
ABSTRACT
Reductive coupling reactions are a facile and versatile way to make carbon – carbon bonds. However, most methods within this class of reactions makes use of reprotoxic, amide-based solvents. Mechanochemistry, a method that does not always require solvent, has been used for reductive coupling methods but still reports the use of at least some amide-based solvent for productive outcomes. This work presents the reactivity trends for the reductive homocoupling of aryl iodides when using various amounts of N,N-dimethylformamide (a commonly used amide-based solvent) and dimethyl carbonate (a greener alternative). The results of these experiments show that dimethyl carbonate may be used in sub-stoichiometric amounts with equal or greater efficiency to N,N-dimethyl formamide. Further, cross-electrophile coupling (XEC) is explored with various solvents, showing that N-butyl pyrrolidinone is efficient as a liquid assisted grinding solvent. These experiments show that under mechanochemical conditions, safer solvents are able to be used with no loss of efficacy for reductive coupling reactions.
GRAPHICAL ABSTRACT
Introduction
Though scientists have conducted chemical reactions by mechanical means as early as the 1800s, mechanochemistry has been intensely studied for organic reactions only since the early 2000s (Citation1). Part of the reason for this increased interest in mechanochemistry for organic synthesis is due to the method’s potential to reduce waste. Mechanochemical reactions do not necessarily require solvent and when they do, the amount of solvent added can be minimal (Citation2). With this amplified attention on mechanochemistry for sustainability, research has progressed so that mechanochemistry can even be used to demonstrate the other twelve principles of green chemistry (Citation3). Another reason for mechanochemistry’s popularity is that this activation technique has been shown to offer different reactivity, and therefore different product outcomes, than other methods (Citation4). Consequently, a plethora of organic reactions have been shown to be successful by mechanochemical activation (Citation5). One such area of organic synthesis that has been studied extensively under mechanochemical conditions is nucleophilic coupling (Citation6).
While nucleophilic coupling has afforded the synthetic community with effective methods to make C–C bonds, this type of synthesis does suffer from drawbacks. For example, the organometallic reagents used for such methods are inherently reactive with acidic and/or electrophilic groups, limiting their applicability to a wide range of substrates. To overcome the challenges posed by nucleophilic coupling, reductive coupling and cross-electrophile coupling (XEC) has seen significant development and promise in recent years (Citation7). Reductive coupling and XEC are attractive because these couplings avoid stoichiometric organometallic substrates and the undesired reactivity and stoichiometric waste that accompanies them. However, challenges remain in the field of electrophilic coupling. Two of the challenges include activation of metal reductants (typically zinc or manganese) and the often-required use of dipolar non-hydrogen-bond donor solvent. These two problems are well-positioned to be alleviated by mechanochemistry. The activation of zinc and manganese by mechanochemistry has been shown to be successful without the use of additives for Negishi and reductive dimerization processes (Citation8). The use of solvent could also potentially be avoided under mechanochemical conditions. When solvent is needed for a particular mechanochemical reaction, liquid assisted grinding (LAG), can be employed to alter the reaction polarity (Citation9). LAG typically uses a very small amount of solvent and is defined by the ratio of solvent volume to sample weight (η), where η lies in the range of 0–2 μL/mg (Citation10).
Few reports of XEC by mechanochemistry have been published (Citation11), so little is known about this class of reactions with this technique. The methods that have been reported utilize the dipolar non-hydrogen-bond donor solvent N,N-dimethylacetamide (DMA). While the quantity of DMA in these reports fits under the LAG definition, DMA is still a reprotoxic solvent that is considered a substance of very high concern, so it should be eliminated where possible. While their work is a productive start to show that mechanochemistry can be a viable method for cross-electrophile coupling, ideally, a mechanochemical reductive coupling reaction would avoid the use of auxiliary materials that are teratogenic and/or problematic by solvent selection guide standards (Citation12). In this work, we investigate the effect of dipolar non-hydrogen-bond donor solvents on the reductive dimerization of aryl iodide derivatives. In particular, we show that unlike previous reports, mechanochemistry can be effective for these reactions without excess reprotoxic dipolar non-hydrogen-bond donor solvent.
Results and discussion
To begin this work, iodobenzene was used as a substrate to explore the optimization for homocoupling. shows a summary of the control reactions performed. Nickel salts, bipyridine ligands, and reducing metals like manganese and zinc have been used successfully for the reductive homocoupling of several aryl halides (Citation13). Because reductive homocoupling is less studied mechanochemically, we started with similar reaction conditions. Catalytic amount of anhydrous NiCl2, 2,2′-bipyridine, and excess manganese in the form of a shot (an irregularly shaped sheet of metal) were used to start our mechanochemical experiments. The reaction was allowed to run for 16 h at the typical operating global temperature for a mechanochemical reaction in our Spex8000M Mixer Mill (28°C). Typically a dipolar non-hydrogen-bond donor solvent like N,N-dimethylformamide (DMF) or DMA is necessary for successful conversion, but we were gratified to find that a moderate conversion (75%) of the biphenyl product (1b) was achieved in the absence of solvent (entry 1). To determine if the 2,2′-bipyridine and/or the manganese shot could be considered auxiliary, control reactions in the absence of these additives were performed (entries 2–3). Minimal conversion (25%) was achieved in the absence of the ligand (entry 2) and no conversion was observed without the manganese shot (entry 3). Finally, different forms of nickel (pellets, powders, etc.) have shown to be effective for other nickel-catalyzed processes in a high-speed ball mill, so Ni powder was also tested in place of NiCl2 (entry 4) (Citation14). Only a small amount (32%) of 1b was observed, possibly because most of the powder was oxidized and could not be effectively reduced by the manganese shot.
Table 1. Initial conditions for homocoupling and control reactions.
With sufficient conditions determined for aryl iodide homocoupling in the absence of solvent (, entry 1), other substrates were tested. To begin investigating the scope for the solventless conditions, 4-iodotoluene was chosen as the first additional substrate. Under identical conditions to those that were successful for iodobenzene, 4-iodotoluene did not yield product (Scheme 1).
Scheme 1. Reaction with conditions from , entry 1 with 4-iodotoluene (2a).
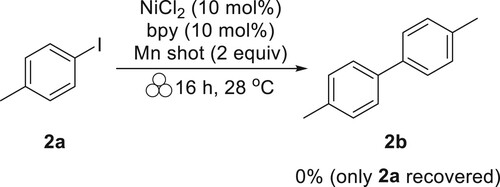
Two differences between the reactions in and Scheme 1 that could cause this difference in outcome were noted. The first difference between 1a and 2a is that 2a has a methyl group in the 4-position, where 1a is unsubstituted. It is possible that the methyl group could influence the reactivity, based on previous mechanochemical Ullman-type reductive coupling (Citation15). The second difference between the substrates is that 1a is a liquid at room temperature, whereas 2a is a solid at room temperature. In mechanochemical nucleophilic coupling reactions, differences in reactivity have been observed for solid versus liquid substrates, where solid substrates have a rate of reaction slower than liquid substrates (Citation16). To determine if the solid state of 4-iodotoluene was inhibiting the reaction efficiency, the same conditions were used, with the addition of various solvents to first dissolve 2a (). The results of these experiments showed that acetonitrile, sometimes used as a greener alternative to DMF (Citation17), converted only a small amount (18%) of the starting material to the substituted biphenyl product (entry 2). The addition of the dipolar non-hydrogen-bond donor solvent DMF was successful at fully converting 2a to the homocoupling product 2b (entry 3). These results follow the trend for other reductive coupling reactions that use similar catalysts and reducing agents, where DMF is more effective than acetonitrile (Citation7). Consequently, it appears that some solvent is necessary to achieve desired results, but not all solvents will do so equally.
Table 2. Reaction of 2a dissolved in polar solvents.
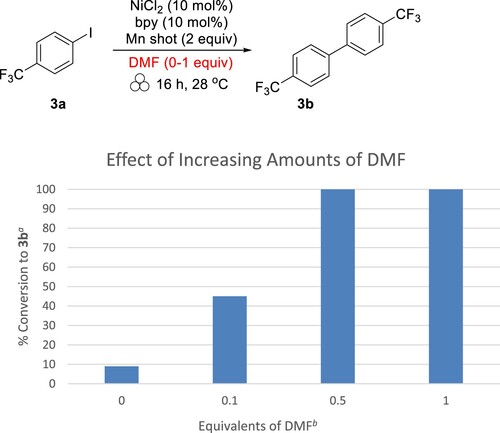
With a method in-place for both solid and liquid substrates, we moved forward with determining the applicability of the method to other aryl iodides. In particular, we wanted to know if DMF would be necessary for other liquid substrates. For this investigation, we chose 4-iodobenzotrifluoride (3a) as the initial substrate since this aryl iodide is a liquid at room temperature (Citation18). The results of various amounts of DMF added to the reaction conditions are shown in . Interestingly, we found that mostly starting material (3a) was recovered for solventless conditions, with only 9% converted to 3b. This is unlike the reaction with iodobenzene. These results indicated that the role of DMF is less important for solubility purposes and more important for providing a polar environment. Consequently, we thought that liquid-assisted grinding (LAG) could be used to reduce the amount of DMF necessary for the reaction. We found that 0.1 equivalents of DMF with respect to 3a increased the conversion of 3b from 9% to 45%. Adding just half of an equivalent of DMF results in complete conversion of 3a to 3b. A stoichiometric amount of DMF also results in complete conversion to product.
Figure 1. Effect of increasing amounts of DMF on reductive homocoupling reaction outcome for 4-iodobenzotrifluoride. aCalculated from 1H NMR. bWith respect to 3a.
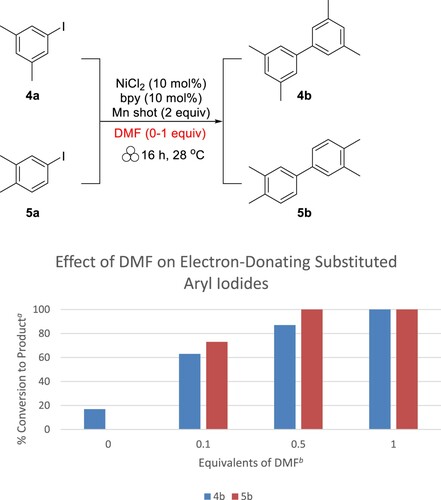
This trend was repeated for two other liquid aryl iodides with electron-donating substituents, 1-iodo-3,5-dimethylbenzene (4a) and 1-iodo-3,4-dimethylbenzene (5a). The results show very similar trends as 3a, which can be seen in . For both aryl iodides, no DMF added resulted in very little (17%, 4b) or no (5b) conversion to product, where 0.5 equivalents of DMF resulted in high conversion (87% for 4b and >99% for 5b). When 1 equivalent of DMF is added to the reaction conditions, both 4a and 5a are fully converted to the products.
Figure 2. Effect of increasing amounts of DMF on additional liquid substrates. aCalculated from 1H NMR. bWith respect to substrate.
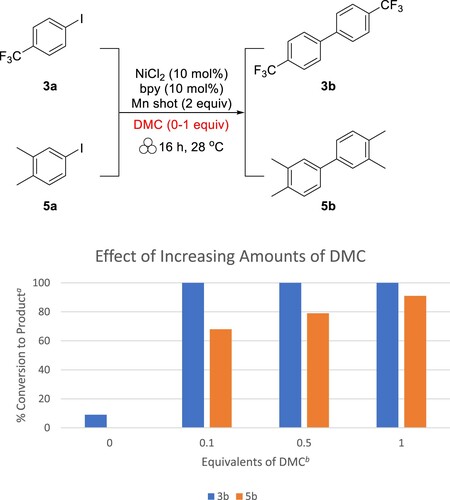
These results present a better understanding of the extent to which solvent is necessary for mechanochemical reductive coupling reactions. However, we were interested to determine if a greener LAG solvent could replace DMF in the reaction. Dimethyl carbonate (DMC) is a greener chemical and solvent that has garnered more interest as a replacement for less desirable solvents (Citation19). DMF is known to be reprotoxic and is considered undesirable by most, if not all solvent-selection guides (Citation20). In contrast, DMC is nontoxic, biodegrades readily, and is noncorrosive to metal, making it a better choice in terms of safety and biodegradability (Citation21). We repeated the experiments for 3a and 5a with DMC instead of DMF (). Complete conversion of 3a to 3b was observed after only 0.1 equivalents of DMC were added to the reaction conditions. The results for the reaction of 5a were slightly less dramatic, with 0.1 equivalents of DMC leading to 68% conversion. However, 1 equivalent of DMC was effective in converting 91% of 5a to 5b and still follows LAG quantities.
Figure 3. Effect of DMC on aryl iodide reductive homocoupling. aCalculated from 1H NMR. bWith respect to substrate.
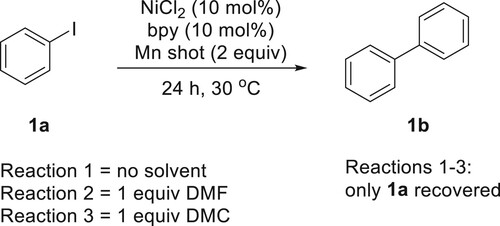
While there are examples of reductive coupling in the absence of amide-based solvents (Citation22), this work demonstrates mechanochemistry’s unique ability to use sub-stoichiometric amounts of a green solvent for a similar process. To further test if these conditions are successful specifically for mechanochemistry, reactions were performed with and without the polar solvents (Scheme 2) under more traditional conditions (stirring). For each reaction, the mixture was stirred for 24 h at 30°C. This temperature is slightly higher than what is observed for the global temperature of a Spex8000M Mixer Mill, which is the mechanochemical instrument used for this work. The vigorous shaking of the reaction vial and ball bearing in a mechanochemical reaction may lead one to believe that the method produces excessive heat upon impact. However, studies have shown that the overall temperature of the reaction jar is more important for the thermal impact of reaction outcome (Citation23). Consequently, we can assume that the heat applied to both activation techniques is nearly equivalent (28°C for mechanochemistry and 30°C for stirring). No reaction was observed for the stirring conditions (Scheme 2), so the unique mechanochemical reactivity must be the driver for a successful outcome. Mechanochemistry may be especially effective because of the mechanochemical activation of the manganese shot. At the end of a mechanochemical reaction, a very fine powder of metal is observed, so the mechanical action could be revealing fresh Mn(0) metal that is available throughout the duration of the reaction.
Scheme 2. ‘Traditional’ control reactions.
While this work is a step forward in understanding the role of solvents in mechanochemical reductive homocoupling, we also wanted to investigate whether the trend holds true for the more useful reaction cross-electrophile coupling. The XEC reaction that was tested was between iodobenzene and heptyl bromide, making it a sp2-sp3 reaction like those performed in other mechanochemical XEC reactions mentioned earlier (Citation11). The catalysts and reagents for this reaction are similar to that of the homocoupling, with few changes/additions. NiCl2 was still used as the catalyst and a Mn shot was still used as the reducing agent, along with the same time and temperature. The ligand used was updated to 1,10-phenanthroline rather than 2,2′-bipyridine and potassium iodide was used as an additive, based on the previous literature (Citation11). The efficacy of the reaction with various solvents was then studied, as depicted in . With no solvent added, a moderate amount of the homocoupling product was obtained. When 1 equivalent of DMC was added, quantitative conversion to products occurred, but the selectivity still heavily favored homocoupling. Since DMC was our greener solvent of choice for the homocoupling, this was slightly disappointing. However, there are other greener solvents that can possibly be used as drop-in replacements for reprotoxic amide-based solvents like DMF and DMA. The solvent that we chose to investigate further was N-butylpyrrolidinone (NBP). NBP has been highlighted in Chemical Reviews as a dipolar non-hydrogen-bond donor solvent that is amide-based, but not reprotoxic or mutagenic. It has even been used in other cross coupling reactions in place of its more structurally similar N-methyl pyrrolidinone (NMP) (Citation24). From the results with DMF, it can be seen that the selectivity was similar, with NBP yielding higher conversion to products.
Table 3. XEC reaction with various solvents.
With NBP as a viable LAG solvent for a greener mechanochemical XEC, we studied the ideal amount of NBP to add for optimal conversion and selectivity. The results are shown in . Increasing the amount of NBP to just 2 equivalents change the selectivity to 6b to 81%. There is no noticeable change with 2.5 equivalents, but for 2.75–4 equivalents, the conversion goes down slightly, but increases the selectivity to 95%. Since there is no difference between 2.75 equivalents, 3, or 4 equivalents, the optimal conditions would be set to 2.75 equivalents of NBP added, which is still within the range of LAG.
Table 4. Varying equivalents of NBP for mechanochemical XEC.
Conclusion
In this work, an ambient and simple reductive homocoupling of aryl iodides by mechanochemistry has been presented. Liquid-assisted grinding with a dipolar non-hydrogen-bond donor solvent is required for both solid and liquid substrates, but DMC can be used as a greener alternative to reprotoxic amide-based solvents. Additionally, preliminary results for XEC under mechanochemical conditions have been successfully conducted. While DMC is not suitable for XEC, the greener alternative NBP may be employed. Considering that most cross-electrophile coupling (XEC) reactions take place in reprotoxic amide-based solvents, this work is a promising step for developing XEC methods that are efficient and avoid undesirable solvents. Currently, the information learned in this work is being used to pursue other greener mechanochemical XEC reactions.
Experimental
General information
Chemicals for this research were purchased from Matrix Scientific (4-iodobenzotrifluoride), Sigma Aldrich (1-iodo-3,4-dimethylbenzene, 1-iodo-3,5-dimethylbenzene, N,N-dimethylformamide, acetonitrile), Eastman Chemical Company (2,2′-bipyridine), TCI (4-iodotoluene), Acros Organics (iodobenzene, dimethyl carbonate), American Elements (Mn shot), LabGuard (NiCl2), and ESPI Metals (Ni powder) and used without further purification. Mechanochemical reactions were done in a Spex8000M Mixer Mill with 15-mL, stainless-steel Form-Tech Scientific jars. Ball bearings used were 3/16″ diameter and 316 stainless-steel alloy from Grainger. A ThermoFisher Scientific Sorvall ST 8 Centrifuge was used to separate the Mn powder created in the reaction. 1H and 13C nuclear magnetic resonance (NMR) spectra were taken on a Bruker Avance III 400 MHz spectrometer (400 and 100 MHz, respectively) using CDCl3 solvent from Sigma Aldrich. Chemical shifts are reported as δ (ppm) and referenced to TMS. Spin–spin coupling constants are recorded as J (Hz) values.
Representative mechanochemical homocoupling procedure
To a 15-mL stainless-steel reaction jar was added 5 stainless-steel balls, followed by the addition of 3a (250 mg, 0.919 mmol), anhydrous NiCl2 (11.9 mg, 0.0919 mmol), 2,2′-bipyridine (14.4 mg, 0.0919 mmol), a Mn shot (101 mg, 1.84 mmol), and the desired amount of dimethyl carbonate (0, 0.10, 0.5, or 1 equivalent) was added. The jar was then closed and sealed with parafilm and secured in the ball mill. The reaction was run at 18 Hz for 16 h. The resulting reaction mixture was washed with approximately 15 mL of ethyl acetate and centrifuged for 2 min at 4000 rpm to separate the Mn powder from the organic supernatant. The supernatant was decanted into a separatory funnel where water was added for liquid–liquid extraction. The organic layer was dried over MgSO4, and the solvent was removed using a rotary evaporator to reveal a white solid.
Following the same experimental procedure, the remaining compounds (1b, 2b, 4b, and 5b) were prepared, with the only difference being the amount of dimethyl carbonate employed.
‘Traditional’ homocoupling procedure
A Mn shot (227 mg, 4.14 mmol) was ground in a mortar and pestle into small particles. NiCl2 (14.6 mg, 11.3 mmol), 2,2′-bipyridine (18.8 mg, 0.120 mmol), and the ground Mn particles were added to a 15-mL reaction vial. Iodobenzene (1.38 mL, 1.23 mmol) was added via 1-mL syringe. The vial was capped and transferred to a ring stand and suspended in a temperature-controlled oil bath set to 30.0°C. The reaction was stirred for 24 h and monitored periodically by thin-layer chromatography using cyclohexane as the developing solvent. No sign of reaction was observed within the 24 h.
Two other reactions were done with the same conditions, with the addition of either 1 equivalent of dimethyl carbonate (0.104 mL, 1.23 mmol) or 1 equivalent of N,N-dimethylformamide (0.0952 mL, 1.23 mmol). Neither reaction showed evidence of product formation within 24 h.
Mechanochemical XEC procedure
To a 15-mL stainless-steel reaction jar was added 5 stainless-steel balls, followed by the addition of 1a (250 mg, 1.23 mmol), anhydrous NiCl2 (15.9 mg, 0.123 mmol), 1,10′-phenanthroline (22.2 mg, 0.123 mmol), potassium iodide (51.1 mg, 0.308 mmol) a Mn shot (101 mg, 1.84 mmol), and 2.75 equivalents N-butylpyrrolidinone (0.45 g, 3.38 mmol) was added. The jar was then closed and sealed with parafilm and secured in the ball mill. The reaction was run at 18 Hz for 16 h. The resulting reaction mixture was washed with approximately 15 mL of ethyl acetate and centrifuged for 2 min at 4000 rpm to separate the Mn powder from the organic supernatant. The supernatant was decanted into a separatory funnel where water was added for liquid–liquid extraction. The organic layer was dried over MgSO4, and the solvent was removed using a rotary evaporator to reveal a yellowish liquid. To purify the heptyl benzene product, column chromatography on silica gel was done with 100% hexanes as eluent.
Spectral data of compounds
The following data were compared to existing literature to confirm their characterization. To view images of the spectra described below, please see the supporting information.
Biphenyl (1b) (Citation25)
Light yellow solid, 1H NMR (CDCl3, 400 MHz) δ: 7.60 (d, J = 8 Hz, 4H), 7.44 (t, J = 8 Hz, 4H), 7.37–7.33 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ: 141.40, 128.91, 127.41, 127.33.
4,4′-dimethyl-1,1′-biphenyl (2b) (Citation20)
White solid, 1H NMR (CDCl3, 400 MHz) δ: 7.48 (d, J = 8 Hz, 4H), 7.24 (d, J = 8 Hz, 4H), 2.39 (s, 6H); 13C NMR (CDCl3, 100 MHz) δ: 138.41, 136.84, 129.56, 126.94, 21.22.
4,4′-bis(trifluoromethyl)biphenyl (3b) (Citation26)
White solid, 1H NMR (CDCl3, 400 MHz) δ: 7.71 (q, J = 8 Hz, 8H); 13C NMR (CDCl3, 100 MHz) δ: 143.26, 130.29 (q, C-F, JC-F = 33 Hz), 127.65, 124.97 (q, C-F, JC-F = 4 Hz), 124.11 (q, C-F, JC-F = 270 Hz).
3,3′,4,4′-tetramethylbiphenyl (4b) (Citation20)
White solid, 1H NMR (CDCl3, 400 MHz) δ: 7.35 (s, 2H), 7.31 (d, J = 8 Hz, 2H), 7.18 (d, J = 8 Hz, 2H), 2.32 (s, 6H), 2.29 (s, 6H); 13C NMR (CDCl3, 100 MHz) δ: 138.04, 136.93, 135.44, 130.12, 128.42, 124.49, 20.10, 19.58.
3,3′,5,5′-tetramethylbiphenyl (5b) (Citation27)
White solid, 1H NMR (CDCl3, 400 MHz) δ: 7.19 (s, 4H), 6.97 (s, 2H), 2.37 (s, 12H); 13C NMR (CDCl3, 100 MHz) δ: 144.63, 138.27, 128.89, 125.28, 21.58.
Heptyl benzene (6b) (Citation28)
Clear liquid, 1H NMR (CDCl3, 400 MHz) δ: 7.29–7.26 (m, 2H), 7.19–7.17 (m, 3H), 2.62–2.58 (m, 2H), 1.63–1.59 (m, 2H), 1.32–1.27 (m, 8H), 0.88 (t, J = 8, 3H); 13C NMR (CDCl3, 100 MHz) δ: 143.13, 128.56, 128.37, 125.69, 36.16, 31.98, 31.71, 29.47, 29.35, 22.83, 14.27.
Supplemental Material
Download MS Word (1.3 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials. Additionally, the raw data that support the findings of this study are openly available in FigShare at 10.6084/m9.figshare.19,674,936.
Additional information
Funding
References
- Takacs, L. The Historical Development of Mechanochemistry. Chem. Soc. Rev. 2013, 42, 7649–7659. DOI:10.1039/c2cs35442j.
- James, S.L.; Adams, C.J.; Bolm, C.; Braga, D.; Collier, P.; Friščić, T.; Grepioni, F.; Harris, K.D.M.; Hyett, G.; Jones, W.; Krebs, A.; Mack, J.; Maini, L.; Orpen, A.G.; Parkin, I.P.; Shearouse, W.C.; Steed, J.W.; Waddell, D.C. Mechanochemistry: Opportunities for New and Cleaner Synthesis. Chem. Soc. Rev. 2012, 41, 413–447. DOI:10.1039/c1cs15171a.
- Ardila-Fierro, K.J.; Hernandez, J.G. Sustainability Assessment of Mechanochemistry by Using the Twelve Principles of Green Chemistry. ChemSusChem. 2021, 14, 2145–2162. DOI:10.1002/cssc.202100478.
- Howard, J.L.; Cao, Q.; Browne, D.L. Mechanochemistry as an Emerging Tool for Molecular Synthesis: What Can It Offer? Chem. Sci. 2018, 9, 3080–3094. DOI:10.1039/C7SC05371A.
- Tan, D.; Friščić, T. Mechanochemistry for Organic Chemists: An Update. Eur. J. Org. Chem. 2018, 18–33. DOI:10.1002/ejoc.201700961.
- (a) Achar, T.K.; Bose, A.; Mal, P. Mechanochemical Synthesis of Small Organic Molecules. Beilstein J. Org. Chem. 2017, 13, 1907–1931. DOI:10.3762/bjoc.13.186. (b) Jiang, Z.J.; Li, Z.H.; Yu, J.B.; Su, W.K. Liquid-Assisted Grinding Accelerating: Suzuki-Miyaura Reaction of Aryl Chlorides under High-Speed Ball-Milling Conditions. J. Org. Chem. 2016, 81, 10049–10055. DOI:10 .1021/acs.joc.6b01938. (c) Grätz, S.; Wolfrum, B.; Borchardt, L. Mechanochemical Suzuki Polycondensation – From Linear to Hyperbranched Polyphenylenes. Green Chem. 2017, 19, 2973–2979. DOI:10 .1039/C7GC00693D. (d) Vogt, C.G.; Grätz, S.; Lukin, S.; Halasz, I.; Etter, M.; Evans, J.D.; Borchardt, L. Direct Mechanocatalysis: Palladium as Milling Media and Catalyst in the Mechanochemical Suzuki Polymerization. Angew. Chem. Int. Ed. 2019, 58, 18942–18947. DOI:10 .1002/anie.201911356. (e) Seo, T.; Ishiyama, T.; Kubota, K.; Ito, H. Solid-State Suzuki-Miyaura Cross-Coupling Reactions: Olefin-Accelerated C-C Coupling Using Mechanochemistry. Chem. Sci. 2019, 10, 8202–8210. DOI:10 .1039/C9SC02185J.
- Goldfogel, M.J.; Huang, L.; Weix, D.J. Cross-Electrophile Coupling: Principles and New Reactions. In Nickel Catalysis in Synthesis: Methods and Reactions: Ogoshi, S., Ed.; Wiley-VCH: Weinheim, 2020.
- Nicholson, W.I.; Howard, J.L.; Magri, G.; Seastram, A.C.; Khan, A.; Bolt, R.R.A.; Morrill, L.C.; Richards, E.; Browne, D.L. Ball-Milling-Enabled Reactivity of Manganese Metal. Angew. Chem. Int. Ed. 2021, 60, 23128–23133. DOI:10.1002/anie.202108752.
- (a) Do, J.; Mottillo, C.; Tan, D.; Štrukil, V.; Friščić, T. Mechanochemical Ruthenium-Catalyzed Olefin Metathesis. J. Am. Chem. Soc. 2015, 137, 2476–2479. DOI:10.1021/jacs.5b00151. (b) Ying, P.; Yu, J.; Su, W. Liquid-Assisted Grinding Mechanochemistry in the Synthesis of Pharmaceuticals. Adv. Synth. Catal. 2021, 363, 1246–1271. DOI:10 .1002/adsc.202001245.
- Friscic, T.; Childs, S.L.; Rizvi, S.A.A.; Jones, W. The Role of Solvent in Mechanochemical and Sonochemical Cocrystal Formation: A Solubility-Based Approach for Predicting Cocrystallisation Outcome. CrystEngComm. 2009, 11, 418–426. DOI:10.1039/B815174A.
- (a) Wu, S.; Shi, W.; Zou, G. Mechanical Metal Activation for Ni-Catalyzed, Mn-Mediated Cross-Electrophile Coupling Between Aryl and Alkyl Bromides. New J. Chem. 2021, 45, 11269–11274. DOI:10.1039/D1NJ01732B. (b) Jones, A.C.; Nicholson, W.I.; Leitch, J.A.; Browne, D.L. A Ball-Milling-Enabled Cross-Electrophile Coupling. Org. Lett. 2021, 16, 6337–6341. DOI:10 .1021/acs.orglett.1c02096.
- Byrne, F.P.; Jin, S.; Paggiola, G.; Petchey, T.H.M.; Clark, J.H.; Farmer, T.J.; Hunt, A.J.; McElroy, C.R.; Sherwood, J. Tools and Techniques for Solvent Selection: Green Solvent Selection Guides. J. Sustain. Chem. Process. 2016, 4, 1–24. DOI:10.1186/s40508-016-0051-z.
- Colon, I.; Kelsey, D.R.J. Coupling of Aryl Chlorides by Nickel and Reducing Metals. Org. Chem. 1986, 51, 2627–2637. DOI:10.1021/jo00364a002.
- Haley, R.A.; Zellner, A.R.; Krause, J.A.; Guan, H.; Mack, J. Nickel Catalysis in a High Speed Ball Mill: A Recyclable Mechanochemical Method for Producing Substituted Cylooctatetraene Compounds. ACS Sustain. Chem. Eng. 2016, 4, 2464–2469. DOI:10.1021/acssuschemeng.6b00363.
- Chen, H.; Fan, J.; Fu, Y.; Do-Thanh, C.; Suo, X.; Wang, T.; Popovs, I.; Jiang, D.; Yuan, Y.; Yang, Z.; Dai, S. Benzene Ring Knitting Achieved by Ambient-Temperature Dehalogenation via Mechanochemical Ullmann-Type Reductive Coupling. Adv. Mater. 2021, 33, 1–8. DOI:10.1002/adma.202008685.
- (a) Tullberg, E.; Peters, D.; Frejd, T.J. The Heck Reaction Under Ball-Milling Conditions. Organomet. Chem. 2004, 689, 3778–3781. DOI:10.1016/j.jorganchem.2004.06.045. (b) Rana, S.; Varadwaj, G.B.B.; Jonnalagadda, S.B. Pd Nanoparticle Supported Reduced Graphene Oxide and Its Excellent Catalytic Activity for the Ullman C-C Coupling Reaction in a Green Solvent. RSC Adv. 2019, 9, 13332–13335. DOI:10 .1039/C9RA01715A.
- Alder, C.M.; Hayler, J.D.; Henderson, R.K.; Redman, A.M.; Shukla, L.; Shuster, L.E.; Sneddon, H.F. Updating and Further Expanding GSK’s Solvent Sustainability Guide. Green Chem. 2016, 18, 3879–3890. DOI:10.1039/C6GC00611F.
- Millipore Sigma. Sigma Aldrich. http://sigmaaldrich.com/US/en/product/Aldrich/365858 (accessed Apr 24, 2022).
- Jordan, A.; Hall, C.G.J.; Thorp, L.R.; Sneddon, H.F. Replacement of Less-Preferred Dipolar Aprotic and Ethereal Solvents in Synthetic Organic Chemistry with More Sustainable Alternatives. Chem. Rev. 2022, 122, 6749–6794. DOI:10.1021/acs.chemrev.1c00672.
- Byrne, F.P.; Jin, S.; Paggiola, G.; Petchey, T.H.M.; Clark, J.H.; Farmer, T.J.; Hunt, A.J.; McElroy, C.R.; Sherwood, J. Tools and Techniques for Solvent Selection: Green Solvent Selection Guides. Sustain. Chem. Process. 2016, 4, 1–24. DOI:10.1186/s40508-016-0051-z.
- Pyo, S.; Park, J.H.; Chang, T.; Hatti-Kaul, R. Dimethyl Carbonate as a Green Chemical. Curr. Opin.Green Sustain. Chem. 2017, 5, 61–66. DOI:10.1016/j.cogsc.2017.03.012.
- (a) Anka-Lufford, L.L.; Huihui, K.M.M.; Gower, N.J.; Ackerman, L.K.G.; Weix, D.J. Nickel-Catalyzed Cross-Electrophile Coupling with Organic Reductants in Non-Amide Solvents. Chem. Eur. J. 2016, 22, 11564–11567. DOI:10.1002/chem.201602668. (b) Molander, G.A.; Wisniewski, S.R.; Traister, K.M. Reductive Cross-Coupling of 3-Bromo-2,1-borazaronaphthalenes with Alkyl Iodides. Org. Lett. 2014, 16, 3692–3695. DOI:10 .1021/ol501495d.
- Andersen, J.M.; Mack, J. Decoupling the Arrhenius Equation via Mechanochemistry. Chem. Sci. 2017, 8, 5447–5453. DOI:10.1039/C7SC00538E.
- Sherwood, J.; Parker, H.L.; Moonen, K.; Farmer, T.J.; Hunt, A.J. N-Butylpyrrolidinone as a Dipolar Aprotic Solvent for Organic Synthesis. Green Chem. 2016, 18, 3990–3996. DOI:10.1039/C6GC00932H.
- Gerleve, C.; Studer, A. Transition-Metal-Free Oxidative Cross-Coupling of Tetraarylborates to Biaryls Using Organic Oxidants. Angew. Chem. Int. Ed. 2020, 59, 15468–15473. DOI:10.1002/anie.202002595.
- Bortnikov, E.O.; Semenov, S.N. Coupling of Alternating Current to Transition-Metal Catalysis: Examples of Nickel-Catalyzed Cross-Coupling. J. Org. Chem. 2021, 86, 782–793. DOI:10.1021/acs.joc.0c02350.
- Tran, H.; McCallum, T.; Morin, M.; Barriault, L. Homocoupling of Iodoarenes and Bromoalkanes Using Photoredox Gold Catalysis: A Light Enabled Au(III) Reductive Elimination. Org. Lett. 2016, 18, 4308–4311. DOI:10.1021/acs.orglett.6b02021.
- Midya, S.P.; Subaramanian, M.; Babu, R.; Yadav, V.; Balaraman, E. Tandem Acceptorless Dehydrogenative Coupling-Decyanation Under Nickel Catalysis. J. Org. Chem. 2021, 86, 7552–7562. DOI:10.1021/acs.joc.1c00592.