
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Naturally occurring sinapic acid is a valuable synthon for many high-value applications (e.g. cosmetic, pharmaceutic) that can be found in numerous plants, especially Brasiccacea species (e.g. rapeseed, mustard). It has been demonstrated that alkaline extraction of matter rich in sinapic acid resulted in the conversion of sinapic acid into 6-hydroxy-5,7-dimethoxy-2-naphtanoic acid (DMNA). Although DMNA could be a potential alternative to petro-based naphtanoic acid used in polymers, untill now, no reliable (bio)synthetic procedure existed to synthesize it at the preparative-scale. Herein, starting from sinapic acid, we describe a very simple one-pot two-step chemo-enzymatic pathway to DMNA – and reaction intermediaries bislactone (BL) and thomasidioic acid (TA) – at gram scale using a simple filtration as purification step. Green metrics – Process Mass Index and EcoScale – have been determined to assess the sustainability of this new process. Finally, the antiradical activities of DMNA, BL and TA have been evaluated.
GRAPHICAL ABSTRACT
Introduction
To achieve efficient and cost-competitive biorefineries, and thus operate the switch from petro- to bioeconomy, Manzanares highlighted the importance of combining the production of low-value/high-volume products (e.g. food, feed, bioenergy) with the production of high-value biobased chemicals (Citation1). p-Hydroxycinnamic acids (p-HCA) represent one of the most widely distributed chemicals in the plant kingdom, along with other phenylpropanoids such as flavonoids, stilbenes, and lignans (Citation1). Sinapic acid (SA) and its analogs are among the most important p-HCA present in Brassicaceae family (e.g. B. carinata, B. napus, B. juncea). They can be extracted and purified from agricultural residues (e.g. mustard, rapeseed press-cakes), (Citation2–6) thus allowing the generation of new applications and additional value to farmers and/or food industries in accordance with the circular economy concept (Citation1).
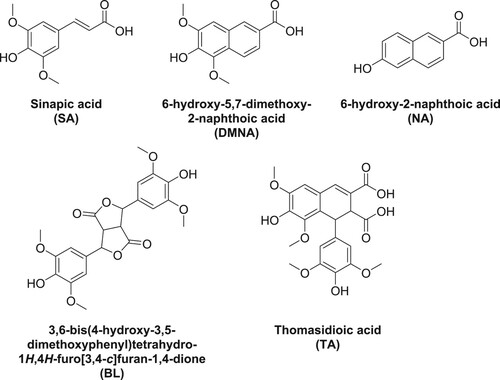
6-Hydroxy-5,7-dimethoxy-2-naphthoic acid (DMNA) was first assumed as a natural product found in the alkaline extract of the heartwood of Ulmus thomasii Sarg () (Citation7). However, the natural occurrence of this compound is questionable due to the observation of Charlton and Lee who suggested that DMNA is an oxidized product of SA under strongly alkaline conditions (Citation8). Interestingly, the structure of DMNA shows a high similarity to that of 6-hydroxy-2-naphthoic acid (NA) (), a petroleum-based chemical commonly used for the production of the high-value-added liquid crystal polymer Vectra® (Citation9–11). Moreover, the antimicrobial activity of some DMNA analogs has been reported (Citation12, Citation13). Considering the aforementioned data, we believe that DMNA could be a potential renewable building block for biobased chemicals and materials that can replace other NA analogs issued from limited fossil resources.
Figure 1. Structure of sinapic acid and its oxidative derivatives; and 6-hydroxy-2-naphthoic acid.
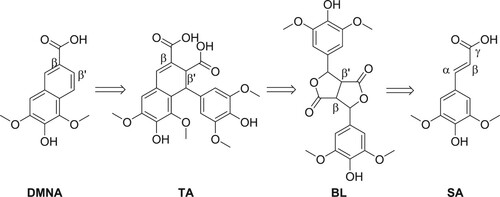
It is known that DMNA is the product of secondary oxidation of a bislactone intermediate (BL) which is formed via the radical-mediated β-β dimerization of SA (Citation14). Contrary to what was reported for sinapyl alcohol (Citation15) or sinapate esters, (Citation16) the β-β dimerization of SA mainly occurs over other dimerization patterns such as β-O-4. Indeed, various reports have shown that the addition of oxidative FeCl3 favors the β-β oxidative dimerization of SA (Citation17–19), the latter allowing the lactonization.
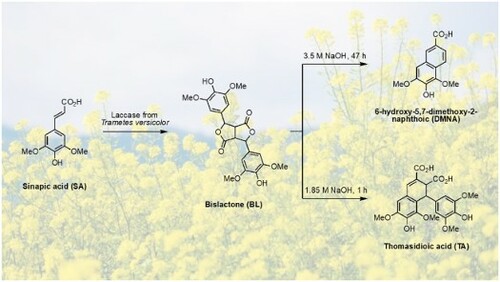
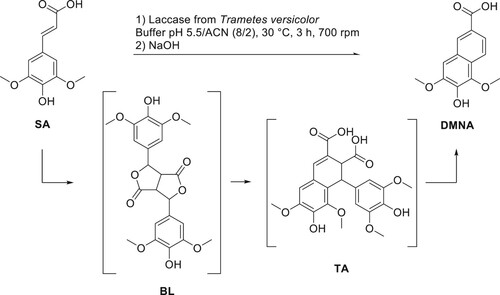
Charlton and Lee have also shown that DMNA can be obtained from thomasidioic acid (TA) (Citation8). Preparation of TA from BL was described both under acidic or alkaline conditions (Citation14, Citation17, Citation20, Citation21). From this information, the retrosynthetic pathway shown in has been selected. Employing the iron(III)-mediated oxidation approach in basic solution (pH 10), Cai et al. produced a maximum of 0.144 mmol.L−1 of DMNA (corresponding to 0.14 mg) from SA solution at 0.446 mmol.L−1 (64% yield) (Citation22). Bunzel et al. reported the isolation of pure DMNA (14 mg, 27% yield) from an experiment aiming at synthesizing TA also through FeCl3-mediated oxidation but in two steps from methyl sinapate (Citation23). Finally, Lee obtained 23 mg of DMNA (72%) from alkaline oxidation of sinapic acid under air (13 mmol.L−1) (Citation14). Another synthetic approach to obtain DMNA was reported by Fuganti and Serra from syringaldehyde, a p-hydroxybenzaldehyde obtained from the oxidation of hard-wood lignin (39% yield) (Citation24). Unfortunately, this method employs a multi-step route that uses hazardous reagents such as ethyl chloroformate.
Scheme 1. Proposed strategy to synthesize DMNA from SA.
Herein, we propose a straightforward and sustainable synthetic method to obtain DMNA from naturally occurring SA. Contrary to earlier strategies (Citation14, Citation22–24), our strategy relies on (1) the use of laccase from Trametes versicolor to form the bis-lactone intermediate (BL), (Citation2) concentrated reaction medium to limit the use of solvent and buffers, and (3) the absence of hazardous reagents.
Results and discussion
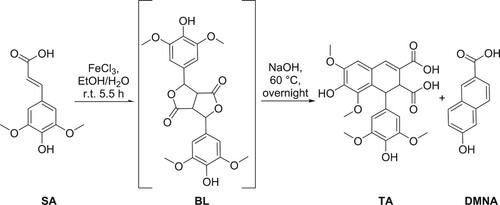
Although SA can be isolated from plant-based products (Citation2–5), its price is rather high, thus the first step of this work consisted in the synthesis of SA using our in-house optimized Knoevenagel-Doebner condensation of lignin-derived syringaldehyde and natural malonic acid (Citation25). SA was then reacted with FeCl3 to provide BL, to which NaOH solution was added to afford TA and DMNA (). Although the three compounds were successfully isolated as proven by NMR analysis (ESI Figure S1 to S6), the method exhibited poor reproducibility and required an extensive purification on silica gel (cyclohexane-ethyl acetate and acetic acid as eluant) and the isolated yields remained low. In addition, all our attempts to perform this reaction on a scale higher than 1 g resulted in a decrease in DMNA yield. Thus, further optimization must be established for efficient production of DMNA at a larger scale.
Scheme 2. Synthesis of BL, DMNA and TA by FeCl3-mediated oxidation of sinapic acid.
Inspired by the study of Tranchimand et al. (Citation26) laccase-mediated synthesis of BL from SA seems a good strategy to obtain the β-β dimer in mild conditions and excellent yield as they reported the total conversion of SA into BL. However, the described conditions – highly diluted medium (5.58 10−3 mol.L−1) and a very high enzyme/substrate ratio of 272 U.mmol−1 – were not applicable for larger-scale production (larger reaction: 20 mL, ca. 25 mg-scale). Building on our previous works on laccase-mediated phenolic dimerization (Citation15, Citation27, Citation28), we decided to set initial conditions at 35 °C, 0.1 mol.L−1, 8 U.mmol−1, buffer pH 4.4 and 12% of acetonitrile as co-solvent. The reaction was monitored by HPLC (, )). A high yield of BL (80%) was obtained after 80 min but a decrease to 70% was recorded at 100 min. This first attempt proved that the reaction can be efficient at a higher concentration and with a reasonable amount of enzyme. To pursue, the impact of temperature ()), percentage of co-solvent ()) and enzyme/substrate ratio ()) were evaluated using an OVAT (one variable at a time) optimization strategy.
Scheme 3. Laccase-mediated synthesis of BL from SA.
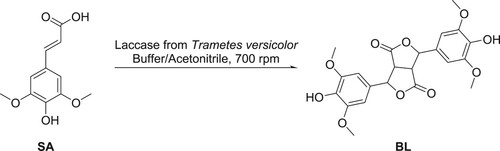
Clearly, in the studied range, the temperature does not have a significant impact on the reaction ((A)). Optimal temperatures for laccase of Trametes versicolor being described between 10 and 30 °C (Citation29), and maintaining the temperature at 25 °C being difficult especially during summer, we decided to fix the temperature at 30 °C for the following experiments. As it was noticed that the yield decreased after a certain time due to further oligomerization and/or degradation, the kinetics of the reaction will be monitored for all subsequent reactions. Unlike temperature, the percentage of co-solvent plays a major role and thus will be further explored in the subsequent Design of Experiments (DoE) ()). Indeed, without acetonitrile, the reaction cannot proceed efficiently. As expected, a decrease of the enzyme/substrate ratio slowed down the kinetics of the reaction but seemed also to limit secondary reactions ((C)). This factor will be also part of the DoE.
Figure 2. Impact of the temperature (A), percentage of co-solvent (B) and enzyme/substrate ratio (C) on the yield of BL over time.
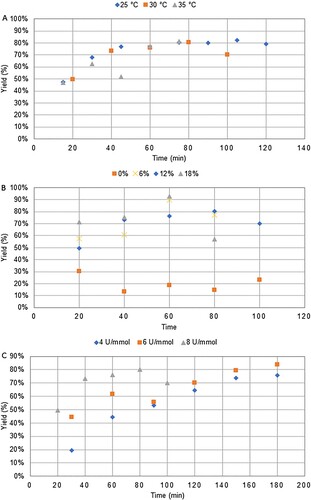
In addition to the aforementioned factors, the impact of increasing concentration will be considered to achieve a preparative-scale, as well as the influence of the pH on the enzymatic reaction. Indeed, it is well-known that enzymes are sensitive to pH. In addition, sinapic acid solubility in water is pH-dependent. Indeed, at pH above 9 (undesirable for laccase), sinapic acid is fully soluble whereas at pH under pH 4 its solubility is near to zero. Thus, the DoE has been conducted on four factors, amount of co-solvent (X1), enzyme/substrate ratio (X2), concentration (X3) and pH (X4) whom range, scale and coded values are given in . Kinetics have been monitored by HPLC for each experiment over 5 h to determine the responses: the productivity (Y1) at the best time (mol.min−1) and the Kcat (min−1) (Y2). A matrix of 20 experiments and 3 repetitions at the central point (cubic centered faces, CCF) has been generated using Modde 12.0 software (ESI Table S1). Reactions have been performed in random order to avoid bias. In addition, since laccase is an oxygen-dependent enzyme (Citation29), to ensure the same level of oxygenation in the medium – and thus reproducibility – the total volume of solution and agitation were fixed at 15 mL and 700 rpm, respectively. As previously mentioned, the temperature was fixed at 30 °C.
Table 1. Independent factors and levels used for CCF design.
The Kcat or enzymatic turnover number is calculated based on the initial speed of the reaction (mol.L−1.min−1) divided by the concentration of the enzyme (mol.L−1). Thus, it indicates the efficiency of the catalytic system. The maximal productivity gives the quantity of substrate formed per minute of reaction if the reaction was stopped at the optimum time or after 5 h for an uncomplete reaction.
After performing the 23 experiments, good regression model (R² (Y1) 0.885 and R² (Y2) 0.941), predictability (Q² (Y1) 0.734 and Q² (Y2) 0.891), model validity and reproducibility were achieved for both responses (ESI Figure S7 and Tabled S2 & S6).
Contour plots were generated to visualized hot spots. As it can be noticed in , the conditions to achieve high productivity did not match with the conditions allowing high Kcat. Indeed, although a high co-solvent percentage was favorable for both responses, higher concentration and enzymatic loading led to higher productivity but lower Kcat values, whereas pH had no impact on Kcat. Considering the coefficients of the model (), pH appeared clearly as an insignificant factor for Kcat, whereas increasing percentage of co-solvent, decreasing concentration and enzymatic loading favored higher turnover of the enzyme. On the other hand, increasing each factor individually increased productivity, with a higher impact of concentration and co-solvent. Based on these findings, one can assume that the conformation of the laccase was not affected in the considered range of pH, thus its activity remained similar for all experiments. Sinapic acid being mainly insoluble in the depicted reactions, higher level of co-solvent probably ensured better dispersion and partial solubility, thus allowing higher availability for the enzyme. Increasing sinapic acid concentration above a certain threshold had deleterious effect on the Kcat. In other words, if one wants to achieve a high productivity at high pH, high co-solvent ratio and high concentration, enzymatic loading has to be high as well to counterbalance the lower Kcat observed at high sinapic acid concentration. Enzymatic loading and concentration helped to refine the model with square and quadratic terms.
Figure 3. Contour plots for productivity (top) and Kcat.
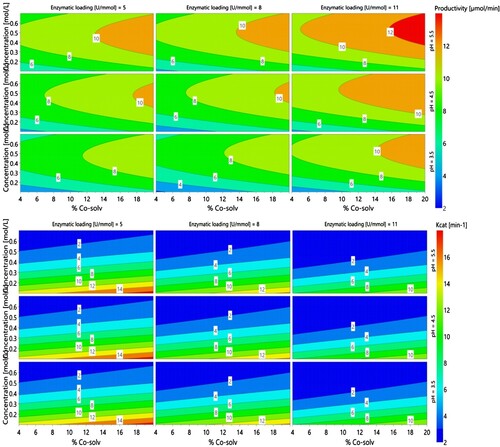
Figure 4. Model coefficients [centered and scaled (SC)], at confidence interval 95% for productivity and Kcat.
![Figure 4. Model coefficients [centered and scaled (SC)], at confidence interval 95% for productivity and Kcat.](/cms/asset/b07c8c40-c404-427f-bcef-3539e6091fa4/tgcl_a_2115317_f0004_oc.jpg)
To validate the model, an experiment was conducted in optimal conditions for maximizing both productivity and Kcat (Table S2). That is to say, 605 mg of sinapic acid (2.7 mmol) in a mixture of 20% of acetonitrile and 80% of buffer phosphate citrate at pH 5.5 (0.18 mol.L−1), enzymatic loading was set at 5 U.mmol−1. Productivity was determined as 7.6 µmol.min−1 for a Kcat of 12.1 min−1 versus the predicted values of 8.8 ± 1.6 µmol.min−1 and 13.4 ± 2.4 min−1. Thus, models of productivity and Kcat were validated. Indeed, experimental results were in the range of the predicted values. Equations of both models are given below for real factors (unscaled and uncentered) (Equations 1 and 2).
(1)
(1)
(2)
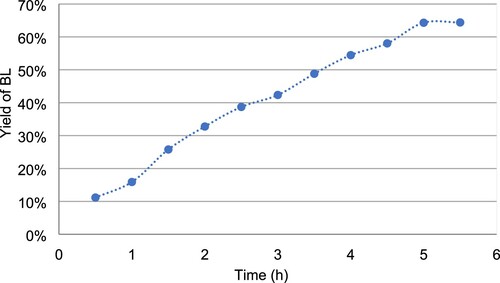
(2) After 3 h under these optimal conditions, SA was completely consumed to form BL without other notable compounds (). It is noteworthy to mention that even with 20% of acetonitrile in the medium, the reaction remains biphasic with the solid (i.e. SA then BL) dispersed in the aqueous phase. At the end of the reaction, a simple filtration followed by washing with osmotic water and drying allows recovering pure BL in good yield (81%) without the need for further extraction or chromatographic purification.
Figure 5. Kinetics over 5 h of bislactone formation in the defined optimal conditions ([SA]I = 0.18 mol.L−1 in buffer pH 5.5 with 20% of acetonitrile and 5 U.mmol−1 of laccase of Trametes versicolor at 30 °C).
![Figure 5. Kinetics over 5 h of bislactone formation in the defined optimal conditions ([SA]I = 0.18 mol.L−1 in buffer pH 5.5 with 20% of acetonitrile and 5 U.mmol−1 of laccase of Trametes versicolor at 30 °C).](/cms/asset/4c5e6ab2-0583-4100-b6af-28f87b47ba56/tgcl_a_2115317_f0005_oc.jpg)
After 3 h of enzymatic reaction under the optimal conditions determined above, sodium hydroxide solution was directly added into the medium, to achieve DMNA synthesis through the intermediary formation of TA. As previously mentioned, under alkaline conditions, BL was converted into TA before DMNA formation (). In consequence, playing with temperature (X5), time (X6) and NaOH equivalent (X7) can change the selectivity. Optimization was conducted on these three factors to select the suitable conditions to yield DMNA or TA (YDMNA and YTA, respectively). The temperature was ranged from 30 to 90 °C (middle 60 °C), time was set at 5, 15 or 25 h, and the stoichiometry in NaOH was 1.67, 5 or 8.33 equivalent based on initial SA loading. In practice, 4.5 mL of 1, 3 or 5 mol.L−1 of NaOH solution have been added to the reaction medium.
Scheme 4. One-pot two steps synthesis of DMNA from SA.
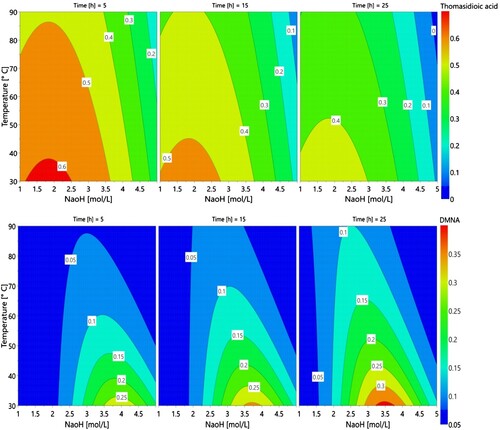
A second CCF design was performed (ESI Table S3). To achieve Gaussian distribution of the results and thus good modeling, transformations by negative logarithm were performed for the TA yield response and by logarithm for DMNA yield response (Citation30). Contour plots, provided through modeling, allow visualizing that low temperatures (< 40 °C) are favorable for both compounds (). It can thus be assumed that high temperature leads to product degradation. Moreover, major impacts were observed with NaOH concentration (ESI Figure S8). Although NaOH was needed to promote the rearrangement toward TA then DMNA, one needs to finely tune its concentration to avoid the formation of unwanted degradation by-products. The optimal values for NaOH concentration were found around 1.85 and 3.50 mol.L−1 for TA and DMNA, respectively. Finally, as expected – DMNA being obtained from TA – the shortest reaction time was favorable for TA, with 72% yield predicted in the best conditions, whereas increasing time led to a slight increase in the predicted yield of DMNA from 25% at 5 h to 36% at 25 h.
Figure 6. Contour plots of thomasidioic acid yield and DMNA yield.
To assess the validity of the modeling (ESI Table S4), experiments were performed in the predetermined best conditions (). Equations of both models are given below for real factors (unscaled and uncentered) (Equations 3 and 4).
(3)
(3)
(4)
(4)
Table 2. Optimal conditions determined by the DoE and external validation for TA (entry 1) and DMNA (entry 2).
Experimental yields recording for both compounds at their respective predicted best conditions were consistent with the predictions, even if ranges were large. However, for the second compound, experimental data did not match the predicted ones (for DMNA 11% vs 2.6% and for TA 6.3% vs 40%). As one can notice, confidence intervals were large probably due to the initial wide range of factors. Nevertheless, satisfying yields were achieved with 67% and 53% for TA and DMNA, respectively.
However, the minimum time recorded during DoE seems too long for TA while the maximum time seems too short for DMNA, therefore both reactions performed to validate DoE were reproduced and monitored by HPLC over time. In addition, a third experiment was also conducted in which heating was stopped after 3 h of enzymatic reaction, allowing the reaction to cool down to room temperature, hoping for a beneficial impact on DMNA yield. In this case, room temperature means uncontrolled temperature, thus a variation between 18 and 21 °C, considering the day and night temperature in the laboratory.
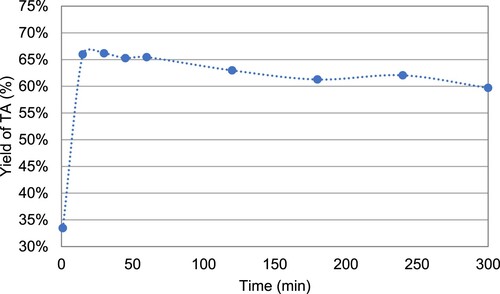
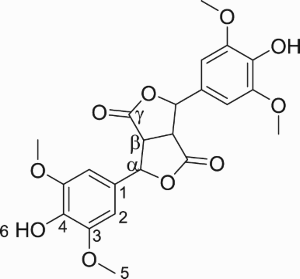
Rearrangement from BL intermediate to TA occurs very fast as shown in . TA reaches its maximum yield after some minutes then decreases slowly. To determine both the yield and purity of TA during the first hours of reaction, assays were stopped after 10 min, 1 and 5 h. The solution was acidified and the resulting precipitate was filtered, washed with water and dried. 343, 334 and 299 mg of TA were recovered for 10 min, 1 and 5 h, respectively. Purity was determined by HPLC and reached 69%, 96% and 90% at 10 min, 1 and 5 h, respectively. In conclusion, TA can be obtained in good yield (55%) and high purity (>95%), without any purification step, if the reaction is stopped 1 h after the addition of NaOH solution.
Figure 7. Evolution of the yield of TA over time after addition of 1.85 M solution of NaOH at the optimal time (3 h) in the enzymatic reaction.
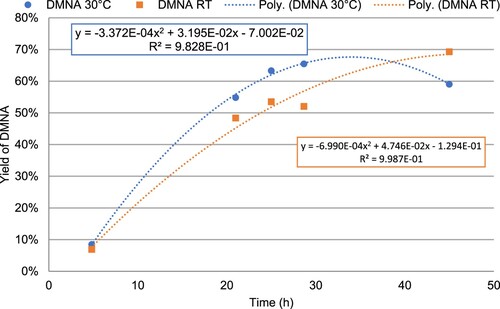
As shown in , decreasing the temperature enhanced DMNA yield. At 30 °C and room temperature, the maximum predicted yields, i.e. 67.8% and 68.7%, were determined at 34.0 and 47.4 h, respectively. Maximum achieved yields were similar. In addition, the shape of the curves shows that degradation occurs slower at room temperature than at 30 °C.
Figure 8. Evolution of DMNA yield over time in function of the temperature (Blue 30 °C and Red room temperature).
To isolate DMNA, an experiment was conducted at room temperature for 47.4 h after the addition of NaOH solution into the initial enzymatic reaction. The reaction was stopped by acidification with 3 M HCl (pH ≈ 2). The resulting precipitate was filtered, rinsed with osmotic water and dried to yield 58% of pure DMNA.
To go further and validate the DMNA synthesis at gram scale, a ten-fold scale-up was attempted. 6.05 g of SA were dimerized through laccase-mediated oxidation (30 °C). After 5.5 h of reaction, although the total consumption of SA was registered, HPLC yield of BL (70%) was notably lower than that recorded in smaller scale synthesis (). This result can be explained by a concomitant degradation or consumption of SA through its oligomerization due to a longer reaction time. Another possible explanation could be a modification of oxygen dissolution in the reaction medium (e.g. oxygen availability for the enzyme) due to the scale-up. Chemical engineering on the reactor can help to prevent such disagreement and reach a higher yield. NaOH solution was then added and the reaction was pursued for 50 h at room temperature. After acidification with concentrated sulfuric acid, a precipitate appeared and NMR analysis revealed pure DMNA (1.54 g, i.e. 46% yield based on the initial amount of SA and 66% based on BL), demonstrating the feasibility of producing DMNA at a gram scale using this synthetic procedure.
Figure 9. Scale-up of laccase-mediated dimerization of SA.
To evaluate the greenness of this new procedure, Process Mass Intensity (PMI) and EcoScale were calculated for each product.
The PMI was defined as the ratio between all reagents entering the process (except water) and the final recovered product (Citation31). In the present case, reagents are SA, laccase, acetonitrile, sodium hydrogenophosphate (Na2HPO4) and citric acid used in the buffer, sodium hydroxide and hydrochloric acid used to stop the reaction ().
Table 3. Calculated PMI for each product. All values are given in mg.
PMI is related to E-factor, defined as the mass ratio of waste to the desired product, as follows PMI = E-factor + 1. R. A. Sheldon showed that industrial fine chemicals have E-factor ranging from 5 to 50 (Citation32). With PMI lower than 25 at laboratory scale, our products generate a low amount of waste compared to that of other fine chemical processes (Table S7).
To go further and not only consider the waste generation but also the cost, the toxicity and the technical setup of the reaction, EcoScale was calculated (Table S8). This metric has been developed by Van Aken as a system of penalty points on 6 criteria allowing us to consider these aspects (Citation33) (Table S5). Data for our products are reported in .
Table 4. Detail of penalty point and final score of EcoScale for BL, TA and DMNA.
The reagents engaged in the reaction are generally safe and cheap, except for laccase which is relatively more expensive. Nevertheless, as laccase is used in a catalytic amount, the price of producing 10 mmol of the desired products remains low. No particular equipment (e.g. addition funnel, inert atmosphere, glove box) is required to conduct the reaction. Following the rules depicted by Van Aken, heating the reaction for a period longer than 1 h induced a 3 points loss (Citation33). We have counted down these points, nevertheless we performed the reactions at mid-temperature (30 °C) during a limited time (3–4 h) and thus its energetical footprint is limited. One can observe that penalty points are mainly due to the yields and not to safety issues. In all cases, satisfying EcoScale scores were achieved.
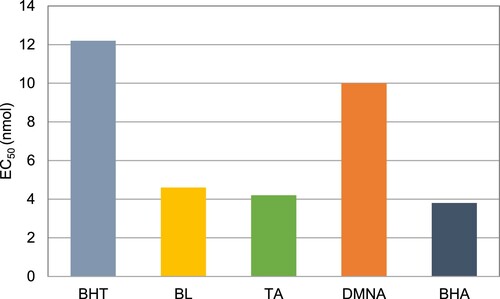
Phenolic compounds, and especially sinapic acid derivatives, exhibit antiradical activities (Citation15, Citation16, Citation34). BL, TA and DMNA activities against radical DPPH were evaluated according to previously reported procedure (Citation34, Citation35) and benchmarked against BHA (butylated hydroxyanisole) and BHT (butylated hydroxytoluene), two well-known antioxidants (). Although BL and TA – EC50 = 4.6 and 4.2 nmol, respectively – can compete with BHA (EC50 = 3.8 nmol), all three chemicals demonstrated better antiradical activities than BHT (EC50 = 12.2 nmol).
Figure 10. Antiradical activities.
Conclusion
DMNA was produced at a gram scale from sinapic acid through a sustainable one-pot two-step process involving laccase-mediated dimerization followed by a sodium hydroxide treatment at room temperature. The product was isolated by simple precipitation and filtration from the reaction medium. The calculated PMI and Ecoscale demonstrated the limited environmental impact of the developed process. Besides DMNA, bislactone (BL) and thomasodioic acid (TA) can be also produced by this method in good to excellent yield (58, 81 and 55%, respectively). DPPH analysis also revealed the great potential of TL, BL and DMNA as antioxidant compounds. With this new ecofriendly gram-scale access to DMNA, as well as BL and TA, one can now investigate further new applications, such as their polymerization (e.g. substitution of naphthoic acid) or their incorporation as antioxidant additives in food or cosmetic matrices. These studies will be published in due course.
Experimental section
Materials
Syringaldehyde, malonic acid, aniline, FeCl3 anhydrous and Laccase from Trametes versicolor, ABTS (2,2'-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)), and tartaric acid were purchased from Sigma-Aldrich. Pyridine was purchased from Alfa Aesar. Di-sodium hydrogen phosphate was purchased from Acros Organics. Sodium tartrate, HPLC grade acetonitrile were purchased from Fisher scientific. Other solvents, citric acid, sodium hydroxide, sulfuric acid and hydrochloric acid were purchased from VWR-Avantor sciences. NMR solvents were purchased from EurIsotope. Melting points were recorded on an MP50, Mettler Toledo. HPLC Chromatograms were recorded at 254 nm on an Ultimate 3000 (Thermofisher) equipped with a DAD detector using an Accucore C18 AQ column (2.6 µm x 3 mm x 100 mm). NMR spectra were recorded on a Fourier 300, Bruker at 300 MHz for 1H (Acetone-d6 residual signal at 2.05 ppm) and 75 MHz for 13C NMR (Acetone-d6 residual signal at 29.8 ppm). Chemical shifts are reported in parts per million (ppm). Multiplicities are reported using the following abbreviations: s, singlet; d, doublet. High-resolution mass spectrometry was performed on an Agilent 1290 system, equipped with a 6545 Q-TOF mass spectrometer and a PDA UV detector. The source was equipped with a JetStream ESI probe operating at atmospheric pressure.
Methods
Laccase activity assay
In a 12-well microplate, 1300 µL of tartaric buffer (pH 4.0) and 150 µL of laccase solution in water (0.5 mg.L−1) or 150 µL of water (for blank) were incubated at 30 °C, then 50 µL of ABTS solution in water (5 mmol.L−1) were introduced. Absorbance at 405 nm was recorded every 8 s for 5 min using an EPOCH2 microplate reader (Biotek). All measurements were performed in triplicate. Laccase activity was determined as 4.2 U.mg−1 (µmol.min−1.mg−1) using Equation 5.
(5)
(5)
Synthesis of 6-hydroxy-5,7-dimethoxy-2-napthoic acid (DMNA) using fecl3
Synthesis of DMNA was performed inspired by the work of Lee (Citation8). 1.0 g of SA (4.5 mmol) and 0.65 g of FeCl3 (4 mmol) were stirred in 5 mL EtOH for 30 min at room temperature. 45 mL of deionized water was added. Immediately turning pink, the mixture was vigorously stirred for 5 h at room temperature. The mixture was adjusted to pH 12 using 3M NaOH and stirred overnight at 60 °C. The solution then became dark brown. The solution was adjusted to pH 2 with 3M HCl and extracted with 3 × 50 mL AcOEt. The organic layers were combined, washed twice with brine (50 mL), dried over anhydrous MgSO4, filtered and evaporated under vacuum to afford a brown residue. Chromatography on silicate-gel with 30/70 AcOEt/Cyclohexane gave a light-yellow solid (0.165 g, 0.6 mmol, 30%).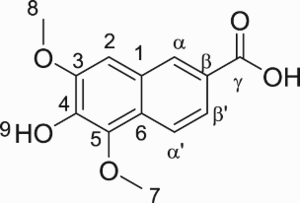
mp: 228 °C. 1H NMR (Acetone-d6) δH (ppm): 8.48 (d, J = 1.7 Hz, 1H, Hα), 8.01 (d, J = 8.7 Hz, 1H, Hα’), 7.91 (dd, J = 8.7 and 1.7 Hz, 1H, Hβ’), 7.32 (s, 1H, H2), 4.02 (s, 3H, H8), 3.99 (s, 3H, H7). 13C NMR (Acetone-d6) δC (ppm): 168.0 (Cγ), 150.9 (C3), 141.3 (C5), 141.1 (C6), 130.6 (Cα), 128.2 (C1), 127.8 (Cβ), 126.7 (C4), 124.2 (Cβ’), 121.6 (Cα’), 104.1 (C2), 60.9 (C7), 56.4 (C8). HRMS [M + H]+ predicted 249.0757, found 249.0759.
Synthesis of bislactone by laccase-mediated dimerization of sinapic acid
Design of experiments
SA (0.33–2.33 g) was suspended in acetonitrile (0.6–3 mL), reaction medium was completed to 10 mL with the appropriated buffer (pH 3.5, 4.5 or 5.5) and heated up at 30 °C. Laccase from Trametes versicolor was weighed (1.7–27.5 mg) and dissolved in 5 mL of the appropriated buffer then the solution was poured into the medium. Aliquots of 100 µL were taken at 15, 30, 45, 60, 90, 120, 150, 180, 240 and 300 min and diluted in acetonitrile (1.4 mL or 2.9 mL) prior to filtration over 0.2 µm regenerated cellulose syringe filter. Samples were analyzed by HPLC to determine the amount of bislactone.
HPLC analysis
Flow rate and temperature were set at 0.8 mL.min−1 and 48 °C, respectively. 1 µL of the sample was injected and the gradient evolved as follow: 50% of formic acid solution 0.1% was maintained all along the run, acetonitrile varied from 5% at 0 min to 15% at 2.5 min, then 27% at 6 min, then 45% at 8.5 min, maintained at 45% during 0.5 min, decreased at 5% from 9 to 10 min and finally maintained at 5% for 1 min (total run 11 min) and complement was brought by MilliQ water. SA and BL were detected at 4.42 and 8.00 min, respectively.
Optimal synthetic conditions
605 mg of sinapic acid (2.7 mmol) were suspended in 3 mL of acetonitrile, then 10 mL of pH 5.5 buffer was added and the reaction was heated up at 30 °C and stirred at 700 rpm. 3.2 mg of laccase from Trametes versicolor (13.5 U) was dissolved in 2 mL of buffer and added to the medium. The reaction was stopped after complete consumption of sinapic acid (3 h) by adding 1 mL of dilute HCl solution (1 M). After filtration, washing with water and drying, pure BL was recovered as a pale-yellow powder (488 mg, 1.1 mmol, 81% yield).
mp: 204 °C. 1H NMR (Acetone-d6) δH (ppm): 7.54 (s, 2H, H6), 6.74 (s, 4H, H2), 5.76 (s, 2H, Hα), 4.13 (s, 2H, Hβ), 3.84 (s, 12H, H5). 13C NMR (Acetone-d6) δC (ppm): 176.1 (Cγ), 149.0 (C3), 137.5 (C1), 129.8 (C4), 104.2 (C2), 83.4 (Cα), 56.7 (C5), 49.1 (Cβ). HRMS [M + H]+ predicted 447.1286, found 447.1283.
Synthesis of thomasidioic acid and DMNA
Design of experiments
605 mg of sinapic acid (2.7 mmol) were suspended in 3 mL of acetonitrile, then 10 mL of buffer pH 5.5 was added and the reaction was heated up at 30 °C and stirred at 700 rpm. 3.2 mg of laccase from Trametes versicolor (13.5 U) was dissolved in 2 mL of buffer and added to the medium. After 3 h, 4.5 mL of the appropriate sodium hydroxide solution was added and the temperature was set at 30, 60 or 90 °C. An aliquot of 100 µL was taken at the scheduled time and diluted in a mixture of acetonitrile and 3M HCl (1300 and 100 µL, respectively).
HPLC method
Flow rate and temperature were set at 0.8 mL.min−1 and 48 °C, respectively. 1 µL of the sample was injected and the gradient evolved as follow: 50% of formic acid solution 0.1% was maintained all along the run, acetonitrile varied from 5% at 0 min to 10% at 1.0 min, then 15% at 3.2 min, then 27% at 6.5 min, then 50% at 10 min, decreased at 5% from 10 to 12 min and finally maintained at 5% for 1 min (total run of 13 min) and complement was brought by MilliQ water. TA and DMNA were detected at 4.93 and 6.72 min, respectively.
Optimal conditions for thomasidioic acid
605 mg of sinapic acid (2.7 mmol) were suspended in 3 mL of acetonitrile, then 10 mL of buffer pH 5.5 was added and the reaction was heated up at 30 °C and stirred at 700 rpm. 3.2 mg of laccase from Trametes versicolor (13.5 U) was dissolved in 2 mL of buffer and added to the medium. After 3 h, 4.5 mL of 1.85 M NaOH was added. After 10 min, reaction medium was acidified with 3M HCl solution (3 mL), and a precipitate appeared. After 2 h, the precipitate was filtered, washed with deionized water and dried to afford thomasidioic acid as a light brown solid (343 mg, 0.77 mmol, 57%).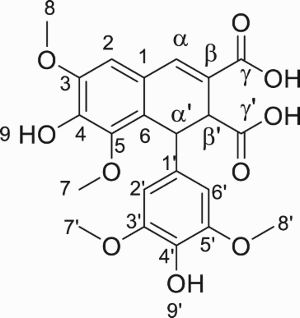
mp: 212 °C. 1H NMR (Acetone-d6) δH (ppm): 8.07 (s, 1H, H9 or H9’), 7.68 (s, 1H, Hα), 7.08 (s, 1H, H9’ or H9), 6.97 (s, 1H, H2), 6.40 (s, 2H, H2’ and H6’), 5.06 (s, 1H, Hα’), 4.00 (d, J = 1.3 Hz, 1H, Hβ), 3.90 (s, 3H, H8), 3.70 (s, 6H, H7’ and H8’), 3.62 (s, 3H, H7). 13C NMR (Acetone-d6) δC (ppm): 173.3 (Cγ’), 168.3 (Cγ), 148.4 (C3), 148.3 (C3’ and C5’), 146.3 (C5), 142.6 (C1), 138.2 (Cα), 135.5 (C1’), 134.4 (Cβ), 124.8 (C6), 124.0 (C4’), 123.9 (C4), 108.7 (C2), 105.9 (C2’ and C6’), 60.3 (C7), 56.5 (C8, C7’ and C8’), 47.2 (Cβ’), 40.2 (Cα’). HRMS [M + H]+ predicted 447.1286, found 447.1287.
Optimal conditions for DMNA
605 mg of sinapic acid (2.7 mmol) were suspended in 3 mL of acetonitrile, then 10 mL of pH 5.5 buffer was added and the reaction was heated up at 30 °C and stirred at 700 rpm. 3.2 mg of laccase from Trametes versicolor (13.5 U) was dissolved in 2 mL of buffer and added to the medium. After 3 h, 4.5 mL of 3.5 M NaOH solution was added. After 47.4 h, reaction media was acidified with 5 M HCl solution (5 mL) and a precipitate appeared. After 18 h, the precipitate was filtered, washed with deionized water and dried to afford DMNA as a pale-yellow solid (195 mg, 0.79 mmol, 58%).
Scale-up of DMNA
6.05 g of sinapic acid (27 mmol) were suspended in 30 mL of acetonitrile, then 110 mL of pH 5.5 buffer was added and the reaction was heated up at 30 °C and stirred at 700 rpm. 32 mg of laccase from Trametes versicolor (13.5 U) was dissolved in 10 mL of buffer and added to the medium. After 3 h, 45 mL of 3.5 M NaOH solution was added. After 50 h, reaction media was acidified with conc. sulfuric acid to pH 3, and a precipitate appeared. After 2 h, the precipitate was filtered, washed with deionized water and dried to afford DMNA as a pale-yellow solid (1.54 g, 6.2 mmol, 46%).
Antiradical assays
Already described method was employed (Citation34, Citation35). Briefly, antiradical activities were determined using the 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay to provide EC50 values (Citation22). To a well containing 10 µL of potential antiradical solution in ethanol (concentrations ranging from 800 to 25 µmol.L−1), 190 µL of DPPH solution (final concentration in the well C = 200 µmol.L−1 in ethanol) was added. Absorbance at 520 nm was recorded every 5 min for 7.5 h using an EPOCH2 microplate reader (Biotek). The use of different concentrations of potential antiradical gave the EC50 value in nmol, which is the quantity needed to reduce half the initial population of DPPH radicals.
Supplemental Material
Download MS Word (1.8 MB)Authors contributions
AF, TN, SF and FA conceptualized this work. Synthetic approach was designed by AF. Experiments were performed by NZ, EV, TN and AF. NZ and EV have been supervised by AF and SF. Formal analysis and visualization were performed by AF. Original draft was written by AF and TN. SF and FA reviewed and edited the initial draft. FA acquired funding.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Manzanares, P. The Role of Biorefinering Research in the Development of a Modern Bioeconomy. Acta Innovations 2020, 37, 47–56. doi:10.32933/ActaInnovations.37.4.
- Flourat, A.L.; Combes, J.; Bailly-Maitre-Grand, C.; Magnien, K.; Haudrechy, A.; Renault, J.; Allais, F. Accessing P-Hydroxycinnamic Acids: Chemical Synthesis, Biomass Recovery, or Engineered Microbial Production? ChemSusChem. 2021, 14, 118–129. doi:10.1002/cssc.202002141.
- Reungoat, V.; Gaudin, M.; Flourat, A.L.; Isidore, E.; Mouterde, L.M.M.; Allais, F.; Ducatel, H.; Ioannou, I. Optimization of an Ethanol/Water-Based Sinapine Extraction from Mustard Bran Using Response Surface Methodology. Food Bioprod. Process. 2020, 122, 322–331. doi:10.1016/J.FBP.2020.06.001.
- Reungoat, V.; Mouterde, L.M.M.; Chadni, M.; Couvreur, J.; Isidore, E.; Allais, F.; Ducatel, H.; Ioannou, I. Simultaneous Extraction and Enzymatic Hydrolysis of Mustard Bran for the Recovery of Sinapic Acid. Food Bioprod. Process. 2021, 130, 68–78. doi:10.1016/J.FBP.2021.09.003.
- Achinivu, E.C.; Flourat, A.L.; Brunissen, F.; Allais, F. Valorization of Waste Biomass from Oleaginous “Oil-Bearing” Seeds Through the Biocatalytic Production of Sinapic Acid from Mustard Bran. Biomass Bioenergy 2021, 145 (105940), 1–7. doi:10.1016/j.biombioe.2020.105940.
- Flourat, A.L.; Willig, G.; Teixeira, A.R.S.; Allais, F. Eco-Friendly Extraction of Sinapine from Residues of Mustard Production. Front. Sustain. Food Syst 2019, 3, 1–10. doi:10.3389/fsufs.2019.00012.
- Hostettler, F.D.; Seikel, M.K. Lignans of Ulmus Thomasii Heartwood-II. Lignans Related to Thomasic Acid. Tetrahedron 1969, 25 (11), 2325–2337. doi:10.1016/S0040-4020(01)82782-4.
- Charlton, J.L.; Lee, K.A. Thomasidioic Acid and 6-Hydroxy-5,7-Dimethoxy-2-Naphthoic Acid: Are They Really Natural Products? Tetrahedron Lett. 1997, 38 (42), 7311–7312. doi:10.1016/S0040-4039(97)01775-9.
- Lukacs, S.J.; Cohen, S.M.; Long, F.H. Optical Properties of a Liquid-Crystalline Random Copolyester. J. Phys. Chem. B 1999, 103 (32), 6648–6652. doi:10.1021/jp984785v.
- Gutierrez, G.A.; Chivers, R.A.; Blackwell, J.; Stamatoff, J.B.; Yoon, H. The Structure of Liquid Crystalline Aromatic Copolyesters Prepared from 4-Hydroxybenzoic Acid and 2-Hydroxy-6-Naphthoic Acid. Polymer (Guildf) 1983, 24 (8), 937–942. doi:10.1016/0032-3861(83)90141-6.
- Heifferon, K.v.; Spiering, G.A.; Talley, S.J.; Hegde, M.; Moore, R.B.; Turner, S.R.; Long, T.E. Synthesis and Characterization of a Nematic Fully Aromatic Polyester Based on Biphenyl 3,4′-Dicarboxylic Acid. Polym. Chem. 2019, 10 (31), 4287–4296. doi:10.1039/c9py00683d.
- Gershon, H.; Parmegiani, R. Antimicrobial Activity of 8-Quinolinols, Salicylic Acids, Hydroxynaphthoic Acids, and Salts of Selected Quinolinols with Selected Hydroxy-Acids. Appl. Microbiol. 1962, 10, 348–353. doi:10.1128/aem.10.4.348-353.1962.
- Dogan, H.N.; Rollas, S.; Erdeniz, H. Synthesis, Structure Elucidation and Antimicrobial Activity of Some 3- Hydroxy-2-Naphthoic Acid Hydrazide Derivatives. Farmaco 1998, 53 (7), 462–467. doi:10.1016/S0014-827X(98)00049-4.
- Lee, K.-A.S. Oxidative Coupling of Sinapic Acid; University of Manitoba, 1997.
- Jaufurally, A.S.; Teixeira, A.R.S.; Hollande, L.; Allais, F.; Ducrot, P.H. Optimization of the Laccase-Catalyzed Synthesis of (±)-Syringaresinol and Study of Its Thermal and Antiradical Activities. ChemistrySelect 2016, 1 (16), 5165–5171. doi:10.1002/slct.201600543.
- Mention, M.M.; Flourat, A.L.; Peyrot, C.; Allais, F. Biomimetic Regioselective and High-Yielding Cu(i)-Catalyzed Dimerization of Sinapate Esters in Green Solvent CyreneTM: Towards Sustainable Antioxidant and Anti-UV Ingredients. Green Chem. 2020, 22 (6), 2077–2085. doi:10.1039/d0gc00122h.
- Ahmed, R.; Lehrer, M.; Stevenson, R. Synthesis of Thomasic Acid. Tetrahedron 1973, 29, 3753–3759.
- Kim, E.; Hyeong, K.L.; Hwang, E.i.; Kim, S.U.; Woo, S.L.; Lee, S.; Jung, S.H. Stereochemistry of Phellinsin A: A Concise Synthesis of α-Arylidene-γ-Lactones. Synth. Commun. 2005, 35 (9), 1231–1238. doi:10.1081/SCC-200054845.
- Andrade e Silva, M.L.; Rodrigues Esperandim, V.; da Silva Ferreira, D.; Guidi Magalhães, L.; Coelho Lima, T.; Cunha, W.R.; Nanayakkara, D.N.P.; Pereira, A.C.; Kenupp Bastos, J. Furofuran Lignans Display Schistosomicidal and Trypanocidal Activities. Phytochemistry 2014, 107, 119–125. doi:10.1016/j.phytochem.2014.08.010.
- Rubino, M.I.; Arntfield, S.D.; Charlton, J.L. Conversion of Phenolics to Lignans: Sinapic Acid to Thomasidioic Acid. J. Am. Oil Chem. Soc. 1995, 72 (12), 1465–1470. doi:10.1007/BF02577839.
- Rubino, M.I.; Arntfield, S.D.; Charlton, J.L. Evaluation of Alkaline Conversion of Sinapic Acid to Thomasidioic Acid. J. Agric. Food Chem. 1996, 44 (6), 1399–1402. doi:10.1021/jf950431e.
- Cai, R.C.; Arntfield, S.D.; Charlton, J.L. Structural Changes of Sinapic Acid During Alkali-Induced Air Oxidation and the Development of Colored Substances. J. Am. Oil Chem. Soc. 1999, 76 (6), 757–764. doi:10.1007/s11746-999-0172-6.
- Bunzel, M.; Ralph, J.; Kim, H.; Lu, F.; Ralph, S.A.; Marita, J.M.; Hatfield, R.D.; Steinhart, H. Sinapate Dehydrodimers and Sinapat-Ferulate Heterodimers in Cereal Dietary Fiber. J. Agric. Food Chem. 2003, 51 (5), 1427–1434. doi:10.1021/jf020910v.
- Fuganti, C.; Serra, S. Studies on the Synthesis of Highly Substituted Naphthol: Preparation of 6-Hydroxy-5,7-Dimethoxy-2-Naphthoic Acid, Isolated from Ulmus Thomasii. J. Chem. Res. Part S 1998, 10, 638–639. doi:10.1039/a803651i.
- Peyrot, C.; Peru, A.A.M.; Mouterde, L.M.M.; Allais, F. Proline-Mediated Knoevenagel-Doebner Condensation in Ethanol: A Sustainable Access to p-Hydroxycinnamic Acids. ACS Sustain. Chem. Eng. 2019, 7 (10), 9422–9427. doi:10.1021/acssuschemeng.9b00624.
- Tranchimand, S.; Tron, T.; Gaudin, C.; Iacazio, G. Synthesis of Bis-Lactone Lignans through Laccase Catalysis. J. Mol. Catal. B: Enzym. 2006, 42 (1–2), 27–31. doi:10.1016/j.molcatb.2006.06.003.
- Mouterde, L.M.M.; Flourat, A.L.; Allais, F. Chemoenzymatic Total Synthesis of a Naturally Ocurring (5-5’)/(8’-O-4’’) Dehydrotrimer of Ferulic Acid. Eur. J. Org. Chem. 2013, 173–179. doi:10.1002/ejoc.201201290.
- Flourat, A.L.; Peru, A.A.M.; Haudrechy, A.; Renault, J.-H.; Allais, F. First Total Synthesis of (β-5)-(β-O-4) Dihydroxytrimer and Dihydrotrimer of Coniferyl Alcohol (G): Advanced Lignin Model Compounds. Front. Chem. 2019, 7. doi:10.3389/fchem.2019.00842.
- Kurniawati, S.; Nicell, J.A. Characterization of Trametes Versicolor Laccase for the Transformation of Aqueous Phenol. Bioresour. Technol. 2008, 99 (16), 7825–7834. doi:10.1016/J.BIORTECH.2008.01.084.
- Teixeira, A.R.S.; Flourat, A.L.; Peru, A.A.M.; Brunissen, F.; Allais, F. Lipase-Catalyzed Baeyer-Villiger Oxidation of Cellulose-Derived Levoglucosenone into (S)-γ-Hydroxymethyl-α,β-Butenolide: Optimization by Response Surface Methodology. Front. Chem. 2016, 4 (16), 1–11. doi:10.3389/fchem.2016.00016.
- Monteith, E.R.; Mampuys, P.; Summerton, L.; Clark, J.H.; Maes, B.U.W.; McElroy, C.R. Why We Might Be Misusing Process Mass Intensity (PMI) and a Methodology to Apply It Effectively as a Discovery Level Metric. Green Chem. 2020, 22 (1), 123–135. doi:10.1039/c9gc01537j.
- Sheldon, R.A. Metrics of Green Chemistry and Sustainability: Past, Present, and Future. ACS Sustain. Chem. Eng. Amer. Chem. Soc. 2018, 2, 32–48. doi:10.1021/acssuschemeng.7b03505.
- van Aken, K.; Strekowski, L.; Patiny, L. EcoScale, a Semi-Quantitative Tool to Select an Organic Preparation Based on Economical and Ecological Parameters. Beilstein J. Org. Chem. 2006, 2 (3), 1–7. doi:10.1186/1860-5397-2-3.
- Peyrot, C.; Mention, M.M.; Brunissen, F.; Allais, F. Sinapic Acid Esters: Octinoxate Substitutes Combining Suitable UV Protection and Antioxidant Activity. Antioxidants 2020, 9 (9), 1–16. doi:10.3390/antiox9090782.
- Rioux, B.; Peyrot, C.; Mention, M.M.; Brunissen, F.; Allais, F. Sustainable Synthesis of P-Hydroxycinnamic Diacids Through Proline-Mediated Knoevenagel Condensation in Ethanol: An Access to Potent Phenolic UV Filters and Radical Scavengers. Antioxidants 2020, 9 (4). doi:10.3390/antiox9040331.