
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
New Dicoumarols 2a-g efficiently synthesized employing p-TSA as a catalyst from the reaction of 4-hydroxycoumarin with aryl aldehydes in water. This method offers direct access to structurally diverse Dicoumarols derivatives in a good yields (65-94%). Upon heating 3,3' -arylidenebis-4-hydroxycoumarins derivative in acetic anhydride, the epoxydicoumarins were formed. 1H, 13C{1H}-NMR, elemental analysis, and infrared spectroscopic techniques were used for the characterization of the obtained compounds. A possible relationship between such hydrogen-bonded structures and the antimicrobial and the antioxidant activities of compounds is suggested. The synthetized compounds 2a-g and 3a-g were subjected to in vitro antimicrobial and antifungal activities against DNA gyrase and Mycobacterium tuberculosis-CYP51 target proteins at the active sites. Compound 3d shows effective inhibitory effect in terms of MIC = 15 µg mL-1. against Salmonella typhimuriumATCC 14028. Compounds 2b, 2c, 3b, 3c, 3e and 3g have recorded an important scavenging activity against the radical DPPH. The EC50 of these compounds was 47.17, 46.90, 50.55,48.27 , 46.55 and 47.54 µg mL-1 respectively. Investigation of the anti-inflammatory activity of the synthesized compounds showed that compounds 2b,2c, 3f, 3b and 3c are the most potent inflammatory activities.
GRAPHICAL ABSTRACT
1. Introduction
The chemistry of oxygenated heterocycles has been the subject of increasing interest. Coumarins are benzo-fused heterocycles containing oxygen atoms and their synthesis is important because of their widespread occurrence in nature (Citation1–3). Coumarin is a fundamental unit of some biological and pharmaceutical products such as warfarin [4-hydroxy-3-(3-oxo-1-phenylbutyl)-2H-1-benzopyran-2-one], acenocoumarol {4-hydroxy-3-[1-(4-nitrophenyl)-3-oxobutyl]-2H-1-benzopyran-2-one}, dicoumarol (3,3'-methylenebis[4-hydroxy-2H-1-benzopyran-2-one]), and phenprocoumon [4-hydroxy-3-(1-phenylpropyl)-2H-1-benzopyran-2-one] (Citation4,Citation5). Through metabolization of coumarin in the sweet clover by molds such as Penicillium nigricans and Penicillium jensi, dicoumarols has resulted. Dicoumarols as a natural anticoagulant drug have been extensively studied because of their application in the pharmaceutical industry (Citation6). Some synthetic strategies have been reported for dicoumarols including the total synthesis starting from salicylaldehyde and formaldehyde (Citation7), biosynthesis of dicoumarols employing micro-organisms such as Penicillium jenseni (Citation8), and Knoevenagel condensation of 4-hydroxycoumarins with carbonyls using several catalysts (Citation9–12).
The chemistry of coumarin derivatives has recently gained much attention from chemists owing to some interesting biological properties (Citation13–15). Dicoumarols are a naturally occurring anticoagulant (Citation16). The most popular strategies towards the synthesis of dicoumarols start from salicylaldehyde and formaldehyde and involve the biosynthesis of dicoumarols using micro-organisms such as Penicillium jenseni (Citation17). The Knoevenagel condensation of 4-hydroxycoumarins with carbonyl compounds using several catalysts gives also dicoumarols (Citation18–20). From an environmental and economic point of view, water as a solvent or media has many advantages and usually results in excellent efficiency and selectivity (Citation21–23).
Most of the existing natural coumarins have been isolated from higher plants, some of them have been discovered in microorganisms, e.g. aminocoumarin antibiotics: novobiocin and chlorobiocin (produced by the actinomycete Streptomyces niveus) (Citation24). The biological activities of coumarin derivatives, in particular their therapeutic application as anticoagulant and antibacterial agents (Citation25), have also been shown to exert efficient anti-proliferative, antifungal, anti-psoriasis, anti-inflammatory, as well as antiviral activities (Citation26–28). The interest in coumarins has recently increased significantly because they play a critical role in the replicative cycle of HIV (Citation29,Citation30). We describe here in this work an eco-friendly method for the synthesis of some new and known dicoumarols in water as a solvent and their epoxydicoumarins 3 to explore their antimicrobial and antioxidant activities. In addition, their antiinflammatory activities were investigated.
2. Results and discussion
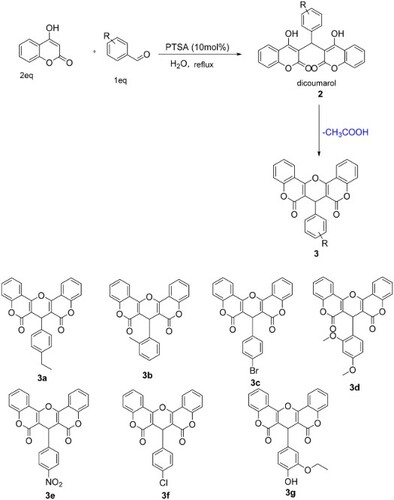
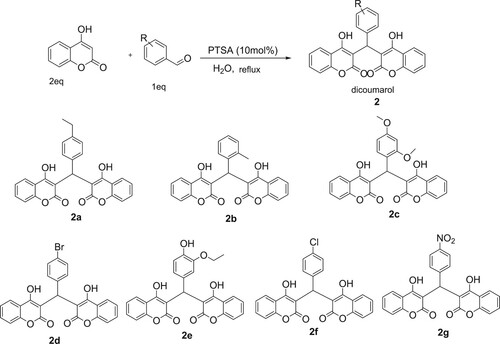
In continuation of our studies on the synthesis and reactivity of coumarin derivatives (Citation26), the present work aimed to synthesize different new substituted dicoumarols and their epoxy derivatives to study their biological activities. In this regard, we found that condensation between 4-hydroxycoumarin and arylaldehydes in the presence of catalytic amounts of p-TSA in water under reflux produces new dicoumarols 2 in good yields (Scheme 1).
Scheme 1. Synthesis of dicoumarols derivatives 2 catalyzed by p-TSA.
The advantages of p-TSA are low corrosivity, simple handling and it is inexpensive. It has been widely used as a catalyst in several organic reactions (Citation27–29). To choose the best conditions for the synthesis of 3 using p-TSA as a catalyst, reaction between 4-hydroxycoumarin and paraethylbenzaldehyde was selected as a model. From , the reaction did not go to completion in the absence of catalyst even after extended reaction times. Higher loadings of catalyst did not afford a marked influence on the product yield nor reaction rate. In another experiment, to illustrate the effect of solvent or media on the reaction progress, several different solvents were employed, the obtained results are illustrated in .
Table 1. Effect of catalyst amount and various solvents on the synthesis of 3a at reflux temperature.
The condensation reaction was accelerated by protonic solvents such as EtOH, MeOH, and H2O.
In addition, this reaction is enhanced using p-TSA (10 mol%) as catalyst under reflux in H2O in 75 min. After determining the optimal reaction conditions, the extension of the scope of this method was studied. For this, various aryl aldehydes were reacted. Results are given in in which it is apparent that aryl aldehydes, including those bearing electron-poor and electron-rich substituents, were able to undergo smoothly this reaction.
Table 2. Synthesis of dicomarols 2 using p-TSA (10 mol%) under reflux in H2O.
The present method provides environmentally safe conditions using water as solvent and p-TSA as a catalyst to obtain the desired products with better yields than previous reported compared with both a previously reported method which has used AcOH as reaction media, (Citation31) and by Fokin et al. (Citation30). Based on their study, it would appear that this reaction type may be placed in the category of ‘on-water’ synthetic reactions. The characterization of these compounds was carried out by mass spectral, IR, 1H NMR, and 13C NMR analyses.
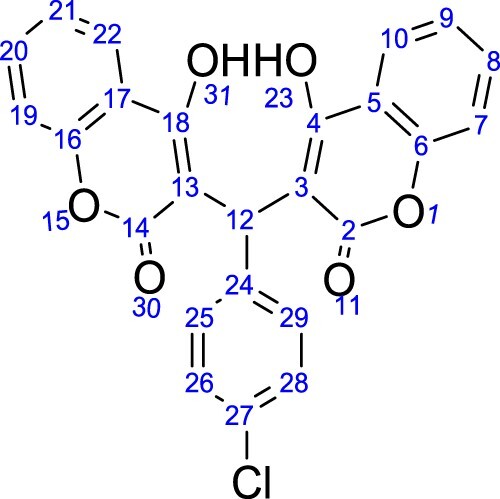
1H-NMR revealed that all the dicoumarol compounds followed the Michael addition reaction. The condensation of 2-ethylbenzaldehyde, 2.4-diethoxybenzaldehyde, 4-nitrobenzaldehyde, 4-bromobenzaldehyde, 3-ethoxy-4-hydroxybenzaldehyde, 4-chlorobenzaldehyde and 2-methylbenzaldehyde gave dicoumarol compounds which were characterized by using spectroscopic and analytical techniques. Elemental analyses established the composition which was further confirmed by MS spectral analysis. The percent abundance coincides with the respective molecular ion peaks. 1H and 13C{1 H}-NMR were carried out for all the compounds using CDCl3. The NMR assignment for dicoumarols is based on .
Figure 1. Atoms numbering used for NMR signals for dicoumarols.
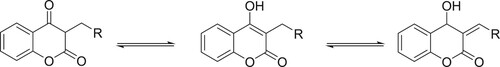
The enolic protons were found to be involved in the formation of hydrogen bonding. A singlet at 11.28–11.56 ppm in different dicoumarol compounds was assigned to those enolic protons because they can be removed easily by adding deuterium oxide (Citation32). A singlet at 8 ppm was assigned to the methylene group of the aldehydes which connects both moieties. This singlet can also be observed at about 9.8–12 ppm because of the possible tautomerism as may be seen in Scheme 2.
Scheme 2. Tautomerism in coumarin structure.
The rest of the NMR spectra coincides with their respective peaks for each proton. The solid-state ATR spectra were recorded for all the compounds in 400–4000 cm−1 spectral region. Since the IR spectra of all the compounds were quite similar therefore discussion is confined to the important vibrations only. The carbonyl stretching frequency varies from 1639 to 1679 cm−1 which may be due to the interaction of the carbonyl group with the hydroxyl group to produce strong hydrogen bonding. Earlier studies revealed that the carbonyl group involved in hydrogen bonding vibrates at a lower frequency (Citation33). The hydrogen bonding may either be intermolecular or intramolecular as shown in Scheme 3.
Scheme 3. Hydrogen bonding in dicoumarol compounds.
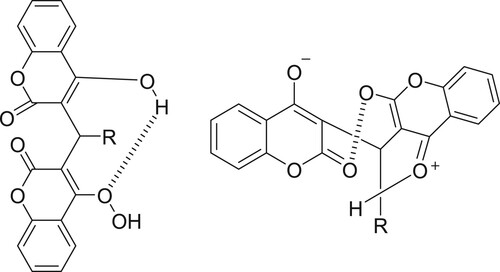
Compounds 2d, 2e, and 2f have carbonyl stretching frequencies in the range 1661–1679 cm−1 whereas compounds 2a and 2 g were observed to have carbonyl stretching frequency at 1639–1651 cm−1 which are around 20 cm−1 lower than the stretching frequency for 2d, 2e, and 2f dichromone structure (structure B) was assigned as given in Scheme 4.
Scheme 4. Dicoumarols and dicoumrone structures.
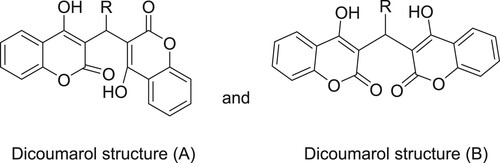
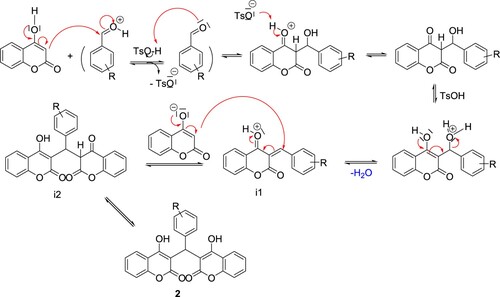
The stretching frequency of C–O of the lactone ring appeared in the range of 1090–1120 cm−1. There is also a strong vibration around 1040 cm−1 in both compounds which is assigned to the complementary band for the ketone carbonyl carbon. Hydroxyl group is involved in hydrogen bond formation therefore the band is weakly observed. It was found that there is a type of competition between the two products which dominates the formation of translactonised product. The two products were separated by column chromatography using alumina and were assigned to those enolic protons because they can be removed easily by adding deuterium oxide (Citation33). 1H-NMR for 2a shows a singlet around 11.28 ppm which was assigned to the proton of the phenolic OH group. Similarly, a singlet around 10.79 ppm was tentatively assigned to the H(10), a proton of the methylene group of the coumarine ring. The rest of the resonances for both compounds were assigned to the protons of the aromatic ring system. The compounds were confirmed to be translactonized by studying the 13C{1 H}-NMR which shows peaks around 192 ppm, assigned to the C=O group of ketone. Similarly the lactone C=O was assigned to the peak appearing around 181 ppm. These two peaks are identical in both compounds. The rest of the spectrum was assigned to their respective carbon atoms. Infrared spectrum reveals absorption bands around 1563 cm−1 which was assigned to the C=O stretching frequency of the ketone. A weak band in the range of 1090–1120 cm−1 was assigned to the stretching frequency of C–O of the lactone ring. The OH group is involved in H-bond formation and the band is weakly observed. There is also a strong vibration around 1040 cm−1 in both compounds, which is assigned to the complementary band for the ketone C=O carbon. We have found that the NMR spectrum of 2b in deuterochloroform shows three singlets at 6.1 bridge (CH: 1H), 11.34 (s, 1H, OH) and 11.57 (s, 1H, OH). The last singlets are independent of concentration and can be removed with the addition of traces of deuterium oxide. We assign these signals to the two strongly deshielded protons of the enolic hydroxyl group which is intramolecularly hydrogen bonded to carbonyl groups and we consider that this NMR evidence confirms the structural assignment for 2b mentioned above. The protons of the ring methylene groups give rise to a single resonance probably due to a rapid tautomeric exchange of their chemical shifts. This indicates the presence of intramolecularly hydrogen-bonded protons and suggests the structure A for the obtained Dicoumarols (Scheme 4). The CH proton found at the linking site of the two coumarin groups was found to be resonating at 6 ppm depending upon the variation in the aromatic aldehydes (Citation34). All the complexes show multiplates in the region of 6–9 ppm, assigned to the resonances caused by aromatic protons. The 13C{1H}-NMR also supports the 1H-NMR spectral analyses, the hydroxyl-based carbon and lactone carbonyl resonances. Based on the mechanistic pathway of the Knoevenagel and Michael reaction, (Citation31,Citation32), we propose a reasonable mechanism involving the protonic acid-catalyzed reaction of arylaldehydes with 4-hydroxycoumarin, as depicted in Scheme 5. First, condensation between 4-hydroxycoumarin and aryl benzaldehyde generates the intermediate i1. Attack of the next 4-hydroxycoumarin molecule through a Michael-type addition to i1, the enolization of adduct i2, gives the final product 2.
Scheme 5. Plausible mechanism for p-TSA catalyzed condensation of 4-hydroxycoumarin with aryl aldehydes.
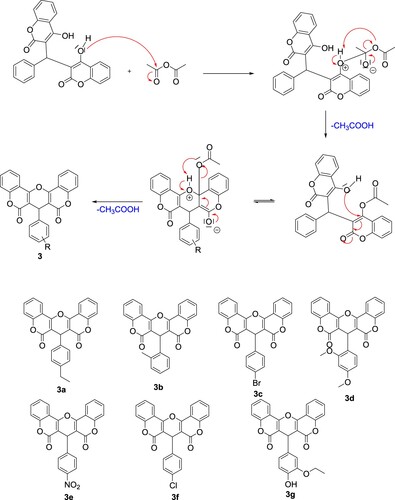
The heating of dicoumarols derivative in acetic anhydride was quickly transformed by these compounds to the corresponding epoxycoumarins 3 (Scheme 6)
Differentiation of compounds 2 and 3 was made mainly by their 1H, 13C and IR spectra. In particular, the main difference in the 1H NMR spectra of compounds 2 and 3 was hydroxylic protons, which appeared as two singlets, respectively, at δ 11.34 and 11.57 ppm in compound 2, whereas in compound 3 they were absent. In the IR spectra, among the characteristic bands confirming the structure of epoxy derivatives, we should pick out the vibrations of the lactone group which appear at 1725 cm−1 and the absence of the vibrations of OH.
Scheme 6. Protocol synthesis of epoxycoumarins 3.
3. Biological activities
3.1. Antibacterial activities
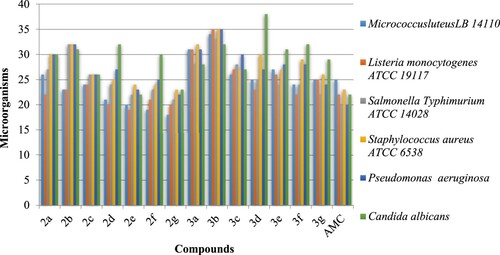
All the synthesized compounds 2–3 were subjected to their antimicrobial activities against selected pathogenic Gram-positive bacteria, Gram-negative bacteria, and a fungal strain. The activities of all the synthesized compounds were compared with a standard drug already used for stopping the pathogenic activities of tested microorganisms. As may be depicted from , the results for the antimicrobial activities of compounds 2b, 2c against Micrococcus luteus LB 14110 and Listeria monocytogenes ATCC 19117 are very close. On the other hand, the dicoumarol 2e was found moderately active against bacteria and fungus. Strong antimicrobial activities were observed against Staphylococcus aureus ATCC 6538 and Pseudomonas Aeruginosa for compound 3b. Compound 3c showed similar activities against Listeria monocytogenes ATCC 19117 and Staphylococcus aureus ATCC 6538 which are also comparable to the drug. Compound 2b was found much active than the standard drug in inhibiting the activities of Bacillus subtilis. Compounds 2b, 3b, and 3d showed greater activities against the Candida albicans.
Figure 2. Zone of bacterial inhibition measured in mm of the synthesized compounds 2–3.
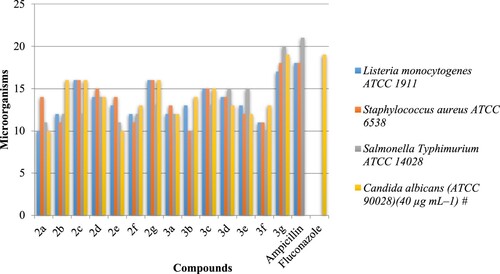
The minimum inhibitory concentration (MIC) against the above organisms was determined by the method of broth dilutions (Citation35). A close survey of MIC values indicates that the compounds 3a–g exhibited a varied range (10–20 µg mL–1.) of antibacterial activity against all the tested bacterial strains. Compound 3b was also found to be active against Listeria monocytogenes ATCC 19117. Compound 3d shows effective inhibitory effect in terms of MIC = 15 µg mL–1 against Salmonella typhimurium ATCC 14028. Compound 3e with NO2 at 4 position of aryl ring showed the moderate activity (MIC = 15 µg mL–1) against Salmonella Typhimurium ATCC 14028. The results are given in .
Figure 3. Minimum Inhibitory Concentration (MIC)* of synthesized compounds 2–3 against L. monocytogenes ATCC 1911, S. aureus ATCC 6538, S. typhimurium ATCC 14028 and C. albicans (ATCC 90028) in µg mL–1. *Values are mean of three replicates. (Standard error ± 1 µg mL–1). # Concentration of solutions for respective activities. *Ampicillin was used as a standard against bacterial species (100 µg mL–1). * Fluconazole was used as a standard against fungi species (10 µg mL–1).
3.2. Antioxidant activity
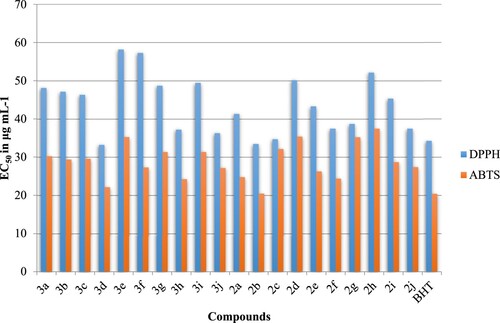
The antioxidant activity of synthesized compounds 2a–g and 3a–g was evaluated by the two methods DPPH (1,1-diphenyl-2-picrylhydrazyl) antiradical assay and ABTS [2.20-azino-bis(3-ethylbenzothiazoline-6-sul phonic acid)] scavenging assay and were expressed as EC50 value (the concentration in µg mL−1 that causes 50% of effect). As shown in , compounds 2b, 2c, 3b, 3c, 3e, and 3 g have recorded an important scavenging activity against the radical DPPH. The EC50 of these compounds was 47.17, 46.90, 50.55,48.27, 46.55, and 47.54 µg mL−1 respectively. Concerning the activity in the free radical ABTS, the EC50 of the synthesized compounds 2a–g and 3a–g was widely variable and it was ranged between 21.48 and 33.17 µg mL−1. The strongest activity in the ABTS assay was for 2a (30.22 µg mL−1), 3b (33.17 µg mL−1), and 3f (30.65 µg mL−1).
Figure 4. Antioxidant activity of the synthesized compounds 2a–g et 3a–g assessed by DPPH and ABTS techniques and expressed as EC50 in μg mL−1. The BHT was used as control.
3.3. Anti-inflammatory activity
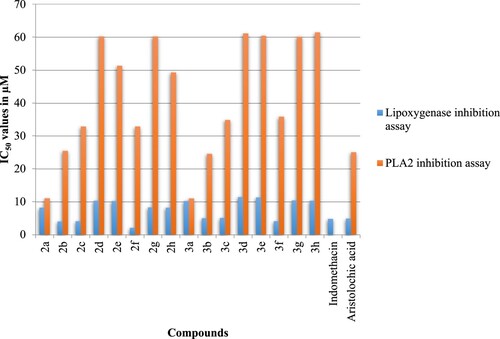
As per our objective, we next examined the anti-inflammatory activities of the synthesized compounds 2a–g and 3a–g by lipoxygenase inhibition and phospholipase A2 (PLA2) inhibition assays. The IC50 values of the standards and test samples in both assays are given in . In both assays, the synthesized compounds 3b and 3c showed potent anti-inflammatory activity in lipoxygenase inhibition assay (5.0–5.1 µM) and PLA2 inhibition assay (24.5–34.9 µM). It should be noted that 3f and 3b have anti-inflammatory activities better than the standards Indomethacin and Aristolochic acid.
Figure 5. IC50 values of the compounds 2a–g and their compounds 3a–g for anti-inflammatory activity.
4. Molecular docking
The antibacterial and antifungal inhibitions are discussed based on the bending mode interactions, scores, and root mean square deviation (RMSD). The docking results are exposed in histograms plotted in . Further, the energy scoring and RMSD values are listed in . It is evident that 2a demonstrates significant inhibitory activities against DNA gyrase, and it is a promising candidate for antifungal therapy. The 2a score is fairly close to that obtained for Ciprofloxacin. On the other hand, 2e and 2a have proved potential inhibition against CYP51, higher than that obtained for fluconazole. Hence, these synthetized compounds exhibit considerable antifungal activities.
Figure 6. Molecular docking scoring of antimicrobial and antifungal activities of 2a–g and 3a–g.
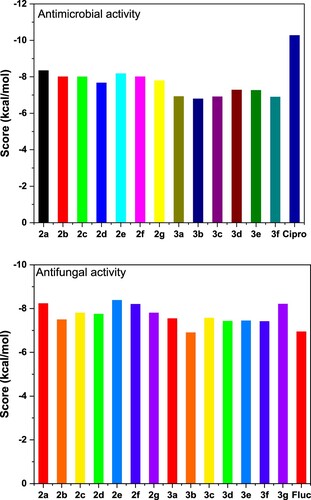
Table 3. Docking results (score and RMSD) of antimicrobial and antifungal activities of 2a–g and 3a–g.
The best 3D poses of 2a and 2e in the protein targets and 2D ligand–protein interactions are depicted in and . In the same context, the best pose through self-docking and the possible interactions between each ligand (CPF and TPF) and targets are in .
Figure 7. 3D best pose of 2a ligand in DNA gyrase (5BTC) target enzyme (top), along with 2D ligand–protein interactions (bottom).
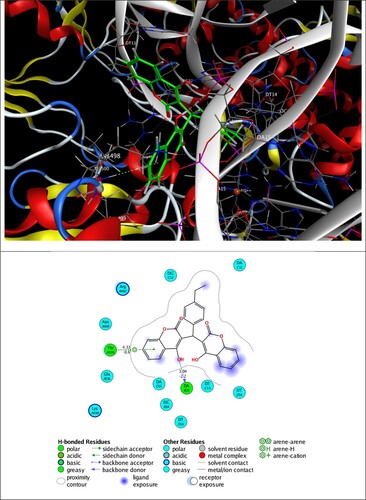
Figure 8. 3D best pose of 2e ligand in Mycobacterium tuberculosis-CYP51 (1EA1) target enzyme (top) along with 2D ligand–protein interactions (bottom).
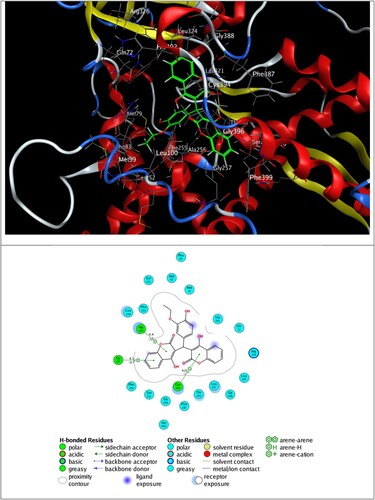
Figure 9. 3D plot of self-docking of ciprofloxacin (CPF) in DNA gyrase (5BTC) target enzyme (top), along with possible interactions (bottom).
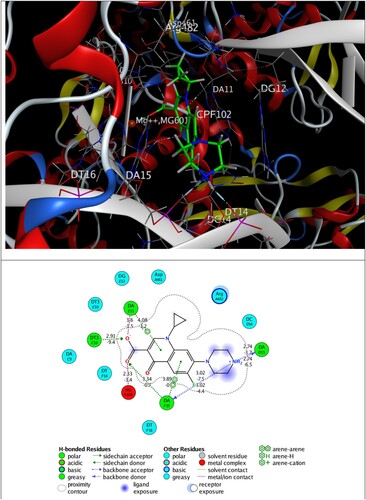
Figure 10. 3D plot of self-docking of fluconazole (TPF) in Mycobacterium tuberculosis-CYP51 (1EA1) target enzyme (top), along with possible interactions (bottom).
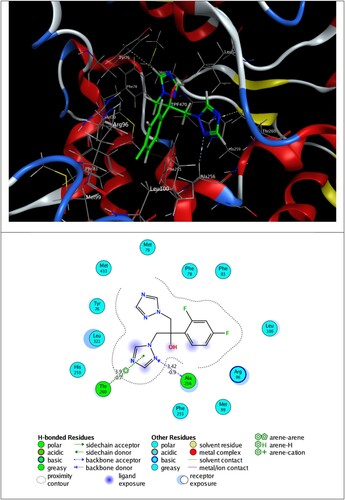
The self-docking calculations show clearly that the co-crystalized entities fit well into their original conformation. CPF ligand and DNA gyrase interact via hydrogen bonding, π–H and π–π interactions, whereas TPF ligand forms hydrogen bonding with Ala 256 and π–H interaction with Thr 260 residues.
The inspection of 2D ligand–protein interactions plotted in and exhibits that 2a interacts with DNA gyrase via hydrogen bonding with Thr A500 and π–H interaction with DAB15, and 2e forms π–H interactions with Ala 256, Gly 257 and Cys 394 residues.
4.1. Theoretical details
Docking simulation of synthetized compounds 2a–g and 3a–g into antimicrobial and antifungal protein targets is carried out using the Molecular Operating Environment (MOE 2015.10) software package (Citation35). Crystal structure of DNA gyrase (PDB ID: 5BTC) as antimicrobial target, and Mycobacterium tuberculosis-CYP51 (PDB ID: 1EA1) as antifungal target were retrieved from protein data bank (Citation36).
For each protein target was 3D protonated, with default parameters of MOE, after removing solvent molecules and the eventual co-crystallized ligand and adding the missing hydrogen atoms. The errors like steric clashes missing loops, missing atom names and picking alternate conformations in the protein crystallographic data were corrected. Then, the energy minimization was performed using MMFF94x force field (gradient: 0.05) (Citation37,Citation38) and the partial charges were assigned. The studied ligands are drawn and minimized with MOE package using the same force field. The ligand pockets are selected as the active site of the proteins, corresponding well to those obtained with Site Finder application in MOE. The dummy site was created with alpha sphere around an active site. The default Triangle Matcher placement method was employed for docking. London dG scoring function was chosen to score top 1000 poses obtained from the Triangle Matcher placement method. The 30 top-ranked poses are tiered by London dG, and then are minimized using MMFF94x force field within a rigid receptor.
The resulting poses were then scored using the Generalized-Born Volume Integral/Weighted Surface area (GBVI/WSA) dG scoring function, which estimates the binding free energy for a given pose of the ligand. The docking score is the binding free energy, calculated by the GBVI/WSA scoring function in the S field, which is the score of the last stage. The lower score indicates the more favorable pose. Ligand Interactions tool was used for 2D and 3D visualizations of protein–ligand interactions.
5. Conclusion
An improved route for the synthesis of dicoumarols containing different groups from simple substrates and p-TSA catalyst has been achieved with a very high atom economy for the preparation of pharmaceutically relevant heterocyclic systems. Importantly, the use of water as a cheap and clean media for reaction should place this chemistry in the category of Green Chemistry. The obtained compounds were characterized by various spectro-analytical techniques and it was confirmed that dicoumarols were confirmed. The prepared compounds exhibit antimicrobial activity against all the tested microorganisms, especially against Pseudomonas aeruginosa, Listeria monocytogenes, and C. albicans. Compound 2a presented potent DNA gyrase inhibition relatively similar to ciprofloxacin. Besides, all synthesized compounds exhibit CYP51 inhibition higher than fluconazole. The compound 2e reveals its high antifungal activities. Therefore, these findings can be considered a hit for further optimization to obtain more active antimicrobial and antifungal agents. Furthermore, these compounds exhibit moderate antioxidant free radical-scavenging activity but strong hydrogen peroxide scavenging activity. The obtained results may open novel avenues to further investigate this class of compounds as antimicrobial agents.
5.1. Experimental
5.1.1. General information
All manipulations were performed using standard Schlenk techniques under an argon atmosphere. Chemicals were purchased from Sigma–Aldrich and were used without further purification. All solvents were purified and dried by the MBRAUN SPS 800 solvent purification system. 1H NMR and 13C NMR spectra were recorded at 400 and 100 MHz, respectively. Chemical shifts, δ, are reported in ppm relative to the internal standard TMS for both 1H and 13C NMR. &e products were characterized by GC (gas chromatography). Quantitative GC analyses were performed with GC-2010 Plus gas chromatography (SHIMADZU) and NMR studies were carried out in high-quality 5 mm NMR tubes. Signals are quoted in parts per million as δ downfield from tetramethylsilane (δ = 0.00) as an internal standard. NMR multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, and m = multiplet signals. IR spectra were recorded on a 398 spectrophotometer. Melting points were determined with a Kofler bench.
5.2. Preparation of dicoumarols 2
A mixture of 4-hydroxycoumarin 1 (20 mmol, 3.2 g), arylaldehydes 2 (10 mmol), and p-TSA (10 mol%) in H2O (20 mL) was refluxed for an appropriate time mentioned in . The progress of the reaction was monitored by TLC (EtOAc/hexane, 1:1). After completion, the mixture was poured on ice and the precipitate was filtered and purified by recrystallization from EtOH/THF (2:1). In some cases, column chromatography is needed (EtOAc/hexane, 1:1).
5.3. 3-[(4-Ethyl-phenyl)-(4-hydroxy-2-oxo-4a,8adihydro-2H-chromen-3-yl)-methyl]-4-hydroxychromen-2-one (2a)
Yield: 87%; m.p. 250°C. IR ν (cm−1): 3081 (OH); 1652 (CO); 1563 (C=C). 1H NMR (400 MHz, CDCl3) (δ, ppm): 1.23 (t, 3H, Hb); 2.64 (q, 2H, Ha); 6.07 (s, 1H, H12); 7.14 (q, 4H,H 29,28,26,25), 7.41 (d, 4H, H9,21,7,19); 7.62 (td, 2H, H8,20); 8.02 (d, 2H, H10,22), 11.28 (s, 1H,H23); 11.51 (s, 1H, H31). 13C NMR (100 MHz, CDCl3) (λ, ppm): 15.53 (Cb); 28.45 (Ca); 36.01 (C12); 104.16 (C3,13); 105.89 (C5,17); 116.73 (C7,19); 124.49 (C10,22); 124.95 (C9,21); 126.53 (C28); 128.23 (C26); 132.38 (C8), 132.90 (C20); 142.91 (C29); 152.42 (C25); 152.62 (C27); 164.63 (C24); 165.78 (C6,16); 166.96 (C2,14); 169.39 (C4,18).
5.4. 4-Hydroxy-3-[(4-hydroxy-2-oxo-4a,8a-dihydro-2Hchromen-3-yl)-p-tolyl-methyl]-chromen-2-one (2b)
Yield: 83%; m.p. 249°C. IR ν (cm−1): 3063 (OH); 1665 (CO); 1606 (C=C). 1H NMR (400 MHz, CDCl3) (δ, ppm): 2.34 (s, 3H, Ha); 6.06 (s, 1H, H12); 7.12 (q, 4H, H 29,28,27,26), 7.41 (d, 4H, H9,21,7,19); 7.62 (td, 2H, H8,20); 8.03 (d, 2H, H10,22), 11.41 (d, 2H, H23,31). 13C NMR (100 MHz, CDCl3) (δ, ppm): 21.10 (Ca); 35.99 (C12); 104.21 (C3,13); 105.88 (C5,17); 116.75 (C7,19); 124.50 (C28); 124.97 (C10,22); 126.49 (C29); 129.46 (C9,21); 132.16 (C27), 132.92 (C8,20); 136.60 (C26); 152.64 (C24); 164.66 (C25); 165.83 (C6,16); 166.97 (C2,14); 169.42 (C4,18).
5.5. 3-[(4-Chloro-phenyl)-(4-hydroxy-2-oxo-4a,8adihydro-2H-chromen-3-yl)-methyl]-4-hydroxychromen-2-one (2c)
Yield: 92%; m.p. 252 °C. IR ν (cm−1): 3066 (OH); 1660 (CO); 1557 (C=C). 1H NMR (400 MHz, CDCl3) (δ, ppm): 6.02 (s, 1H, H12); 7.13 (dd, 2H,H 29, 25), 7.25–7.28 (m, 2H, H28,26); 7.40 (d, 4H, H9,21,7,19); 7.62 (td, 2H, H8,20), 8.02 (dd, 2H, H10,22), 11.30 (s, 1H, H23), 11.53 (s, 1H, H31). 13C NMR (100 MHz, CDCl3) (δ, ppm): 35.94 (C12); 103.83 (C3); 105.38 (C13); 116.47 (C5); 116.80 (C17); 116.95 (C7,19); 124.54 (C10,22); 125.11 (C9,21); 128.10 (C8), 128.89 (C20); 132.85 (C28,26); 133.15 (C29); 133.98 (C25); 152.41 (C27); 152.66 (C24); 164.75 (C6,16), 166.12 (C2); 166.96 (C14); 169.32 (C4,18).
5.6. 4-Hydroxy-3-[(4-hydroxy-2-oxo-4a,8a-dihydro-2H-chromen-3-yl)-(2,4-dimethoxy-phenyl)-methyl]-chromen-2-one (2d)
Yield: 75%; m.p. 216°C. IR ν (cm−1): 3077 (OH); 1679 (CO); 1548 (C=C). 1H NMR (400 MHz, CDCl3) (δ, ppm): 3.52 (s, 3H, Hd); 3.68 (s, 3H, Hb); 6.14 (s, 1H, H12); 6.33–6.37 (m, 2H,H 29,28,27,26), 7.10 (dd, 1H, H9,21,7,19); 7.18–7.22 (m, 4H, H8,20); 7.45 (td, 2H, H10,22), 7.80 (dd, 2H, H23,31). 13C NMR (100 MHz, CDCl3) (δ, ppm): 32.31 (C12); 54.95 (Cd); 55.48 (Cb); 98.50 (C26); 103.48 (C3,13); 103.93 (C28); 115.29 (C24); 120.23 (C5,17); 122.69 (C7,19); 123.19 (C10), 123.98 (C22); 129.23 (C9,21); 130.44 (C8,20); 152.33 (C29); 157.92 (C6); 158.33 (C16); 164.09 (C2,14); 167.14 (C4,18).
5.7. 4-Hydroxy-3-[(4-hydroxy-2-oxo-4a,8a-dihydro-2Hchromen-3-yl)-(4-nitro-phenyl)-methyl]-chromen-2-one (2e)
Yield: 94%; m.p. 237°C. IR ν (cm−1): 3070 (OH); 1661 (CO); 1548 (C=C). 1H NMR (400 MHz, CDCl3) (δ, ppm): 6.12 (s, 1H, H12); 7.42 (t, 6H,H 9,21,7,19,29,25), 7.67 (t, 2H, H8,20); 8.00 (d, 1H, H10); 8.08 (d, 1H, H22), 8.19 (d, 2H, H28,26), 11.37 (s, 1H, H23), 11.56 (s, 1H, H31). 13C NMR (100 MHz, CDCl3) (δ, ppm): 36.62 (C12); 103.37 (C3,13); 104.86 (C5,17); 116.33 (C7); 116.77 (C19); 116.83 (C10); 116.90 (C22); 123.96 (C28,26); 124.56 (C9), 124.60 (C21); 125.26 (C13); 125.32 (C25); 127.68 (C8); 133.46 (C20); 143.47 (C27), 146.97 (C24), 152.42 (C6); 152.67 (C16); 164.92 (C2); 166.52 (C14); 167.08 (C4); 169.19 (C18).
5.8. 3-[(4-Bromo-phenyl)-(4-hydroxy-2-oxo-4a,8a-dihydro-2H-chromen-3-yl)-methyl]−4-hydroxychromen-2-one (2f)
Yield: 85%; m.p. 265°C. IR ν (cm−1): 3070 (OH); 1661 (CO); 1548 (C=C). 1H NMR (400 MHz, CDCl3) (δ, ppm): 6.02 (s, 1H, H12); 7.10 (dd, 2H,H29,25), 7.41–7.45 (m, 6H, H9,21,8,20,7,19); 7.64 (td, 2H, H28,26); 8.04 (dd, 2H, H10,22), 11.31 (s, 1H, H23), 11.53 (s, 1H, H31). 13C NMR (100 MHz, CDCl3) (δ, ppm): 36.02 (C12); 103.77 (C3,13); 106.33 (C5); 116.80 (C17); 120.94 (C7); 124.56 (C19); 125.13 (C27); 128.48 (C10,22); 131.85 (C9,21), 133.18 (C8,20); 134.57 (C29,25); 152.42 (C28,26); 152.68 (C24); 164.78 (C6, 16); 166.17 (C2), 166.98 (C14), 169.34 (C4,18).
5.9. 3-[(4-Hydroxy-5-ethoxy-phenyl)-(4-hydroxy-2-oxo-4a,8a-dihydro-2H-chromen-3-yl)-methyl]−4-hydroxychromen-2-one (2g)
Yield: 79%; m.p. 215°C. IR ν (cm−1): 3074 (OH); 1639 (CO); 1563 (C=C). 1H NMR (400 MHz, CDCl3) (δ, ppm): 1.36 (t, 3H, Hc), 3.95 (q, 2H, Hb), 6.06 (s, 1H, H12); 6.66–6.73 (m, 2H,H29,25), 6.87 (d, 1H, H28); 7.41 (d, 4H, H9,21,7,19); 7.63 (td, 2H, H8,20), 8.02 (s,2 H, H10,22), 11.40 (d, 2H, H23,31). 13C NMR (100 MHz, CDCl3) (δ, ppm): 14.85 (Cc); 35.94 (C12); 64.82 (Cb); 110.64 (C3,13); 114.57 (C28,25); 116.78 (C5,17, 7,19); 119.61 (C29,10,22); 124.51 (C9,21,8,20); 125.01 (C24); 126.88 (C27), 132.95 (C26); 144.92 (C2,14); 146.07 (C6,16); 152.53 (C4,18).
5.10. Synthesis of epoxydicoumarins
Dicoumarols were dissolved by heating in acetic anhydride. The reaction mixture was boiled for 2 h, kept in the refrigerator for 24 h, and the precipitate was filtered off and dried.
5.11. 7-(4-Ethoxy-phenyl)-7,14b-dihydro-4aH-pyrano[3,2-c;5,6-c’]dichromene-6,8-dione (3a)
Yield: 96%; m.p. 269°C. IR ν (cm−1): 1666 (CO); 1576 (C=C). 1H NMR (400 MHz, CDCl3) (δ, ppm): 1.16 (t, 3H, Hb); 2.57 (q, 2H, Ha); 5.14 (s, 1H, H12); 7.10 (d, 2H,H28,26), 7.34–7.39 (m, 4H, H29,25,7,19); 7.45 (t, 2H, H9,21); 7.64 (td, 2H, H8,20), 8.09 (dd, 2H, H10,22). 13C NMR (100 MHz, CDCl3) (λ, ppm): 15.48 (Cb); 28.61 (Ca); 34.63 (C12); 106.68 (C3,13); 113.65 (C5,17); 117.29 (C7,19); 122.46 (C10,22); 124.72 (C9,21); 128.21 (C28); 128.92 (C26); 132.84 (C8,7’), 138.44 (C29,25); 143.80 (C24,27); 152.86 (C6,16); 153.58 (C4,18); 160.31 (C2,14).
5.12. 7-(p-Tolyl)-7,14b-dihydro-4aH-pyrano[3,2-c;5,6-c’]dichromene-6,8-dione (3b)
Yield: 88%; m.p. 254°C. IR ν (cm−1): 1661 (CO); 1562 (C=C). 1H NMR (400 MHz, CDCl3) (δ, ppm): 2.27 (s, 3H, Ha); 5.12 (s, 1H, H12); 7.08 (d, 2H,H 28,27), 7.33 (d, 2H, H26,29); 7.38 (d, 2H, H7,19); 7.45 (t, 2H, H9,21); 7.64 (td, 2H, H8,20), 8.09 (dd, 2H, H10,22). 13C NMR (100 MHz, CDCl3) (δ, ppm): 21.24 (Ca); 34.64 (C12); 106.65 (C3,13); 113.63 (C5,17); 117.30 (C7,19); 122.46 (C28); 124.72 (C10,22); 128.91 (C29); 129.41 (C9,21); 132.85 (C27), 137.56 (C8,20); 138.27 (C24,26,25); 152.87 (C6,16); 153.56 (C4,18); 160.29 (C2,14).
5.13. 7-(4-Chloro-phenyl)-7,14b-dihydro-4aH-pyrano[3,2-c;5,6-c’]dichromene-6,8-dione (3c)
Yield: 77%; m.p. 258°C. IR ν (cm−1): 1665 (CO); 1487 (C=C). 1H NMR (400 MHz, CDCl3) (λ, ppm): 4.88 (s, 1H, H12); 7.32 (d, 2H,H 29,25), 7.43 (d, 2H, H28,26); 7.50–7.55 (m, 4H, H9,21,7,19); 7.77 (td, 2H, H8,20), 8.40 (dd, 2H, H10,22). 13C NMR (100 MHz, CDCl3) (δ, ppm): 51.78 (C12); 122.26 (C3,13); 130.40 (C5,17, 7,19); 134.06 (C10,22); 140.78 (C9,21, 8,20); 142.42 (C28,26); 145.58 (C29,25); 148.32 (C27); 150.72 (C24), 169.54 (C6,16); 170.87 (C4,18); 177.09 (C2,14).
5.14. 7-(2,4-Dimethoxy-phenyl)-7,14b-dihydro-4aH-pyrano[3,2-c;5,6-c’]dichromene-6,8-dione (3d)
Yield: 90%; m.p. 283°C. IR ν (cm−1): 1669 (CO); 1587 (C=C). 1H NMR (400 MHz, CDCl3) (δ, ppm): 3.54 (s, 3H, Hd); 3.74 (s, 3H, Hb); 5.11 (s, 1H, H12); 6.31 (d, 1H,H28), 6.50 (dd, 1H, H26); 7.36 (d, 2H, H7,19); 7.44 (td, 2H, H9,21), 7.53 (d, 1H, H29),), 7.61 (td, 2H, H8,20), 8.08 (dd, 2H, H10,22). 13C NMR (100 MHz, CDCl3) (δ, ppm): 32.43 (C12); 55.43 (Cd); 55.67 (Cb); 99.40 (C26); 104.69 (C3,13); 104.73 (C28); 113.82 (C24); 117.21 (C5,17); 120.19 (C7,19); 122.18 (C10,22); 124.57 (C9,21), 132.44 (C8,20); 133.49 (C29); 152.81 (C6,16); 154.12 (C27); 159.24 (C25); 160.51 (C4,18), 160.80 (C2,14).
5.15. 7-(4-Nitro-phenyl)-7,14b-dihydro-4aH-pyrano[3,2-c;5,6-c’]dichromene-6,8-dione (3e)
Yield: 79%; m.p. 285°C. IR ν (cm−1): 1666 (CO); 1608 (C=C). 1H NMR (400 MHz, CDCl3) (δ, ppm): 5.03 (s, 1H, H12); 7.51–7.58 (6, 4H,H 9,21,7,19), 7.72–7.81 (m, 4H, H8,20,29,25); 8.12 (d, 2H, H10,22); 8.42 (dd, 1H, H28,26). 13C NMR (100 MHz, CDCl3) (λ, ppm): 35.02 (C12); 104.21 (C3,13); 112.91 (C5,17); 116.68 (C7,19); 123.26 (C10,22); 123.49 (C28,26); 125.07 (C9,21); 130.64 (C29,25); 133.49 (C8,20), 146.66 (C27); 148.76 (C24); 152.20 (C6,16); 153.75 (C4,18); 159.68 (C2,14).
5.16. 7-(4-Chloro-phenyl)-7,14b-dihydro-4aH-pyrano[3,2-c;5,6-c’]dichromene-6,8-dione (3f)
Yield: 80%; m.p. 304°C. IR ν (cm−1): 1665 (CO); 1606 (C=C). 1H NMR (400 MHz, CDCl3) (δ, ppm): 5.13 (s, 1H, H12); 7.32–7.49 (m, 8H,H9,21,8,20,7,19,29,25), 7.67 (t, 2H, H28,26); 8.10 (d, 2H, H10,22). 13C NMR (100 MHz, CDCl3) (δ, ppm): 34.39 (C12); 106.65 (C3,13); 113.13 (C5,17); 117.10 (C7,19); 122.21 (C27); 124.54 (C10,22); 130.50 (C9,21); 131.50 (C8,20); 132.83 (C29,25), 139.89 (C28,26,24); 152.63 (C6,16); 153.51 (C4,18); 159.89 (C2,14).
5.17. 7-(4-Hydroxy-5-methoxy-phenyl)-7,14b-dihydro-4aH-pyrano[3,2-c;5,6-c’]dichromene-6,8-dione (3g)
Yield: 93%; m.p. 295°C. IR ν (cm−1): 1667 (CO); 1606 (C=C). 1H NMR (400 MHz, CDCl3) (δ, ppm): 1.37 (t, 3H, Hc), 4.10 (q, 2H, Hb), 5.15 (s, 1H, H12); 6.74 (dd, 1H,H29), 6.87 (d, 1H, H25); 7.29 (d, 1H, H26); 7.93 (dd, 2H, H9,21), 7.45 (td,2 H, H7,19), 7.64 (td, 2H, H8,20), 8.09 (dd,2 H, H10,22). 13C NMR (100 MHz, CDCl3) (δ, ppm): 14.75 (Cc); 34.64 (C12); 64.57 (Cb); 106.21 (C3,13); 113.50 (C29); 115.46 (C26); 117.31 (C27); 119.78 (C5,17); 122.43 (C7,19); 122.71 (C25); 124.79 (C10,22), 133.01 (C9,21); 139.58 (C8,20); 139.61 (C24); 150.36 (C27), 152.83 (C28), 153.82 (C6,16); 160.28(C4,18); 169.03 (C2,14).
6. Materials and methods
6.1. Biological activities
6.1.1. Antimicrobial activity
6.1.1.1. Microorganisms, media and growth conditions
For the antibacterial activity assays, the microorganisms used as indicators were the two Gram-positive bacteria Staphylococcus aureus (S. aureus) ATCC6538 and Listeria monocytogenes (L. monocytogenes) ATCC 19117 and the three Gram-negative bacteria Escherichia coli (E. coli) ATCC 8739, Pseudomonas aeruginosa (P. aeruginosa) ATCC 49189 and Salmonella typhimurium (S. typhimurium) ATCC 14028. Antifungal activity was determined against C. albicans ATCC 10231. All indicator microorganisms were obtained from the International Culture Collections (ATCC) and local culture collection of the Laboratory of Microorganisms and Enzymatic Biotechnology and Biomolecules of the Center of Biotechnology of Sfax-Tunisia.
The indicator microorganisms were grown overnight in Luria Bertani (LB) medium (g L−1: peptone 10; yeast extract 5 and NaCl 5, pH 7.2) under aerobic conditions and constant agitation (200 rpm) at 30°C for L. monocytogenes ATCC 19117 and S. typhimurium ATCC 14028 and at 37°C for E. coli ATCC 8739, S. aureus ATCC6538 and P. aeruginosa ATCC 49189 and then diluted 1:100 in LB media and incubated for 5 h under constant agitation (200 rpm) at the appropriate temperature. albicans ATCC 10231 was cultured at 30°C on Sabouraud medium (g L−1: dextrose 40, peptone 10, pH 5.6) under aerobic conditions and constant agitation (200 rpm) and then diluted 1:50 in Sabouraud medium and incubated for 5 h under constant agitation (200 rpm) at 30°C.
6.1.1.2. Agar well diffusion method
According to Guven et al. (Citation39), the agar well diffusion method was employed for the determination of the antimicrobial activity of the synthesized compounds with some modifications. Briefly, 15 mL of molten agar (45°C) was poured into sterile Petri dishes (Ø 90 mm). Fifty microliters of 5 h old culture of the five tested bacteria and 100 μL of 5-h-old culture of the fungus C. albicans were evenly spread onto the surface of the agar plates of LB agar medium for bacteria and Sabouraudagar medium for C. albicans. Once the plates had been aseptically dried, 5 mm wells were punched into the agar with a sterile cork borer. Then, 100 μL of a solution of each synthesized compound at 500 μg mL−1 was dispensed into the wells. After staying at 4°C for 2 h, the plates were incubated at the appropriate temperature for 24 h for bacterial strains and 48 h for C. albicans. The antimicrobial activity was assayed by measuring the diameter of the inhibition zone formed around each well in millimeters.
6.1.1.3. Minimum inhibitory concentration (MIC)
MIC of the synthesized compounds and the standards ampicillin, kanamycin and fluconazole (stock solutions at 20 mg/mL−1) against the five tested bacteria and the fungus C. albicans were determined according to Sellem et al. (Citation40). The test was performed in sterile 96-well microplates with a final volume in each microplate well of 100 μL. Stock solutions of synthesized compounds and standards were serially diluted with dimethyl sulfoxide (DMSO). To each test well, a cell suspension was added to a final inoculum concentration of 106 CFU mL−1 of indicator microorganism. The plates were then incubated at appropriate growth conditions of the corresponding indicator microorganism. The MIC was defined as the lowest concentration of the synthesized compounds and standards at which the microorganism did not demonstrate visible growth after incubation. Twenty-five microliters of thiazolyl blue tetrazolium bromide (MTT) at 0.5 mg mL−1 was added to the wells and incubated at room temperature for 30 min. The colorless tetrazolium salt acted as an electron acceptor and was reduced to a red-colored formazan product by the indicator microorganisms. When microbial growth was inhibited, the solution in the well remained clear after incubation with MTT.
For the antimicrobial activity determination (inhibition zones and CMIs), each experiment was carried out simultaneously three times under the same conditions. The obtained diameters of the inhibition zones reported in mm and the MIC values reported in μg mL−1 were quite similar, and the reported results are the average of the three experiments.
6.1.2. Antioxidant activity
Antioxidant activity was expressed as EC50 (the concentration that causes 50% of the effect). The free radical scavenging activity of the synthesized compounds was assessed by 2.2-diphenyl-1-picrylhydrazyl (DPPH) and 2.2'-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) radical assays. Butylated hydroxytoluene (BHT) was used as a control, and it is well known as a strong antioxidant compound.
6.1.2.1. (DPPH) radical scavenging activity
The DPPH assay is the simplest and most widely reported method for screening antioxidant activity. The procedure involves measuring the decrease in absorbance of DPPH at its absorption maximum of 517 nm. This assay determines the scavenging of stable radical species according to the method of Kirby and Schmidt (Citation41) with slight modifications. Briefly, the synthesized compounds were dissolved in dimethylsulfoxide (DMSO)/water (1/9; v/v) and diluted with ultrapure water to different concentrations (1, 0.5, 0.250, 0.125, 0.0625, 0.03125 mg mL−1). Then, 500 µL of a 4% (w/v) solution of DPPH radicals in ethanol was mixed with 500 µL of the samples. The mixture was incubated for 30 min in the dark at room temperature. The scavenging capacity was determined spectrophotometrically by monitoring the decrease in absorbance at 517 nm against a blank. The percentage of antiradical activity (% ArA) was calculated as follows:
6.1.2.2. (ABTS) radical scavenging activity
This manipulation was carried out according to the protocol proposed by Re et al. (Citation42) with some modifications. A total of 20 μL of a solution of synthesized compounds or control was mixed with 18 μL of ABTS solution and mixed vigorously for 30 s. A control consisting of ethanol and ultrapure water was prepared (5:5) v/v to which the previous solution was added. The samples were white with ethanol and ultrapure water (5:5) v/v. Samples and controls were incubated for 6 min in the dark, and then the OD was measured at 734 nm. The calculation is performed according to the following formula:
Ac: absorbance of the control, Ae: absorbance of the sample.
For the antioxidant activity detection (DPPH and ABTS), each experiment was carried out simultaneously three times under the same conditions. The obtained values in μg mL−1 were quite similar, and the reported results are the average of the three experiments.
6.1.3. Molecular docking
Molecular docking is a very valuable and popular tool used for drug discovery and design to create new therapies for diseases. This technique is used to predict and examine the conformations and binding interactions of a ligand in the active site of a target enzyme. Herein, molecular docking calculations were carried out to evaluate in silico the antimicrobial and antifungal activities of the synthesized compounds 2a–g and 3a–g. DNA gyrase and Mycobacterium tuberculosis-CYP51 are selected to explore the antimicrobial and antifungal activities, respectively. DNA gyrase protein has long been known as an efficient target for antibacterial drugs evaluation such as nalidixicacid and ciprofloxacin, and its inhibition results in the disruption of DNA synthesis and, subsequently, cell death (Citation35,Citation43).
Additionally, CYP51 is considered the key target enzyme of azole antifungal agents. Exposure of Malassezia species to azole antifungal agents leads to a depletion of ergosterol and an accumulation of 14α-methylated sterols, which interferes with the functions of ergosterol in fungal membranes, disorders the structure of the membrane and other functions, and causes inhibition of fungal growth.
The crystal structures of DNA gyrase (PDB ID: 5BTC) and Mycobacterium tuberculosis-CYP51 (PDB ID: 1EA1), with their co-crystallized ligands ciprofloxacin (CPF) and fluconazole (TPF), are retrieved from the Protein Data Bank. Each co-crystallized ligand has been self-docked to check the accuracy of the docking results. Ciprofloxacin is a bactericidal antibiotic of the fluoroquinolone drug class. In addition, fluconazole is used to prevent and treat a variety of fungal and yeast infections. It belongs to a class of drugs called azole antifungals.
Author contributions
Naceur Hamdi conceptualized the project’s primary principles, drafted the analysis methods, conducted the scientific investigation, formal analysis, data curation, and acquired funding. Donia Bensalah, Sadeq M. Al-Hazmy worked on the project's concept, design, and monitoring and evaluation throughout the project. Naceur Hamdi reviewed and edited the first draft of the paper, which was written by Naceur Hamdi and Sadeq M. Al-Hazmy. Both the authors contributed to the final revision of the manuscript.
Availability of data and materials
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Acknowledgments
The authors extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education and Qassim University, Saudi Arabia, for funding this research work through the project number (QU-IF-05-04-28440). The authors also thank Qassim University for its technical support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Karimi, A.R.; Sedaghatpour, F. Novel Mono- and Bis(Spiro-2-Amino-4H-Pyrans): Alum-Catalyzed Reaction of 4-Hydroxycoumarin and Malononitrile with Isatins, Quinones, or Ninhydrin. Synthesis. (Mass) 2010, 2010 (10), 1731–1735. doi:10.1055/s-0029-1219748.
- Musa, M.A.; Cooperwood, J.S.; Khan, M.O.F. A Review of Coumarin Derivatives in Pharmacotherapy of Breast Cancer. Curr. Med. Chem. 2008, 15, 2664–2679. doi:10.2174/092986708786242877.
- Kontogiorgis, C.; Detsi, A.; Hadjipavlou-Litina, D. Coumarinbased Drugs: A Patent Review (2008–Present). Expert Opin. Ther. Pat. 2012, 22, 437–454. doi:10.1517/13543776.2012.678835.
- (a) Lin, Y.; Shen, X.; Yuan, Q.; Yan, Y. Microbial Biosynthesis of the Anticoagulant Precursor 4-Hydroxycoumarin. Nat. Commun. 2013, 4, 3603/1–3603/8. (b) Dicks, A.P. A Review of Aqueous Organic Reactions for the Undergraduate Teaching Laboratory. Green Chem Lett Rev 2009, 2 (1), 9–21. doi: 10.1080/17518250902820182.
- Beinema, M.; Brouwers, J.R.B.J.; Schalekamp, T.; Wilffert, B. Pharmacogenetic Differences Between Warfarin, Acenocoumarol and Phenprocoumon. Thromb. Haemostasis 2008, 100, 1052–1057. doi:10.1160/TH08-04-0116.
- Madari, H.; Panda, D.; Wilson, L.; Jacobs, R.S. A Unique Microtubule Stabilizing Natural Product that is Synergistic with Taxol. Cancer Res. 2003, 63, 1214–1220.
- Cherkupally, S.R.; Mekala, R. P-TSA Catalyzed Facile and Efficient Synthesis of Polyhydroquinoline Derivatives Through Hantzsch Multi-Component Condensation. Chem. Pharm. Bull. 2008, 56, 1002–1004. doi:10.1248/cpb.56.1002.
- Bellis, D.M.; Spring, M.S.; Stoker, J.R. The Biosynthesis of Dicoumarol. Biochem. J. 1967, 103, 202–206. doi:10.1042/bj1030202.
- (a) Hamdi, N.; Puerta, M.C.; Valerga, P. Synthesis, Structure, Antimicrobial and Antioxidant Investigations of Dicoumarol and Related Compounds. Eur. J. Med. Chem. 2008, 43, 2541–2548. (b) Karmakar, B.; Nayak, A.; Banerji, J. Sulfated Titania Catalyzed Water Mediated Efficient Synthesis of Dicoumarols. A Green Approach. Tetrahedron Lett. 2012, 53, 2541–2548. doi:10.1016/j.ejmech.2008.03.038.
- (a) Kolos, N.N.; Gozalishvili, L.L.; Yaremenko, F.G.; Shishkin, O.V.; Shishkina, S.V.; Konovalova, I.S. Aroylbis(4-Hydroxycoumarin-3-yl)Methanes in Reactions with 1,2-Diaminobenzenes. Russ. Chem. Bull. 2007, 56, 2277–2283. (b) Sadeghi, B. Synthesis of Silica-Supported Perchloric Acid Nanoparticles (HClO4-SiO2 NPs). Efficient Synthesis of Biscoumarin Derivatives in Water. J. Chem. Res. 2013, 37, 171–173.
- (a) Siddiqui, Z.N.; Farooq, F. Zn(Proline)2: A Novel Catalyst for the Synthesis of Dicoumarols. Catal. Sci. Technol. 2011, 1, 810–816. (b) Rammohan, A.; Munikishore, R.; Gunasekar, D.; Deville, A.; Bodo, B. A new di-C-Prenylated Coumarin from Sophora Interrupta. Nat. Prod. Res. 2015, 29 (1), 82–85. doi: 10.1080/14786419.2014.959010.
- Ziarani, G.M.; Hajiabbasi, P. Recent Application of 4-Hydroxycoumarin in Multi-Component Reactions. Heterocycles 2013, 87, 1415–1439. doi:10.3987/REV-13-768.
- Weinmann, I. History of the Development and Applications of Coumarin and Coumarin-Related Compounds. In Coumarins: Biology, Applications and Mode of Action: O'Kennedy, R., Thornes, R.D., Eds.; John Wiley & Sons, Inc.: Los Angeles, CA, USA, 1997; pp 1–22.
- Marcu, M.G.; Schulte, W.T.; Neckers, L. Novobiocin and Related Coumarins and Depletion of Heat Shock Protein 90-Dependent Signaling Proteins. J. Natl. Cancer Inst. 2000, 92, 242–248. doi:10.1093/jnci/92.3.242.
- Borges, F.; Roleira, F.; Milhazes, N.; Santana, L.; Uriarte, E. Simple Coumarins and Analogues in Medicinal Chemistry: Occurrence, Synthesis and Biological Activity. Curr. Med. Chem. 2005, 12, 887–916. doi:10.2174/0929867053507315.
- Chiang, C.C.; Hsu, L.Y.; Tsai, H.J.; Yao, C.W.; Chang, T.C. Synthesis and Antimicrobial Evaluation of Coumarin Derivatives. J. Chiong Chang Inst. Techol 2008, 37, 15–22.
- Ćavar, S.; Kovač, F.; Maksimović, M. Synthesis and Antioxidant Activity of Selected 4-Methylcoumarins. Food Chem. 2009, 117, 135–142. doi:10.1016/j.foodchem.2009.03.087.
- Jung, J.C.; Park, O.S. Synthetic Approaches and Biological Activities of 4-Hydroxycoumarin Derivatives. Molecules 2009, 14, 4790–4803. doi:10.3390/molecules14114790.
- Symeonidis, T.; Chamilos, M.; Hadjipavlou-Litina, D.J.; Kallitsakis, M.; Litinas, K.E. Synthesis of Hydroxycoumarins and Hydroxybenzo[f]- or [h]Coumarins as Lipid Peroxidation Inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 1139–1142. doi:10.1016/j.bmcl.2008.12.098.
- Kostova, I. Studying Plant-Derived Coumarins for Their Pharmacological and Therapeutic Properties as Potential Anticancer Drugs. Expert Opin. Drug Discovery 2007, 2, 1605–1618. doi:10.1517/17460441.2.12.1605.
- Nolan, A.K.; Doncaster, R.J.; Dunstan, S.M.; Scot, A.K.; Frenkel, D.; Siegel, D.; Ross, D.; Barnes, J.; Levy, C., Leys, D., et al. Synthesis and Biological Evaluation of Coumarin-Based Inhibitors of NAD(P)H: Quinone Oxidoreductase-1 (NQO1). J. Med. Chem. 2009, 52, 7142–7156. doi:10.1021/jm9011609.
- Mahajan, D.H.; Pannecouque, C.; De Clercq, E.; Chikhalia, K.H. Synthesis and Studies of New 2-(Coumarin-4-Yloxy)-4,6-(Substituted)-s-Triazine Derivatives as Potential Anti-HIV Agents. Arch. Pharm. 2009, 342, 281–290. doi:10.1002/ardp.200800149.
- Zhao, H.; Neamati, N.; Hong, H.; Mazumder, A.; Wang, S.; Sunder, S.; Milne George, W.A.; Pommier, Y.; Burke, T.R., Jr. Coumarin-Based Inhibitors of HIV Integrase. J. Med. Chem. 1997, 40, 242–249. doi:10.1021/jm960450v.
- Hooper, C.; Wolfson, S.; McHugh, L.; Winters, B.; Swartz, N. Effects of Novobiocin, Coumermycin A1, Clorobiocin, and their Analogs on Escherichia coli DNA Gyrase and Bacterial Growth. Antimicrob. Agents Chemother. 1982, 22 (4), 662–71. doi:10.1128/AAC.22.4.662.
- Pan, Y.; Liu, T.; Wang, X.; Sun, J. Research Progress of Coumarins and their Derivatives in the Treatment of Diabetes. J. Enzyme Inhib. Med. Chem. 2022, 37 (1), 616–628. doi:10.1080/14756366.2021.2024526.
- Slimani, I.; Hamzaoui, S.; Mansour, L.; Harrath, A.H.; Hamdi, N. One-Pot, Simple and Efficient Synthesis of Novel Bioactive 4-Aryl-1,2-Dihydro-6-(4-Hydroxy-2-oxo-2H-Chromen-3-yl)-2-Oxopyridin-3-Carbonitriles via Multi-Component Approach. J. King Saud Univ. Sci. 2020, 32, 1212–1217. doi:10.1016/j.jksus.2019.11.023.
- Le Bras, G.; Hamze, A.; Messaoudi, S.; Provot, O.; P.-B.L. Calvez; Brion, J.-D.; Alami, M. Synthesis of Isocoumarin via pTSA-Catalyzed Annulation of Diarylalkynes. Synthesis. 2008, 40, 1607–1611.
- da Silva, C.R.B.; Gonçalves, V.L.C.; Lachter, E.R.; Mota, C.J.A. Etherification of Glycerol with Benzyl Alcohol Catalyzed by Solid Acids. J. Braz. Chem. Soc. 2009, 20, 201–204. doi:10.1590/S0103-50532009000200002.
- Mobinikhaledi, A.; Moghanian, H.; Deinavizadeh, M. pTSA-catalyzed Condensation of Xylenols and Aldehydes Under Solvent-Free Conditions: One-Pot Synthesis of 9H-Xanthene or Bisphenol Derivatives. C. R. Chim. 2013, 16, 1035–1041. doi:10.1016/j.crci.2013.03.008.
- Chanda, A.; Fokin, V.V. Organic Synthesis “on Water”. Chem. Rev. 2009, 109, 725–748. Heravi, M.M.; Sadjadi, S.; Mokhtari Haj, N.; Oskooie, H.A.; Bamoharram, F.F. Role of Various Heteropolyacids in the Reaction of 4-Hydroxycoumarin, Aldehydes and Ethylcyanoacetate. Catal. Commun. 2009, 10, 725–748. doi:10.1021/cr800448q.
- Heravi, M.M.; Sadjadi, S.; Mokhtari Haj, N.; Oskooie, H.A.; Bamoharram, F.F. Role of Various Heteropolyacids in the Reaction of 4-Hydroxycoumarin, Aldehydes and Ethylcyanoacetate. Catal. Commun. 2009, 10, 1643–1646. doi:10.1016/j.catcom.2009.04.031.
- Singh, P.; Kumar, P.; Katyal, A.; Kalra, R.; Dass, S.K.; Prakash, S.; Chandra, R. Phosphotungstic Acid: An Efficient Catalyst for the Aqueous Phase Synthesis of bis-(4-Hydroxycoumarin-3-yl)Methanes. Catal. Lett. 2010, 134, 303–308.
- Zhao, H.; Yan, B.; Peterson, L.B.; Blagg, B.S.J. 3-Arylcoumarin Derivatives Manifest Anti-Proliferative Activity Through Hsp90 Inhibition. ACS. Med. Chem. Lett. 2012, 3 (4), 327–331. doi:10.1021/ml300018e.
- Rehman, S.; Ikram, M.; Baker, R.J.; Zubair, M.; Azad, E.; Min, S.; Riaz, K.; Mok, K.H.; Rehman, S.-U. Synthesis, Characterization, in Vitro Antimicrobial, and U2OS Tumoricidal Activities of Different Coumarin Derivatives. BMC Chemistry 2013, 7, 68. doi:10.1186/1752-153X-7-68.
- Jiang, Y.; Zhang, J.; Cao, Y.; Xiaoyun, C.; Zou, Y.; Wu, Q.; Zhang, D.; Jiang, Y.; Sun, Q. Synthesis, in Vitro Evaluation and Molecular Docking Studies of New Triazole Derivatives as Antifungal Agents. Bioorg. Med. Chem. Lett. 2011, 21 (15), 4471–4475. doi:10.1016/j.bmcl.2011.06.008.
- Bano, S.; Alam, M.S.; Javed, K.; Dudeja, M.; Kumar Das, A.; Dhulap, A. Synthesis, Biological Evaluation and Molecular Docking of Some Substituted Pyrazolines and Isoxazolines as Potential Antimicrobial Agents. Eur. J. Med. Chem. 2015, 95, 96–103. doi:10.1016/j.ejmech.2015.03.031.
- http://www.rcsb.org.
- Molecular Operating Environment (MOE), 2015.10; Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite 910, Montreal, QC, Canada, H3A 2R7, 2015.
- Güven, K.; Yücel, E.; Cetintaş, F. Antimicrobial Activities of Fruits of Crataegus. and Pyrus. Species. Pharm. Biol. 2006, 44 (2), 79–83. doi:10.1080/13880200600591253.
- Sellem, I.; Kaaniche, F.; Chakchouk, A.M.; Mellouli, L. Anti-Oxidant, Antimicrobial and Anti-Acetylcholinesterase Activities of Organic Extracts from Aerial Parts of Three Tunisian Plants and Correlation with Polyphenols and Flavonoids Contents. Bangladesh J Pharmacol 2016, 11 (2), 531–544. doi:10.3329/bjp.v11i2.26001.
- Kirby, A.J.; Schmidt, R.J. The Antioxidant Activity of Chinese Herbs for Eczema and of Placebo Herbs—I. J. Ethnopharmacol. 1997, 56 (2), 103–108. doi:10.1016/S0378-8741(97)01510-9.
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant Activity Applying an Improved ABTS Radical Cation Decolorization Assay. Free Radical Biol. Med. 1999, 26 (9–10), 1231–1237. doi:10.1016/S0891-5849(98)00315-3.
- Bano, S.; Alam, M.S.; Javed, K.; Dudeja, M.; Kumar Das, A.; Dhulap, A. Synthesis, Biological Evaluation and Molecular Docking of Some Substituted Pyrazolines and Isoxazolines as Potential Antimicrobial Agents. Eur. J. Med. Chem 2015, 95, 4471–4475. doi:10.1016/j.bmcl.2011.06.008.