur. J. Med. Chem. 2015, 97, 419–443; b) Bansal, Y.; Silakari, O. Bioorg. Med. Chem. 2012, 20, 6208–6236; c) Carvalho, L. C.; Fernandes, E.; Marques, M. M. B. Chem. - Eur. J. 2011, 17, 12544–12555; d) Min, J.; Won, J.; Kang, Y. S.; Nagase, S. J. Photoch. Photobio. A. 2011, 219, 148–153; e) O’Hagan, D. J. Fluorine. Chem. 2010, 131, 1071–1081; f) Asensio, J. A.; Gomez-Romero, P. Fuel Cells. 2005, 5, 336–343; g) Prajapat, P.; Kumawat, M.; Talesara, G. L.; Kalal, P.; Agarwal, S.; Kapoor, C. S. Chem. Biol. Interface, 2018, 8, 1–10; h) Sethiya, A.; Sahiba, N.; Soni, J.; Gandhi, D.; Agarwal, S. Current. Org. Chem. 2018, 22, 2681–2716; h) Adalat, B.; Rahim, F.; Taha, M.; Alshamrani, F. J.; Anouar, E. H.; Uddin, N.; Shah, S. S. A.; Ali, Z.; Zakaria, Z. A. Molecules 2020, 25, 4828–4843.
- a) Tumelty, D.; Cao, K.; Homes, C. P. Org. Lett. 2001, 3, 83–86; b) Wu, Z.; Rea, P.; Wickam, G. Tetrahedron Lett. 2000, 41, 9871–9874; c) Huang, W.; Scarborough, R. M. Tetrahedron Lett. 1999, 40, 2665–2668; d) Wang, Y.; Sarris, K.; Sauer, D. R.; Djuric, S. W. Tetrahedron Lett. 2006, 47, 4823–4826.
- a) Nale, D. B.; Bhanage, B. M. Synlett. 2015, 26, 2835–2842; b) Xiang, S.-K.; Tan, W.; Zhang, D.-X.; Tian, X.-L.; Feng, C.; Wang, B.-Q.; Zhao, K.-Q.; Yang, H. Org. Biomol. Chem. 2013, 11, 7271–7275; c) Cano, R.; Ramón, D. J.; Yus, M. J. Org. Chem. 2010, 76, 654–660; d) Menteşe, E.; Doğan, I. S.; Kahveci, B. Chem. Heterocycl. Com. 2013, 49, 1136–1140; e) Habibi, A.; Valizadeh, Y.; Mollazadeh, M.; Alizadeh, A. Int. J. Org. Chem. (Irvine) 2015, 5, 256–263; f) Sharma, J.; Soni, P. K.; Bansal, R.; Halve, A. K. Current. Org. Chem. 2018, 22, 2280–2299.
- a) Chari, M. A.; Shobha, P. S. D.; Mukkanti, K. J. Heterocycl. Chem. 2010, 47, 153–155; b) Maiti, D. K.; Halder, S.; Pandit, P.; Chatterjee, N.; Joarder, D. D.; Pramanik, N.; Saima, Y.; Patra, A.; Maiti, P. K. J. Org. Chem. 2009, 74, 8086–8097; c) Bahrami, K.; Khodaei, M. M.; Naali, F. J. Org. Chem. 2008, 73, 6835–6837; d) Radatz, C. S.; Silva, R. B.; Perin, G.; Lenardão, E. J.; Jacob, R. G.; Alves, D. Tetrahedron Lett. 2011, 52, 4132–4136; e) Bonacci, S.; Iriti, G.; Mancuso, S.; Novelli, P.; Paonessa, R.; Tallarico, S.; Nardi, M. Catalysts 2020, 10, 845–856; f) Di Gioia, M. L.; Cassano, R.; Costanzo, P.; Herrera Cano, N.; Maiuolo, L.; Nardi, M.; Nicoletta, F. P.; Oliverio, M.; Procopio, A. Molecules 2019, 24, 2885–2898.
- a) Chen, Y.-X.; Qian, L.-F.; Zhang, W.; Han, B. Angew. Chem. Int. Ed. 2008, 47, 9330–9333; b) Kumar, A.; Maurya, R. A.; Ahmad, P. J. Comb. Chem. 2009, 11, 198–201; c) Bahrami, K.; Khodaei, M. M.; Kavianinia, I. Synthesis. (Mass) 2007, 4, 547–550; d) Saha, D.; Saha, A.; Ranu, B. C. Green Chem. 2009, 11, 733–737; e) Karthik, M.; Suresh, P. New. J. Chem. 2018, 42, 17931–17938.
- a) Sharghi, H.; Beyzavi, M. H.; Doroodmand, M. M. Eur. J. Org. Chem. 2008, 2008, 4126–4138; b) Yang, D.; Fu, H.; Hu, L.; Jiang, Y.; Zhao, Y. J. Org. Chem. 2008, 73, 7841–7844; c) Dezfoolinezhad, E.; Ghodrati, K.; Badri, R. New J. Chem. 2016, 40, 4575–4587; d) Smitha, G.; Sreekumar, K. RSC Adv. 2016, 6, 18141–18155; e) Feng, F.; Ye, J.; Cheng, Z.; Xu, X.; Zhang, Q.; Ma, L.; Lu, C.; Li, X. RSC Adv. 2016, 6, 72750–72755; f) Pizzetti, M.; Luca, E. D.; Petricci, E.; Porcheddu, A.; Taddei, M. Adv. Synth. Catal. 2012, 354, 2453–2464; g) Kumar, M.; Sharma, S.; Bhatt, V.; Kumar, N. Adv. Synth. Catal. 2015, 357, 2862–2868; h) Jin, H.; Xu, X.; Gao, J.; Zhong, J.; Wang, Y. Adv. Synth. Catal. 2010, 352, 347–350; i) Ravi, O.; Shaikh, A.; Upare, A.; Singarapu, K. K.; Bathula, S. R. J. Org. Chem. 2017, 82, 4422–4428. j) Li, M.; Tang, Y.; Gao, H.; Rao, G.; Mao, Z. Asian J. Org. Chem. 2020, 9, 1027–1031.
- a) Cave, G. W. V.; Raston, C. L.; Scott, J. L. Chem. Commun. 2001, 0, 2159–2169; b) Rodriguez, B.; Bruckmann, A.; Rantanen, T.; Bolm, C. Adv. Synth. Catal. 2007, 349, 2213–2233; c) Bruckmann, A.; Krebs, A.; Bolm, C. Green Chem. 2008, 10, 1131–1141; d) Hernandez, J. G.; Juaristi, E. J. Org. Chem. 2010, 76, 1464–1467; e) Shearouse, W. C.; Mack, J. Green Chem. 2012, 14, 2771–2775; f) Landeros, J. M.; Juaristi, E. Eur. J. Org. Chem. 2017, 2017, 687–694; g) Chakraborty, B. J. Heterocyclic Chem. 2019, 56, 3414–3422.
- a) Hernández, J. G.; Friščić, T. Tetrahedron Lett. 2015, 56, 4253–4265; b) Wang, G.-W. Chem. Soc. Rev. 2013, 42, 7668–7700.
- a) Konnert, L.; Gauliard, A.; Lamaty, F.; Martinez, J.; Colacino, E. Acs. Sustain. Chem. Eng. 2013, 1, 1186–1191; b) Stolle, A.; Szuppa, T.; Leonhardt, S. E. S.; Ondruschka, B. Chem. Soc. Rev. 2011, 40, 2317–2329.
- (10) a) Rodŕıguez, B.; Rantanen, T.; Bolm, C. Angew. Chem. Int. Ed. 2006, 45, 6924–6926; b) Almasi, D.; Alonso, D. A.; Balaguer, A. N.; Najera, C. Adv. Synth. Catal. 2009, 351, 1123–1131; c) Hernández, J. G.; García-López, V.; Juaristi, E. Tetrahedron 2012, 68, 92–97; d) Joerres, M.; Acena, J. L.; Soloshonok, V. A.; Bolm, C. ChemCatChem. 2015, 7, 1265–1269.
- Mack, J.; Shumba, M. Green Chem. 2007, 9, 328–330.
- a) Schneider, F.; Szuppa, T.; Stolle, A.; Ondruschka, B.; Hopf, H. Green Chem. 2009, 11, 1894–1899; b) Schneider, F.; Ondruschka, B. ChemSusChem. 2008, 1, 622–625; c) Fulmer, D. A.; Shearouse, W. C.; Medonza, S. T.; Mack, J. Green Chem. 2009, 11, 1821–1825; d) Thorwirth, R.; Stolle, A.; Ondruschka, B. Green Chem. 2010, 12, 985–991; e) EL-Sayed, T.; Aboelnaga, A.; Hagar, M. Molecules 2016, 21, 1111–1118.
- a) Sharma, H.; Singh, N.; Jang, D. O. Green Chem. 2014, 16, 4922–4930; b) Thakuria, H.; Das, G. Arkivoc 2008, 15, 321–328; c) Azarifar, D.; Pirhayati, M.; Maleki, B.; Sanginabadi, M.; Yami, N. R. J. Serb. Chem. Soc. 2010, 75, 1181–1189; d) Daw, P.; Ben-David, Y.; Milstein, D. ACS Catal. 2017, 7, 7456–7460.
- a) Yan, X.; Long, R.; Luo, F.; Yang, L.; Zhou, X. Tetrahedron Lett. 2017, 58, 54–58; b) Yan, X.; Sun, H.; Xiang, H.; Yu, D. G.; Luo, D.; Zhou, X. Chem. Commun. 2018, 54, 8606–8609; c) Luo, F.; Yang, J.; Li, Z.; Xiang, H.; Zhou, X. Adv. Synth. Catal. 2016, 358, 887–893; d) Ke, F.; Chen, X.; Li, Z.; Xiang, H.; Zhou, X. RSC Adv. 2013, 3, 22837–22284; e) Yao, X.; Weng, X.; Wang, K.; Xiang, H.; Zhou, X. Green Chem. 2018, 20, 2472–2476.
- Banerjee, M.; Chatterjee, A.; Kumar, V.; Bhutia, Z. T.; Khandare, D. G.; Majik, M. S.; Roy, B. G. RSC Adv. 2014, 4, 39606–39611; b) Kumar, V.; Khandare, D. G.; Chatterjee, A.; Banerjee, M. Tetrahedron Lett. 2013, 54, 5505–5509; c) Zhang, C.; Zhang, L.; Jiao, N. Green Chem. 2012, 14, 3273–3276.
- Cortés, E.C.; Mendoza, R.S.; Gutiérrez, M.S.; De Cortés, O.G.M. J. Heterocyclic Chem 2004, 41, 273–276.