
ABSTRACT
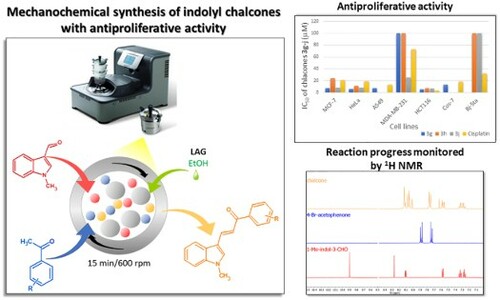
An efficient eco-friendly one-step mechanochemical synthesis of indole hybrid chalcones via the Claisen–Schmidt condensation of 1-methylindole-3-carboxaldehyde with various substituted acetophenones is described using liquid assisted grinding in a high-energy ball mill. The notable advantages of the present method are small organic solvent required (100 μL ethanol) and mainly shorter reaction time (15–60 min) as compared to the conventional method. Antiproliferative activity of synthesized chalcones was studied on several human cancer cell lines (MCF-7, HeLa, A549, MDA-MB-231, HCT116) and non-cancer lines (Cos-7 or BJ-5ta). 3,4,5-Trimethoxyphenyl 3g, 2,5-dimethoxyphenyl 3h, and 2,4,5-trimethylphenyl 3j chalcones showed considerable cytotoxicity, particularly, chalcones 3h and 3j were identified as the most potent and selective anticancer agents with IC50 values 7.9 and 6.6 μM, respectively, against the colon cancer HCT116 cell line.
GRAPHICAL ABSTRACT
Introduction
Mechanochemistry utilizing tools of high-energy ball milling has emerged as an efficient approach applicable in different fields (Citation1) such as catalysis (Citation2), polymers (Citation3), metal–organic frameworks area (Citation4), co-crystal preparation (Citation5), extractions (Citation6), pharmacology (Citation7), materials science (Citation8), nanomaterials (Citation9,Citation10), waste treatment (Citation11) and also organic synthesis (Citation12–15). Organic mechanochemistry is the most rapidly developing branch of mechanochemistry, due to its environmental friendliness (reactions are performed in the absence of solvents, without external heating, and in a short time), the possibility to prepare products that are not accessible using a solution-based synthesis and scalability (Citation16).
Chalcones (1,3-diarylprop-2-en-1-ones) are widely distributed in plants, acting as precursors for the biosynthesis of flavonoids and isoflavonoids. Thanks to easy derivatization and synthesis, chalcones are used as an effective template in medicinal chemistry for drug development. The natural and synthetic chalcones have shown numerous interesting biological activities such as anticancer (Citation17–19), antibacterial (Citation20), antiviral (Citation21), and anti-inflammatory (Citation22). Many techniques and processes have been reported with various catalysts and reaction conditions for the synthesis of these medically important compounds: Claisen–Schmidt condensation, Suzuki reaction, Heck reaction, Friedel–Crafts acylation, Wittig reaction, and Photo-Fries rearrangement of phenyl cinnamate (Citation23). The Claisen–Schmidt condensation between acetophenone and aryl aldehyde derivatives, using either strong base or acid as catalyst, is the most frequently used reaction in chalcone preparation. The disadvantage of these reactions is the long duration and often the formation of complex reaction mixtures containing also, in addition to the expected product, by-products and starting materials. The yield could vary ranging from 10 to near 100% conversion, depending on reactants and catalyst (Citation23). The classical Claisen–Schmidt reaction is generally carried out using aqueous sodium or potassium hydroxide at 50 °C over several hours (Citation24).
Many eco-friendly methods for chalcone preparation including grinding (Citation25), solvent-free synthesis (Citation26), microwave- (Citation27), and ultrasonic- (Citation28) assisted approaches have been described. Mechanochemical reactions have been studied for organic synthesis mainly in the last few decades. Efficient grinding (in a mortar and pestle) of aryl aldehydes and acetophenones with anhydrous Ba(OH)2 (Citation25), KOH (Citation29), and I2 (Citation30) in the absence of any solvent was described. The single-screw drill device was applied for the synthesis of chalcones with 10 mol% KOH (Citation31).
In the present research, following our study of mechanochemical synthesis and isomerization of N-substituted indole-3-carboxaldehyde oximes (Citation32), we focused on the preparation of 1-methylindole-chalcones by rapid, green synthesis using a ball mill. We investigated the effect of the amount of catalyst, milling time, type of milling material, and mainly the effect of solvent addition (liquid-assisted grinding, LAG) on the product-forming. The desired chalcones were prepared with high yields, which are superior to those reached by conventional methods. Our new method has the advantage of using equimolar catalysts and short reaction times . The product can be isolated simply by washing with eco-friendly citric acid and subsequent crystallisation. The universality of the proposed approach was corroborated by the condensation of 1-methylindol-3-carboxaldehyde with different substituted acetophenones. The methoxy groups and halogens were selected because it was proven that indole hybrid chalcones with these substituents exhibit antitumor effect (Citation33–35). Most of the synthesized products exhibited antiproliferative activity.
Results and discussion
Grinding experiments
The reaction of 1-methylindole-3-carboxaldehyde (1) with 4-bromoacetophenone (2a) on which the high-energy milling conditions were optimized is outlined in Scheme 1 and the results are summarized in .
Scheme 1. Claisen-Schmidt reaction of 1-methylindole-3-carboxaldehyde with 4-bromoacetophenone under ball milling conditions.
Table 1. Optimization of the milling condition for (2E)-1-(4-bromophenyl)-3-(1-methyl-1H-indol-3-yl)prop-2-en-1-one (3a).
The preparation of chalcone 3a by the classical method in abs. EtOH, catalyzed by piperidine with reflux for 24 h was described with a 92% yield (Citation36). Also, KOH catalyzed Claisen–Schmidt condensation in EtOH at r.t. during 24 h, without exact yield, was published (Citation37,Citation38). In order to compare the reaction output with the one reported in the mentioned studies, the reaction was performed under the same reaction conditions (entry 1). The reaction progress of all experiments was monitored by 1H NMR spectroscopy. For the initial experiment, an agate milling chamber and the same stoichiometry of KOH (3 equiv) as in the conventional approach were used (entries 2, 3). After 15 min of milling, the ratio of starting aldehyde 1 to desired chalcone 3a was 60:40. Another 30 min of grinding changed the ratio to 51:49 (entry 3). In addition to the chalcone, unidentifiable by-products were formed in both cases. Although excess catalyst is usually used in classical Claisen–Schmidt reactions, we experimented with a stoichiometric amount of catalyst (entry 4). In this case, the ratio of aldehyde 1 and chalcone 3a formed was better than in previous cases, exactly 37:48. In addition to unreacted acetophenone 2a (10%), 1,5-diketone 4a (5%), a product of Michael addition of acetophenone 2a to the enone system of chalcone was also identified in the reaction mixture. The similar product was isolated as a secondary product in the synthesis of naphthalen-1-yl chalcones (Citation39). These 1,3,5-triarylpentane-1,5-diones are quite useful intermediates for the synthesis of nitrogen heterocycles (Citation40) and other important complex molecules (Citation41). They can be prepared by a one-pot process by the Michael addition of acetophenones to the Claisen–Schmidt condensation product of acetophenone and arylaldehydes (Citation42,Citation43).
After determining the optimal chemical conditions, we focused on the solvent effect on the grinding. Liquid-assisted grinding (LAG), an older name: solvent-drop grinding, is an extension of traditional solvent-free mechanochemical technique (neat grinding) by which a small amount of liquid is added to enhance or control reactivity (Citation44). The parameter ŋ is an empirical indicator of how mechanochemical reactivity is influenced by the ratio of the liquid additive to the weight of reactants [ŋ = V (liquid; μL)/m (reagents, mg)]. If ŋ > 12 μL/mg, this indicates a typical solution reaction, while the ŋ in the LAG is usually in the range of 0−2 μL/mg and the result of the reaction appears to be independent of reactive solubility (Citation44). Experiments were performed with liquid additivities EtOH (entry 5−8) and MeOH (entry 9), usually used in conventional synthesis, and aprotic solvent EtOAc (entry 10). Compared to neat grinding, the amount of chalcone increased from 48% to 82%, already at the addition of 50 μL (ŋ = 0.14) and to 90−92% using 100–400 μL (ŋ from 0.29–1.14) of EtOH. The solvent amount of 100 and 200 µL, where the chalcone ratio was 91% and 90%, respectively, was proven to be optimal, and after working up the reaction mixture with environmentally friendly citric acid, the highest yield of product was obtained. At 400 µL the amount of isolated product has already decreased. Methanol proved to be less suitable (entry 9), but still more advantageous than neat grinding. The addition of an aprotic solvent (entry 10) caused an even lower chalcone 3a ratio than in neat milling, but on the other hand, it promoted dione 4a formation.
To potentially reduce the necessary milling time, the reaction with 100 μL of EtOH was run for only 5 min, however, not all the reactants were consumed (entry 11). All the reactions mentioned so far were performed at a milling speed of 600 rpm. To investigate the effect of this parameter on the reaction progress, the reactions were performed also at 400 (entry 12) and 800 rpm (entry 13). No significant differences were found and the chalcone amount ranged from 90 to 92% in all the cases.
Finally, the effect of a milling chamber and ball materials were investigated. The suitability of the best conditions for grinding (entry 6) in the steel vessel (entry 14) was checked. It was also investigated whether neat grinding (entry 15) and liquid-assisted grinding with EtOAc (entry 16) in a steel vessel would give a similar amount of dione 4a as in the agate chamber. When maintaining all the reaction parameters as in the ideal experiment, the condensation in the steel chamber (entry 14) proceeded similarly as in the agate one, but to determine the ratio of reactants and products, it was necessary to purify the resulting mixture by flash chromatography from contamination from the steel milling medium hampering the proper measurement by NMR. Neat grinding in the steel chamber (entry 15) yielded dione 4a in the highest yield of all experiments. When using ethyl acetate (entry 16), the reaction progress was very low.
The different reaction pathway when using neat grinding (entries 4, 15) can be explained by the fact that there is an insufficient diffusion rate of the reagents toward each other. This is a typical problem in solvent-free synthesis. A local increase of temperature at impact sites during high-energy milling can lead to local melting that speeds up the reaction but can also lead to product degradation (Citation45) or side-reactions as in our case. The small amounts of liquid additives (LAGs) typically speed up mechanochemical transformations. The possible mechanism of such an effect is the formation of a thin interface layer on the grain boundaries of solid reagents or solutions of both reagents at the interface (Citation45).
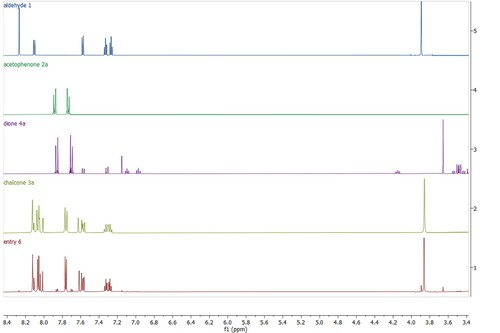
The reaction progress was monitored by 1H NMR spectroscopy (), as the chemical shifts of the protons of starting aldehyde (H-2, 8.27 ppm, N-CH3, 3.89 ppm) and acetophenone (H-2,6, 7.88 ppm; H-3,5, 7.74 ppm) and protons of products chalcone (H-2'’,6''and H-3, 8.09−8.00 ppm; H-3'’,5'’, 7.76 ppm; N-CH3, 3.86 ppm) and dione (H-2'’,6'’, 7.86 ppm; H-3'’,5'’, 7.70 ppm; H-2, 7.15 ppm; H-5’, 7.09 ppm; H-6’, 6.97 ppm) are significantly different.
Figure 1. 1H NMR spectra of starting aldehyde 1 and acetophenone 2a, products chalcone 3a and dione 4a and the reaction mixture under the best experimental milling conditions (, entry 6).
To examine the universality of the proposed approach, we performed the LAG (best conditions determined from previous experiments, , entry 6) of 1-methylindole-3-carboxaldehyde (1) with acetophenones 2b−j catalyzed by KOH at a ratio of 1:1:1 in agate vessel at 600 rpm, using 100 μL EtOH as a solvent (Scheme 2). The preparation of chalcones 3 was published previously using Claisen–Schmidt condensation with an excess of catalyst (KOH or piperidine) at r.t. during 24 h, and at 60−80°C during 12 h, respectively. The published conventional syntheses of chalcones 3 are listed in Table S1 in Supplementary data.
Scheme 2. Claisen-Schmidt reaction of 1-methylindole-3-carboxaldehyde with substituted acetophenones under ball milling conditions.
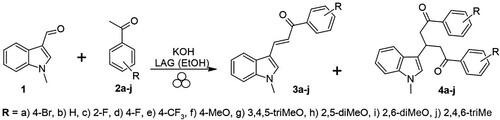
The results in terms of yield are presented in . It can be seen that a milling time of 15 min was insufficient for unsubstituted acetophenone 2b (entry 1), however, the additional 15 min of treatment resulted in the formation of a very high amount of chalcone 3b and a trace amount of dione 4b. In contrast to the conventional synthesis of 3b (Citation37,Citation38), our approach provided a significant reduction in reaction time from 24 h to 0.5 h and a reduction in the amount of solvent and basic catalyst (KOH) from 3 equiv to 1 equiv. The electron-withdrawing groups 2-F, 4-F, and 4-CF3, as well as the investigated 4-Br, increased the reactivity of acetophenones 2a,c−e. In all the cases, a reaction time of 15 min was sufficient (entries 3−5) and a small amount of dione 4a,c−e (3−8%) was also formed. Acetophenone 4f (entry 6) with a methoxy group in position 4 had approximately the same reactivity as the investigated ketone 4a. Three methoxy groups in positions 3,4,5 reduced the reactivity (entries 7, 8), and in contrast to the unsubstituted acetophenone, chalcone 3g was formed in the only ratio of 73% after 30 min in this case. If the methoxy group was in position 2 (2,5-dimethoxyactophenone 2h as reactant, entries 9–11), the ratio of chalcone 3h was reduced to 57%; this decrease was even more marked in the case of the two methoxy groups next to the carbonyl group (2,6-dimethoxy acetophenone, 3i, entries 12–14), with a ratio of 34%, presumably due to steric reasons. The progress of the reaction after 15, 30 min, or 60 min was approximately the same and did not change with further grinding. Acetophenone 2j with three electron-donor and less bulky methyl groups at positions 2,4,6 gave chalcone 3j in a good ratio of 85% (entries 15,16). It should be noted that all chalcones except 3h and 3j (oily compounds, extracted with EtOAc from the mill) were isolated as solids after grinding and were washed with citric acid. The ratios to control the reaction progress were determined by NMR spectroscopy from the crude milled mixtures taken from the mill after indicated milling times. To avoid crystallisation losses, yields are reported based on the ratio (obtained by 1H NMR spectroscopy) of the desired chalcone in powder isolated after treatment of the mixture after grinding (neutralization by washing with a citric acid solution, water, and drying). However, also in small-scale reactions (1−2 mmol), losses occur during extraction from the grinding chamber as well as during filtration.
Table 2. Synthesis of indole hybride chalcones by ball milling conditions.Table Footnotea
Antiproliferative activity
The in vitro growth inhibition activities of the prepared chalcones 3a–h,j, and dione 4a were tested against human breast adenocarcinoma (MCF-7), human cervical adenocarcinoma (HeLa), human alveolar adenocarcinoma (A549), human mammary gland adenocarcinoma (MDA-MB-231), human colorectal carcinoma (HCT116), green monkey kidney fibroblast (Cos-7) and human immortalized foreskin fibroblast cell line (BJ-5ta) using colorimetric MTT assay. The determined IC50 values of the tested compounds in comparison with reference drug cisplatin are listed in . The data are presented as the mean ± SD of three independent experiments.
Table 3. Antiproliferative activity of derivatives 3a−h,j, 4a, and reference drug cisplatin toward selected cancer and normal cell lines expressed as IC50 (μM) ± SD from three replicates after 72 h incubation.
The results of the studies on antiproliferative activity of tested compounds in show that almost none of the tested compounds affected the triple-negative breast cancer cell line MDA-MB-231, but most of them exhibit average antitumor activity on all other cell lines. Monohalogenated and monomethoxylated chalcones have a moderate antiproliferative effect against all tested cell lines, and unfortunately, also against non-cancer ones. Chalcone 3e with the CF3 group also shows excellent selective activity with an IC50 12.3 μM on colon cancer HCT116 cell line and no toxicity to Cos-7 in the tested range of concentration. The 3,4,5-trimethoxy-substituted chalcone 3g exhibits the highest toxicity against all examined cancer lines (except for the already mentioned MDA-MB-231). The disadvantage is its relatively high toxicity to non-cancer cell line Cos-7. Trimethylated chalcone 3j demonstrated high activity against the tested cancer cell lines including the triple-negative breast cancer cell line MDA-MB-231 (it was the only active substance against this line in the whole study). Moreover, its antiproliferative activity is selective, as it is non-toxic to non-cancer foreskin fibroblast cell line BJ-5ta. The tests have also shown that the most sensitive cell line to synthesized chalcones was colon cancer HCT116, where the three chalcones 3g−j acted with an IC50 below 8.0 μM. The 1,5-diketone 4a (dione) was not active against any tested cell lines in the used range of concentration.
Summary
We developed a convenient, simple, efficient, and eco-friendly green procedure for the synthesis of chalcones from 1-methylindole-3-carboxaldehyde and various acetophenones under liquid-assisted high-energy ball milling conditions. In just 15 min of treatment, eight different chalcones can be easily obtained. The major advantages of this protocol are the high yields, short reaction times, simple work-up procedure, safety, and environmental concerns. To our knowledge, this method represents the first example of the synthesis of indole hybride chalcones by milling.
The preliminary anticancer activity study of 1-methylindole hybride chalcones reveals that 2,5-dimethoxyphenyl and 2,4,6-trimethylphenyl moiety is beneficial for anticancer activity and selectivity.
Experimental section
Chemistry
Materials and methods
All reagents used in the milling process were obtained commercially (from Sigma-Aldrich, Acros Organics, Merck, and Centralchem Slovakia) and used without further purification, except for 1-methylindole-3-carboxaldehyde (1) prepared according to the literature (Citation46) and 2,5-dimethoxy-, 2,6-dimethoxyacetophenone (2h and 2i) prepared by methylation of 2,5-dihydroxy- and 2,6-dihydroxyacetophenone, respectively (Citation47). The reactions were monitored by thin-layer chromatography (TLC) using TLC-sheets ALUGRAM-SIL G/ UV254 (Macherey Nagel, Germany), eluent: hexane/EtOAc 2:1, hexane/acetone 2:1 or hexane/EtOAc 2:1. Purification by flash chromatography was performed using Silica gel 60 Å (0.0040–0.063 mm, Merck, Germany). NMR spectra were recorded on a VNMRS spectrometer (Varian) operating at 600 MHz for 1H and 150 MHz for 13C at 299.15 K and on a Mercury Plus spectrometer (Varian) 400 MHz operating at 400 MHz for 1H and 100 MHz for 13C. Chemical shifts (δ in ppm) are given from the internal solvent DMSO-d6.
Experimental procedure for mechanochemical synthesis
The experiments were performed with agate or steel milling media, a planetary ball mill Pulverisette 7 premium line (Fritsch, Germany) with the 20 mL milling vessel. Eighty agate milling balls with a diameter 5 mm were used and the whole mass of reactants was 0.350 g. In the case of milling in steel, 24 g of steel milling balls with a diameter 3 mm were used and the whole mass of reactants was also 0.350 g. The number of balls and mass of reactants in both cases was selected to maintain a ball-to-powder ratio of 71. The atmosphere in both cases was air. Almost all experiments were performed at a milling speed of 600 rpm, only in entry 12 and 13, the milling speed 400 rpm and 800 rpm, respetively, was used. Further details regarding the experimental conditions are listed in and . After a certain milling time, a sample (7−10 mg) was taken from the reaction mixture for a 1H NMR experiment to monitor the reaction progress. After milling, the reaction mixtures were either scraped from the milling chamber manually or extracted with ethyl acetate. The latter was done for entries 11 and 16 in . In the case of manual scraping, the product was transferred to beaker. Water was then added to the chamber, the product residues were dispersed using a spatula and subsequently transferred to the beaker where the majority of the product was already present. The water solution of product was then neutralized with citric acid (0.2–0.4 g) until the pH value of 5–6 was reached. This was done to remove the residual KOH and the residual starting aldehyde 1. The product was then filtered, dried and subjected to NMR measurement, and crystallized to give pure compounds for product identification.
For the experiments in steel milling media, the product before NMR measurement was purified from media wear using flash chromatography over silica gel (0.5 g), using EtOAc as eluent.
When the liquid acetophenones 2h, 2j were used the product of the milling process was oily and was extracted with ethyl acetate from the vessel (2 × 15 mL), then washed with a citric acid solution, water (25 mL) and brine (10 mL), dried over Na2SO4, evaporated. The residue after evaporation was subjected to chromatographic separation over silica gel (eluent: hexane/EtOAc 1:2 for 2h and hexane/EtOAc 2:1 for 2j).
Spectral data for the products
(2E)-1-(4-Bromophenyl)-3-(1-methyl-1H-indol-3-yl)prop-2-en-1-one (3a)
1H NMR (400 MHz, DMSO-d6): δ = 8.12 (s, H-2’), 8.12 (d, 1H, J 7.8 Hz, H-4’), 8.06 (d, J 8.6 Hz, H-2'’,6'’), 8.03 (d, 1H, J 15.5 Hz, H-3), 7.76 (d, 2H, J 8.5 Hz, H-3'’,5'’), 7.61 (d, 1H, J 15.5 Hz, H-2), 7.57 (d, 1H, J 8.0 Hz, H-7’), 7.32 (dd, 1H, J 8.0, 7.6 Hz, H-6’), 7.28 (dd, 1H, J 8.1, 7.6 Hz, H-5’), 3.86 (s, 3H, NCH3). 13C NMR (100 MHz, DMSO-d6): δ = 187.7 (C-1), 139.0 (C-3), 138.1 (C-7'a), 137.5 (C-1'’), 137.1 (CH, C-2’), 131.7 (C-3'’,5'’), 130.2 (C-2'’,6'’), 126.4 (C-4'’), 125.6 (C-3'a), 122.9 (C-6’), 121.5 (C-5’), 120.6 (C-4’), 114.8 (CH, C-2), 111.8 (C-3’), 110.9 (C-7’), 33.1 (N-CH3).
1,5-bis-(4-Bromophenyl)-1-(1-methyl-1H-indol-3-yl)pentane-1,5-dione (4a)
1H NMR (400 MHz, DMSO-d6) δ = 7.86 (d, 4H, J 8.6 Hz, H-2'’,6'’), 7.70 (d, 4H, J 8.6 Hz, H-3'’,5'’), 7.57 (dt, 1H, J 8.0, 0.9 Hz, H-4’), 7.31 (dt, 1H, J 8.2, 0.8 Hz, H-7’), 7.15 (s, 1H, H-2’), 7.09 (ddd, 1H, J 8.2, 7.0, 1.1 Hz, H-6’), 6.97 (ddd, 1H, J 8.0, 7.0, 1.0 Hz, H-5’), 4.15 (p, 1H, J 6.9 Hz, H-3), 3.66 (s, 3H, N-CH3), 3.52 (dd, 2H J 17.0, 6.6 Hz, Ha-2,4), 3.45 (dd, 2H, J 17.0, 7.3 Hz, Hb-2,4). 13C NMR (100 MHz, DMSO-d6) δ = 198.2 (C-1), 136.6 (C-7'a), 135.7 (C-1'’), 131.8 (C-3'’,5'’), 130.0 (C-2'’,6'’), 127.2 (C-4'’), 126.6 (C-2’), 126.4 (C-3'a), 121.0 (C-6’), 119.0 (C-4’), 118.4 (C-5’), 116.5 (C-3’), 109.6 (C-7’), 44.4 (C-2,4), 32.3 (NCH3), 27.9 (C-3).
(2E)-3-(1-Methyl-1H-indol-3-yl)-1-(phenyl)prop-2-en-1-one (3b)
1H NMR (400 MHz, DMSO-d6): δ = 8.14–8.09 (m, 3H, H-4’, H-2'’,6'’), 8.11 (s, H-2’), 8.02 (d, 1H, J 15.4 Hz, H-3), 7.64 (d, 1H, J 15.4 Hz, H-2), 7.64–7.62 (m, 1H, H-4'’), 7.58–7.54 (m, 3H, H-7’, H-3'’,5'’), 7.32 (dd, 1H, J 7.8, 7.1 Hz, H-6’), 7.28 (dd, 1H, J 8.1, 7.1 Hz, H-5’), 3.86 (s, 3H, NCH3). 13C NMR (100 MHz, DMSO-d6): δ = 188.7 (C-1), 138.5 (C-3), 138.5 (C-1'’), 138.0 (C-7'a), 136.8 (C-2’), 132.5 (C-4'’), 128.7 (C-3'’,5'’), 128.2 (C-2'’,6'’), 125.6 (C-3'a), 122.8 (C-6’), 121.4 (C-5’), 120.4 (C-4’), 115.3 (C-2), 111.8 (C-3’), 110.9 (C-7’), 33.1 (NCH3).
(2E)-1-(2-Fluorophenyl)-3-(1-methyl-1H-indol-3-yl)prop-2-en-1-one (3c)
1H NMR (400 MHz, DMSO-d6): δ = 8.07 (s, 1H, H-2’), 7.94 (dd, 1H, J 7.7, 0.9 Hz, H-4’), 7.88 (dd, 1H, J 15.6, 1.5 Hz, H-3), 7.76 (td, 1H, J 7.6, 1.8 Hz, H-6'’), 7.50 (dddd, 1H, J 8.4, 7.2, 5.3, 1.8 Hz, H-4'’), 7.57 (dd, 1H, J 8.2, 0.9 Hz, H-7’),7.38–7.23 (m, H-2, H-6’, H-5’, H-5'’, H-3'’), 3.84 (3H, NCH3). 13C NMR (100 MHz, DMSO-d6): δ = 188.1 (d, JCF 2.2 Hz, C-1), 160.1 (d, JCF 249.7 Hz, C-2'’), 139.3 (C-3), 138.2 (C-7'a), 137.4 (C-2’), 133.5 (d, JCF 8.6 Hz, C-4'’), 130.4 (d, JCF 3.0 Hz, C-6'’), 127.8 (d, JCF 13.9 Hz, C-1'’), 125.4 (C-3'a), 124.8 (d, JCF 3.3 Hz, C-5'’), 123.0 (C-6’), 121.7 (C-5’), 120.2 (C-4’), 119.4 (d, JCF 5.0 Hz, C-2), 116.6 (d, JCF 22.9 Hz, C-3'’), 111.4 (C-3’), 111.1 (C-7’), 33.1 (CH3).
(2E)-1-(4-Fluorophenyl)-3-(1-methyl-1H-indol-3-yl)prop-2-en-1-one (3d)
spectral data identical with literature (Citation35)
(2E)-3-(1-Methyl-1H-indol-3-yl)-1-(4-trifluoromethylphenyl) prop-2-en-1-one (3e)
1H NMR (400 MHz, DMSO-d6): δ = 8.29 (d, 2H, J 8.2 Hz, H-2″,6″), 8.15 (s, 1H, H-2′), 8.13 (dd, 1H, J 8.0, 1.0 Hz, H-4′), 8.06 (d, 1H, J 15.4 Hz, H-3), 7.92 (d, 2H, J 8.2 Hz, H-3″,5″), 7.63 (d, 1H, J 15.4 Hz, H-2), .7.58 (dt, 1H, J 8.2, 0.8 Hz, H-7′), 7.33 (ddd, 1H, J 8.0, 7.1, 1.0 Hz, H-6′), 7.29 (ddd, 1H, J 8.2, 7.1, 1.1 Hz, H-5′), 3.87 (s, 3H, NCH3). 13C NMR (100 MHz, DMSO-d6): δ = 188.0 (C-1), 141.9 (C-1″), 139.7 (C-3), 138.2 (C-7′a), 137.5 (C-2′), 131.8 (q, JCF 31.8 Hz, C-4″), 128.9 (C-2″,6″), 125.7 (q, JCF 3.7 Hz, C-3″,5″), 125.6(C-3′a), 124.0 (q, JCF 272,6 Hz, CF3), 123.0 (C-6′), 121.7 (C-5′), 120.7 (C-4′), 114.9 (C-2), 111.9 (C-3′), 111.0 (C-7′), 33.2 (NCH3).
(2E)-1-(4-Methoxyphenyl)-3-(1-methyl-1H-indol-3-yl)prop-2-en-1-one (3f)
1H NMR (400 MHz, DMSO-d6): δ = 8.13 (d, 2H, J 8.9 Hz, H-2″,6″), 8.10 (d, 1H, J 8.1 Hz, H-4′), 8.09 (s, 1H, H-2′), 7.97 (d, 1H, J 15.4 Hz, H-3), 7.65 (d, 1H, J 15.4 Hz, H-2), 7.56 (d, 1H, J 8.1 Hz, H-7′), 7.34–7.24 (m, 2H, H-6′, H-5′), 7.08 (d, 2H, J 8.8 Hz, H-3″, 5″), 3.86 (s, 3H, NCH3), 3.86 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6): δ = 187.1 (C-1), 162.7 (C-4″), 138.0 (C-7′a), 137.5 (C-3), 136.3 (C-2′), 131.2 (C-1″), 130.4 (C-2″,6″), 125.6 (C-3′a), 122.7 (C-6′), 121.4 (C-5′), 120.5 (C-4′), 115.3 (C-2), 113.9 (C-3″,5″), 111.8 (C-3′), 110.9 (CH, C-7′), 55.5 (OCH3), 33.1 (NCH3).
(2E)-3-(1-Methyl-1H-indol-3-yl)-1-(3,4,5-trimethoxyphenyl)prop-2-en-1-one (3g)
1H NMR (600 MHz, DMSO-d6): δ = 8.13 (s, H-2’), 8.07 (dd, 1H, J 7.9, 1.2 Hz, H-4’), 8.01 (d, 1H, J 15.4 Hz, H-3), 7.61 (d, 1H, J 15.4 Hz, H-2), 7.56 (dd, 1H, J 8.2, 1.1 Hz, H-7’), 7.36 (s, 2H, H-2'’,6'’), 7.31 (ddd, 1H, J 8.2, 7.1, 1.2 Hz, H-6’), 7.27 (ddd, 1H, J 7.9, 7.1, 1.3 Hz, H-5’), 3.91 (s, 6H, OCH3), 3.87 (s, 3H, OCH3), 3.76 (s, 3H, NCH3). 13C NMR (150 MHz, DMSO-d6): δ = 187.8 (C-1), 152.9 (C-3'’,5'’), 141.3 (C-4'’), 138.0 (C-7'a), 137.9 (C-3), 136.1 (C-2’), 134.0 (C-1'’), 125.8 (C-3'a), 122.8 (C-6’), 121.4 (C-5’), 120.4 (C-4’), 115.5 (C-2), 111.8 (C-3’), 110.9 (C-7’), 105.7 (C-2'’,6'’), 60.2, 56.2 (2×OCH3), 33.1 (NCH3).
(2E)-1-(2,5-Dimethoxyphenyl)-3-(1-methyl-1H-indol-3-yl)prop-2-en-1-one (3h)
1H NMR (400 MHz, DMSO-d6): δ = 8.00 (s, H-2’), 7.89 (dd, 1H, J 7.9, 0.9 Hz, H-4’), 7.74 (d, 1H, J 15.8 Hz, H-3), 7.56 (dd, 1H, J 8.1, 0.9 Hz, H-7’), 7.31 (d, 1H, J 15.8 Hz, H-2), 7.30 (ddd, 1H, J 8.0, 7.0, 1.1 Hz, H-6’), 7.26 (t, 1H, J 8.0, 7.1, 1.0 Hz, H-5’), 7.14 (d, 1H, J 9.0 Hz, H-3'’), 7.09 (dd, 1H, J 9.0, 3.1 Hz, H-4'’), 7.05 (d, 1H, J 3.1 Hz, H-6'’), 3.84 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 3.75 (s, 3H, NCH3). 13C NMR (100 MHz, DMSO-d6): δ = 191.1 (C-1), 153.1 and 151.6 (C-2'’, C-5'’), 138.0 (C-7'a), 137.6 (C-3), 136.4 (C-2’), 130.1 (C-1'’), 125.4 (C-3'a), 122.7 (C-6’), 121.4 (C-5’), 121.0 (C-2), 120.0 (C-4’), 117.7 (C-4'’), 114.0 (C-6'’), 113.9 (C-3'’), 111.5 (C-3’), 110.9 (C-7’), 56.4 and 55.5 (OCH3), 33.0 (NCH3).
(2E)-1-(2,6-Dimethoxyphenyl)-3-(1-methyl-1H-indol-3-yl)prop-2-en-1-one (3i)
1H NMR (400 MHz, DMSO-d6): δ = 7.91 (s, H-2’), 7.83 (d, 1H, J 7.9 Hz, H-4’), 7.53 (d, 1H, J 8.1 Hz, H-7’), 7.37 (t, 1H, J 8.4 Hz, H-4'’), 7.35 (d, 1H, J 16.0 Hz, H-3), 7.29 (ddd, 1H, J 8.2, 7.1, 1.2 Hz, H-6’), 7.22 (ddd, 1H, J 8.0, 7.1, 1.1 Hz, H-5’), 6.77 (d, 1H, J 16.1 Hz, H-2), 6.75 (d, 2H, J 8.4 Hz, H-3'’,5'’), 3.79 (s, 3H, NCH3), 3.71 (s, 6H, OCH3). 13C NMR (100 MHz, DMSO-d6): δ = 193.6 (C-1), 156.8 (C-2'’,6'’), 139.2 (C-3), 138.1 (C-7'a), 136.2 (C-2’), 130.4 (C-4'’), 125.3 (C-3'a), 123.1 (C-2), 122.8 (C-6’), 121.5 (C-5’), 120.1 (C-4’), 118.5 (C-1'’), 110.9 (C-3’), 110.9 (C-7’), 104.4 (C-3'’,5'’), 55.8 (2×OCH3), 33.0 (NCH3).
(2E)-3-(1-Methyl-1H-indol-3-yl)-1-(2,4,6-trimetylphenyl)prop-2-en-1-one (3j)
1H NMR (600 MHz, DMSO-d6): δ = 7.94 (s, H-2’), 7.87 (dd, 1H, J 7.9, 1.0 Hz, H-4’), 7.54 (dd, 1H, J 8.2, 0.9 Hz, H-7’), 7.33 (d, 1H, J 16.2 Hz, H-3), 7.30 (ddd, 1H, J 8.2, 7.1, 1.0 Hz, H-6’), 7.23 (ddd, 1H, J 8.1, 7.1, 1.0 Hz, H-5’), 6.93 (d, 2H, J 0.7 Hz, H-3'’,5'’), 6.85 (d, 1H, J 16.2 Hz, H-2), 3.79 (s, 3H, NCH3), 2.29 (s, 3H, Ar-CH3), 2.12 (s, 6H, 2×Ar-CH3). 13C NMR (150 MHz, DMSO-d6): δ = 199.4 (C-1), 140.5 (C-3), 138.1 (C-7'a), 137.8 (C-1'’), 137.2 (C-4'’), 136.8 (C-2’), 133.3 (C-2'’,6'’), 128.1 (C-3'’,5'’), 125.2 (C-3'a), 122.9 (C-6’), 122.2 (C-2), 121.6 (C-5’), 120.2 (C-4’), 110.9 (C-3’), 110.9 (C-7’), 33.0 (N-CH3), 20.7 (Ar-CH3), 18.9 (2×Ar-CH3).
Cell culture
Cell lines MCF-7 (human breast adenocarcinoma), A549 (human alveolar adenocarcinoma), COS-7 (green monkey kidney fibroblast), and Caco-2 (human colorectal carcinoma) were cultured in high glucose DMEM with sodium pyruvate (GE Healthcare, Piscataway, NJ, United States). HeLa (human cervical adenocarcinoma), MDA-MB-231 (human mammary gland adenocarcinoma), HCT116 (human colorectal carcinoma), and Jurkat (human leukemic T cell lymphoma) cell lines were maintained in RPMI 1640 medium (Biosera, Kansas City, MO, United States). The growth media were supplemented with 10% fetal bovine serum, 1X HyClone™ Antibiotic/Antimycotic Solution (GE Healthcare, Little Chalfont, UK). The BJ-5ta cell line was cultured in a 4:1 combination of DMEM with pyruvate (GE Healthcare) and M199 (Sigma Aldrich, St. Louis, MO, USA) supplemented with 10% FBS, hygromycin B (0.01 mg/ml) and NaHCO3 (2 mg/ml). Cell cultures were maintained under standard 5% CO2 conditions at 37 °C. Prior to each experiment, cell viability was verified by trypan blue staining, which was greater than 95%
MTT viability assay
The effect of prepared compounds on the viability of cancer cells was evaluated by MTT assay (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (5 mg/mL, Sigma-Aldrich Chemie, Steinheim, Germany). The cells were seeded at a density of 5 × 103 cells/well in 96-well microplates (SARSTEDT, Nümbrecht, Germany). After 24 h, the cells were treated with tested chalcones and cisplatin in concentrations of 1, 5, 10, 50, and 100 μM. 72 h later, cells were incubated with 10 μL of MTT per well at 37 °C. During 4 h incubation, soluble MTT was metabolized to formazan crystals. To dissolve the formazan, 100 μL/well of a 10% sodium dodecyl sulfate was added to each well and incubated for 12 h. The absorbance was measured at 540 nm using the automated Cytation™ 3 Cell Imaging Multi-Mode Reader (Biotek, Winooski, VT, United States), and IC50 was determined for each tested compound. Three independent experiments were performed.
Supplemental Material
Download MS Word (1.3 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Tan, D.; García, F. Chem. Soc. Rev. 2019, 48 (8), 2274–2292.
- Groote, R.; Jakobs, R.T.M.; Sijbesma, R.P. Polym. Chem. 2013, 4 (18), 4846–4859.
- May, P.A.; Moore, J.S. Chem. Soc. Rev. 2013, 42 (18), 7497–7506.
- André, V.; Quaresma, S.; da Silva, J.L.F.; Duarte, M.T. Beilstein J. Org. Chem. 2017, 13, 2416–2427.
- Braga, D.; Maini, L.; Grepioni, F. Chem. Soc. Rev. 2013, 42 (18), 7638–7648.
- Wu, K.; Ju, T.; Deng, Y.; Xi, J. Trends Food Sci. Technol. 2017, 66, 166–175.
- Colacino, E.; Porcheddu, A.; Halasz, I.; Charnay, C.; Delogu, F., Guerra, R., et al. Green Chem. 2018, 20 (13), 2973–2977.
- Huot, J.; Ravnsbæk, D.B.; Zhang, J.; Cuevas, F.; Latroche, M.; Jensen, T.R. Prog. Mater. Sci. 2013, 58 (1), 30–75.
- Baláž, P.; Achimovicová, M.; Baláž, M.; Billik, P.; Zara, C.Z., Criado, J.M., et al. Chem. Soc. Rev. 2013, 42 (18), 7571–7637.
- Muñozo Batista, M.J.; Rodríguez-Padrón, D.; Puente-Santiago, A.R.; Luque, R. ACS. Sustain. Chem. Eng. 2018, 6, 9530–9544.
- Baláž, M. Environmental Mechanochemistry - Recycling Waste into Materials Using High-Energy Ball Milling. 2021. 619 p.
- James, S.L.; Adams, C.J.; Bolm, C.; Braga, D.; Collier, P., Friščić, T., et al. Chem. Soc. Rev. 2012 Jan, 41 (1), 413–447.
- Yang, X.; Wu, C.; Su, W.; Yu, J. European. J. Org. Chem. 2022, 2022 (8), e202200147.
- Espro, C.; Rodríguez-Padrón, D. Curr Opin Green Sustain. Chem. 2021, 30, 100478.
- Kubota, K.; Ito, H. Trends Chem. 2020, 2 (12), 1066–1081.
- Colacino, E.; Isoni, V.; Crawford, D.; García, F. Trends Chem. 2021, 3 (5), 335–339.
- Gao, F.; Huang, G.; Xiao, J. Med. Res. Rev. 2020, 40 (5), 2049–2084.
- Mahapatra, D.K.; Bharti, S.K.; Asati, V. Eur. J. Med. Chem. 2015, 98, 69–114.
- Michalkova, R.; Mirossay, L.; Gazdova, M.; Kello, M.; Mojzis, J. Cancers 2021, 13 (11), 2730.
- Dan, W.; Dai, J. Recent developments of chalcones as potential antibacterial agents in medicinal chemistry [Internet]. Eur J Med Chem 2020, 187, 111980.
- Elkhalifa, D.; Al-Hashimi, I.; Al Moustafa, A.E.; Khalil, A. J. Drug Target 2021, 29 (4), 403–419.
- ur Rashid, H.; Xu, Y.; Ahmad, N.; Muhammad, Y.; Wang, L. Bioorg Chem. 2019, 87, 335–365.
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chem. Rev. 2017, 117 (12), 7762–7810.
- Chopra, P.K.P.G. IJREAS 2016, 6 (5), 173–185.
- Kumar, S.; Lamba, M.S.; Makrandi, J.K. Green Chem Lett Rev. 2008, 1 (2), 123–125.
- Dhruva Kumar, S.; Sandhu, J.S. Green Chem Lett Rev. 2010, 3 (4), 283–286.
- Siddiqui, Z.N.; Farooq, F.; Musthafa, T.N.M. Green Chem Lett Rev. 2011, 4 (1), 63–68.
- Tiwari, V.; Ali, P.; Meshram, J. Green Chem Lett Rev. 2011, 4 (3), 219–224.
- Rateb, N.M.; Zohdi, H.F. Synth. Commun. 2009, 39 (15), 2789–2794.
- Wang, H.; Zeng, J. Can. J. Chem. 2009, 87 (9), 1209–1212.
- Gomes, C.; Vinagreiro, C.S.; Damas, L.; Aquino, G.; Quaresma, J., Chaves, C., et al. ACS Omega 2020, 5 (19), 10868–10877.
- Baláž, M.; Kudličková, Z.; Vilková, M.; Imrich, J.; Balážová, L.; Daneu, N. Molecules 2019, 24 (18), 3347.
- Kumar, D.; Kumar, N.M.; Akamatsu, K.; Kusaka, E.; Harada, H.; Ito, T. Bioorganic Med. Chem. Lett. 2010, 20 (13), 3916–3919.
- Valdameri, G.; Gauthier, C.; Terreux, R.; Kachadourian, R.; Day, B.J., Winnischofer, S.M.B., et al. J. Med. Chem. 2012, 55 (7), 3193–3200.
- Kudličková, Z.; Takáč, P.; Sabolová, D.; Vilková, M.; Baláž, M., Béres, T., et al. Med. Chem. Res. 2021, 30 (4), 897–912.
- Sum, T.J.; Sum, T.H.; Galloway, W.R.J.D.; Twigg, D.G.; Ciardiello, J.J.; Spring, D.R. Tetrahedron 2018, 74 (38), 5089–5101.
- Yang, M.R.; Qin, Y.J.; Chen, C.; Zhang, Y.L.; Li, B.Y., Liu, T.B., et al. RSC Adv. 2016, 6 (36), 30412–30424.
- Zhang, Y.L.; Qin, Y.J.; Tang, D.J.; Yang, M.R.; Li, B.Y., Wang, Y.T., et al. Chem. Med. Chem. 2016, 11 (13), 1446–1458.
- Cabrera, M.; Cerecetto, H.; Mercedes, G. Med. Chem. Commun. 2016, 7, 2395–2409.
- Saikia, P.; Gogoi, S.; Boruah, R.C. J. Org. Chem. 2015, 80 (13), 6885.
- Schmidt, E.Y.; Trofimov, B.A.; Bidusenko, I.A.; Cherimichkina, N.A.; Ushakov, I.A., Protzuk, N.I., et al. Org. Lett. 2014, 16 (15), 4040–4043.
- Wang, C.; Jiang, W.-Q.; Zou, J.-P.; Zeng, R.-S. Chinese J. Chem. 2006, 24, 1427–1430.
- Takahashi, H.; Arai, T.; Yanagisawa, A. Synlett. 2006, 17, 2833–2835.
- Ying, P.; Yu, J.; Su, W. Adv. Synth. Catal. 2021, 363 (5), 1246–1271.
- Zaikin, P.A.; Dyan, O.T.; Elanov, I.R.; Borodkin, G.I. Molecules 2021, 26 (19), 1–10.
- Suzdalev, K.F.; Babakova, M.N. J. Heterocycl. Chem. 2015, 53 (4), 1200–1206.
- Akama, T.; Jarnagin, K.; Plattner, J.J.; Pulley, S.R.; White, W.H.; Zhang, Y.-K., et al. 1 -Hydroxy-benzooxaboroles as antiparasitic agents (2014) Patent n. WO2014149793A1.