
ABSTRACT
In this work, we synthesized a series of six unsymmetrical benzimidazolium salts 2 and their pyridine-enhanced precatalyst preparation stabilization and initiation (PEPPSI)-themed palladium N-heterocyclic carbene complexes [PdCl2(NHC)(Py)]. All the products were isolated in satisfactory yields (75-85%). The synthesis of these novel palladium PEPPSI complexes involved in reacting NHC precursors with PdCl2 in pyridine at 60°C in the presence of excess K2CO3. The structures of all compounds have been characterized by 1H NMR, 13C NMR, HRMS and IR spectroscopy, as well as elemental analysis techniques, which support the proposed structures. The catalytic activity of the six complexes was assessed in the Suzuki-Miyaura cross-coupling of phenylboronic acid and aryl halides. The reactions required only a low catalyst loading (0.1 mol%) and were carried out under mild aerobic conditions in a green, water-based solvent mixture.
GRAPHICAL ABSTRACT
1. Introduction
Over the past few decades, the chemical industry has developed growing interest in inexpensive and environmentally friendly reaction media such as water based on considerations of safety, availability and environmental impact (Citation1). With palladium-catalyzed cross-coupling reactions being widely applied for the synthesis of fine chemicals, functional materials and industrial starting materials (Citation2), the development of water soluble Pd compounds as highly efficient catalysts for crosscoupling reactions in aqueous reaction media has become an important field of research (Citation3). Since the first example was described by Calabrese and co-workers in 1990 (Citation4), considerable efforts have been undertaken to develop water-soluble Pd catalysts for cross-coupling reactions, especially by modifying traditional Pd phosphine catalysts (Citation5). However, most of the phosphine ligands are air and moisture sensitive, restricting the reuse of the catalyst and leading to undesirable residues under aqueous reaction conditions (Citation6). In addition, the syntheses of phosphine ligands are generally demanding and toxic intermediates are involved (Citation7). Therefore, introducing more stable and less toxic ligands is desirable. N-heterocyclic carbenes (NHCs) have recently gained a strong foothold as ancillary ligands for homogeneous catalysis, sometimes displacing phosphine ligands (Citation8,Citation9). NHCs are advantageous due to their high thermal stability and non-toxic chemical properties. Oxidative stability (Citation10,Citation11), strong σ-donor ability (Citation12) and weak π-acceptor ability, tunable electronic and steric properties, and easy process handling (Citation6). Commonly used transition metal NHC ligands are generated from imidazolium, imidazolinium, and benzimidazolium salts in the presence of strong bases. NHC-Pd complexes have been successfully used as efficient catalysts for C–C bond formation (Citation13–16). In particular, the work of Organ and colleagues plays an important role in the cross-coupling reaction (Citation17). They synthesized a series of Pd-PEPPSI complexes with different NHC ligands. The catalytic properties of these substances resulted in their ability to react efficiently with a diverse range of substrates. According to theoretical analyses, the N-heterocyclic carbene (NHC) ligands are responsible for the origin of the Pd-PEPPSI complex's reactivity (Citation18–22). Principally, the properties of NHC ligands in transition-metalcatalysis can be effectively described by three factors: (1) r-donation from the carbene to the metal center; (2) p-backbonding from the metal to the empty p-orbitals of the NHC; and (3) steric impact (Citation23,Citation24). Since the first report of Pd-NHC systems in C–C bond forming reaction by Hermann and coworkers (Citation25); several research groups such as of Nolan (Citation26–28), Organ (Citation29–31), Szostak (Citation32,Citation33) etc. have developed a plethora of well-defined Pd-NHC-based catalytic systems for Suzuki–Miyaura reactions. Although majority of these NHC complexes were very stable in air and moisture, the catalytic cross-coupling reactions were mainly performed in an oxygen free environment, as the actual catalytic species Pd (0) was highly air sensitive (Citation29). In recent years, a new type of Pd-NHC systems, known as Pd-PEPPSI (PEPPSI: Pyridine enhanced pre-catalyst preparation, stabilization and initiation) has surfaced in literature that showed remarkable activity as catalysts in many cross-coupling reactions including Suzuki–Miyaura reaction (Citation34,Citation35). Unlike conventional Pd-NHC systems, these Pd-PEPPSI complexes are air and water stable and can promote cross-coupling reactions in water or mixed aqueous environment (Citation36,Citation37). Moreover, due to the presence of a weakly bound pyridine moiety makes the Pd-PEPPSI system more efficient as the labile pyridine moiety can act as a ‘through-away ligand’ and can create a vacant coordination site for the substrate binding and are thus expected to enhance catalytic activities. In fact, in 2006, Organs’ group for the first time introduced PdPEPPSI system in Suzuki–Miyaura reactions (Citation38). Since then, a number of highly efficient Pd-PEPPSI complexes have been reported for Suzuki–Miyaura reactions, however majority of the systems suffered from limitations like narrow substrate scope, harsh reaction conditions, high metal loadings (Citation39–41), etc. Until now, only a couple of examples are known that remained successful in activating Suzuki reaction at room temperature in an aqueous environment (Citation42). Thus, there is a need to develop high performance Pd-PEPPSI-based catalytic systems for Suzuki–Miyaura reaction in room temperature in an aqueous environment. In 1990, Casalnuovo et al., reported the Suzuki reaction between aryl halides and boron reagents in water (Citation43). This reaction has been crucial in the advancements of aqueous phase cross-coupling reactions (Citation44). As a consequence, numerous catalysts capable of promoting Suzuki coupling under mild conditions in pure water or in combination with co-solvents are currently known (Citation45).
Catalytic tests performed by Szostak et al. (Citation46) and in our group (Citation47) showed that complexes 1–3 displayed a high catalytic activity in various palladium-catalyzed cross-coupling reactions. In particular, both studies highlighted that they efficiently promoted the Suzuki–Miyaura cross-coupling of aryl halides with phenylboronic acids. To minimize its environmental impact, we optimized the experimental procedure for these reactions by working with a low catalyst loading (0.25 mol%) at 40°C in a green, water-based solvent mixture using K2CO3 as an innocuous base in the presence of air. Under these mild, aerobic conditions, aryl bromides and iodides bearing either electron-donating or withdrawing substituents afforded a wide range of biaryl derivatives. Although less reactive, aryl chlorides were also successfully activated under more forcing conditions, provided that an electron-withdrawing group was present on their aromatic ring. Encouraged by the good results obtained in Suzuki–Miyaura cross-coupling reactions, we decided to investigate more challenging catalytic applications that could take advantage of the environmental-friendly Pd-NHC PEPPSI complexes 3. Herein, we describe the synthesis and characterization of the benzimidazoles 2 and their palladium PEPPSI complexes 3. Thus, the aim of this study is to synthesize novel benzimidazoles 2 and their palladium PEPPSI complexes 3 featuring unsymmetrical ligands with various combinations on their nitrogen atoms. The catalytic activity of these novel complexes will be assessed in the Suzuki–Miyaura cross-coupling of phenylboronic acid with aryl halides in aqueous media, even at room temperature. Using water as a solvent for such reactions is not only cost-effective, but also beneficial for the environment, since it is widely available, inexpensive, and easily separable from organic compounds.
2. Experimental section
2.1. Materials
All reactions for the preparation benzimidazolium salts 2 and palladium-(NHC)-PEPPSI complexes 3 were carried out under air. PdCl2, pyridine and solvents were purchased from Sigma-Aldrich and used as obtained. Elemental analyses were performed by Elementar Vario EL III Carlo Erba 1108. The melting points of the complexes and NHC precursors were determined using Stuart automatic melting point apparatus (SMP-40). IR spectra were recorded with a PerkinElmer Spectrum 100 GladiATR FT/IR spectrophotometer. 1H, 13C NMR spectra were recorded in CDCl3 or DMSO-d6 solutions operating on a Bruker Advance 400 MHz NMR spectrometer and chemical shifts were reported relative to tetramethylsilane for 1H, 13C NMR spectra as standard. Signals are quoted in parts per million as δ downfield from tetramethylsilane (δ 0.00) as an internal standard. Coupling constants (J values) are given in hertz. NMR multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, m = multiplet signal. The HRMS (ESI) electrospray ionization mass spectra were recorded on a Shimadzu LCMS-IT-Toff spectrometer in CH3CN/CHCl3. All reactions were monitored on a Agilent 6890N GC system by GC-FID with a HP-5 column of 30 m length, 0.32 mm diameter and 0.25 μm film thickness. Column chromatography was performed using silica gel 60 (70–230 mesh). Solvent ratio is given as v/v.
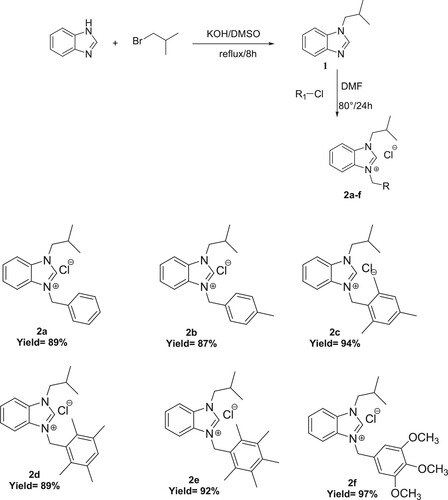
2.2. General procedure for the synthesis of NHC precursors, 2a-f
Compounds 1 (1.1 mmol) and aryl chloride (1 mmol) were dissolved in 5 mL DMF in a pre-dried Schlenck tube. The temperature of the medium is brought to 80°C. with stirring and the reaction is monitored by 1 H-NMR for 24 h. At the end of the reaction, the reaction mixture was diluted with ether to precipitate the product. After filtration, the white solid obtained was washed with diethyl ether, dried in vacuo, and then recrystallized from DCM/DE (1:3).
2.2.1. 1-(Isobutyl)-3-(benzyl)benzimidazolium chloride 2a
Yield: 89%, m.p. = 128.3°C, IR ν(CN): 1650.9 cm−1, 1H NMR (CDCl3, 400MHz) δ (ppm) = 12.07 (s, 1H, H2); 7.71–7.30 (m, 9H, H4, 5, 6, 7, 3″, 4″, 5″, 6″, 7″); 5.95 (s, 2H, H1″); 4.44 (d, 2H, H1′); 2.44 (sept, 1H, H2′); 1.06 (d, 6H, CH3 (a,b)). 13CNMR(CDCl3, 101MHz) δ (ppm) = 144.05 (C2); 132.94 (C2″); 131.73 (C8); 131.12 (C9); 129.36 (C4″;6″); 129.18 (C5″); 128.30 (C3″;7″); 127.11 (C5); 127.08 (C6); 113.88 (C7); 113.15 (C4); 54.44 (C1′); 51.38 (C1″); 28.89 (C2′); 19.86 (Ca,b). Anal. Calc. for C18H21ClN2 (%):C, 71.86%; H, 7.03%; N, 9.31%. found (%): C, 71.9%; H, 7.1%; N, 9.2%.
2.2.2. 1-(Isobutyl)-3-(4-methylbenzyl)benzimidazolium chloride 2b
Yield: 87%, m.p. = 176.1°C, C19H23ClN2, M = 314,85g mol−1. IR ν(CN):1550 cm−1. 1H NMR (CDCl3, 400MHz) δ (ppm) = 11.81 (s, 1H, H2); 7.30–7.49 (m, 7H, H4, 7, 3′′, 4′′, 5′′, 6′′, 7′′); 5.86 (s, 2H, H1′′); 4.36 (d, 2H, H1′, J = 8 Hz); 2.44 (sept, 1H, H2′, J = 8 Hz); 2.42 (s, 3H, CH3(d)); 2.38 (s, 3H, CH3(c)); 1.04 (d, 6H, CH3(a,b), J = 8 Hz). 13CNMR(CDCl3, 101MHz) δ (ppm) = 143.96 (C2); 139.16 (C2″); 131.75 (C9); 131.09 (C8); 129.99 (C4″;6″); 129.91 (C5″); 128.31 (C3″;7″); 127.05 (C5;6); 113.97 (C7); 113.12 (C4); 54.39 (C1′); 51.24 (C1″); 28.90 (C2′); 21.19 (Cc); 19.86 (Ca,b). Anal. Calc. for C19H23ClN2 (%):C, 72.48%; H, 7.36%; N, 8.89% . found (%): C, 72.5; H, 7.4; N, 8.9%. DART-TOF-MS: m/z = 340, 267.
2.2.3. 1-(isobutyl)-3-(2,4,6-trimethylbenzyl)benzimidazolium Chloride 2c
Yield: 94%, m.p. = 218.3°C, C21H27ClN2, M = 342.90g mol−1. IR ν(CN):1469cm−1. 1H NMR (CDCl3, 400MHz) δ (ppm) = 11.61 (s, 1H, H2); 7.64 (d, 1H, H7, J = 8 Hz); 7.51 (t, 1H, H6, J = 8 Hz); 7.37 (t, 1H, H5, J = 8 Hz); 7.10 (d, 1H, H4, J = 8 Hz); 6.88 (s, 2H, H4″,6″); 5.90 (s, 2H, H1″); 4.42 (d, 2H, H1′, J = 8 Hz); 2.37 (sept, 1H, H2′, J = 8 Hz); 2.28 (s, 6H, CH3(c,e)); 2.25 (s, 3H, CH3(d)); 1.00 (d, 6H, CH3(a,b), J = 8 Hz), 13CNMR(CDCl3, 101MHz) δ (ppm) = 144.2 (C2); 130.2 (C8); 139.6 (C2″); 137.9 (C5″,7″); 131.8 (C3″); 131.3 (C9); 127.1 (C6″); 126.9 (C4″); 125.2 (C5,6); 114.0 (C7); 113.1 (C4); 54.3 (C1′); 47.5 (C1″); 28.9 (C2′); 21.1 (Cd); 20.3 (Ca,b); 19.8 (Cc,e). Anal. Calc. for C21H27ClN2 (%):73.56%; H, 7.94%; N, 8.17%. found (%): C, 73.5; H, 7.9; N, 8.1%.
2.2.4. 1-(Isobutyl)-3-(2.3.5.6-tetramethylbenzyl)benzimidazolium chloride 2d
Yield: 89%, m.p. = 176.3°C, C22H29ClN2, M = 491.51 g mol−1. IR ν(CN) = 1660.1 cm−1. 1H NMR (CDCl3, 400MHz) δ (ppm) = 11.44 (s, 1H, H2); 7.07–7.69 (m, 5H, H4, 5, 6, 7, 5″), 5.97 (s, 2H, H1″); 4.49 (d, 2H, H1′, J = 8 Hz); 2.38 (sept, 1H, H2′, J = 8 Hz); 2.25 (s, 12H, CH3(c,d,e,f)); 1.02 (d, 6H, CH3 (a,b), J = 8 Hz). 13CNMR(CDCl3, 101MHz) δ (ppm) = 143.9 (C2); 135.0 (C2″); 134.0 (C4″,6″); 133.5 (C3″, 7″); 131.8 (C9); 131.3 (C8), 127.9 (C5″); 127.0 (C5); 126.8 (C6); 113.8 (C7); 113.0 (C4); 54.2 (C1′); 48.0 (C1″); 28.8 (C2′); 20.5 (Ca,b); 19.7 (Cd,e); 16.12 (Cc,f). Anal. Calc. for C22H29ClN2 (%):74.03%; H, 8.18%; N, 7.84%. Found (%): C, 8.2; H, 7.2; N, 7.9%.
2.2.5. 1-(Isobutyl)-3-(2.3.4.5.6-pentamethylbenzyl)benzimidazolium chloride 2e
Yield: 92%, m.p. = 198.2°C, C23H31ClN2, M = 370,95g mol−1. IR ν(CN) = 1546 cm−1. 1H NMR (CDCl3, 400MHz) δ (ppm) = 11.29 (s, 1H, H2); 7.22–7.70 (m, 4H, H4, 5, 6, 7); 5.94 (s, 2H, H1″); 4.51 (d, 2H, H1′); 2.38 (sept, 1H, H20); 2.28 (s, 3H, CH3(e)); 2.28 (s, 6H, CH3(d,f)); 2.24 (s, 6H, CH3(c,g)); 1.05 (d, 6H, CH3 (a,b), J = 4 Hz), 13CNMR(CDCl3, 101MHz) δ (ppm) = 143.78 (C2); 137.21 (C2″); 133.87 (C8;9); 133.54 (C3″;7″); 131.87 (C6″); 131.39 (C4″); 127.00 (C6); 126.84 (C5); 125.15 (C5″);113.98 (C7); 113.06 (C4); 54.27 (C1′); 48.50 (C1″); 28.88 (C2′); 19.79 (Ca,b); 17.34 (Ce); 17.12 (Cd,f); 16.98 (Cc,g). Anal. Calc. for C23H31ClN2 (%):C, 74.46; H, 8.42; N, 7.55 . found (%): C, 74.5; H, 8.5; N, 7.4%.
2.2.6. 1-(Isobutyl)-3-(3,4,5 trimethoxylbenzyl)benzimidazolium chloride 2f
Yield: 97%, m.p = 250.9°C, C27H31N2Br, M = 463.45 g mol-1. IR ν(CN) = 1562.47 cm-1. 1H NMR (CDCl3, 400MHz) δ (ppm) = 12.02 (s, 1H, H2); 7.57–7.74 (m, 4H, H4,5,6,7); 6.88 (s, 2H, H3″,7″); 5.81 (s, 2H, H1″); 4.40 (d, 2H, H1′, J = 4 Hz); 3.84 (s, 3H, CH3(d)); 3.78 (s, 6H, CH3(c,e)); 2.39 (sept, 1H, H2′, J = 4 Hz); 1.01 (d, 6H, CH3(a,b)). 13CNMR(CDCl3, 101MHz) δ (ppm) = 153.9 (C4″,6″); 144.1 (C2,2″); 138.6 (C5″); 131.7 (C9); 131.2 (C8); 128.6 (C6); 127.2 (C5); 113.8 (C7); 113.2 (C4); 106.1 (C3″,7″); 60.9 (Cd); 56.7 (Cc,e); 54.5 (C1′); 51.6 (C1″); 28.9 (C2′); 19.9 (Ca,b). DART-TOF-MS: m/z = 267, 225. Anal. Calc. for C27H31N2Br (%):C, 69.97; H, 6.74; N, 6.04 . Found (%): C, 69.9; H, 6.7; N, 6.1%.
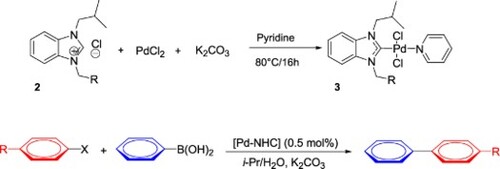
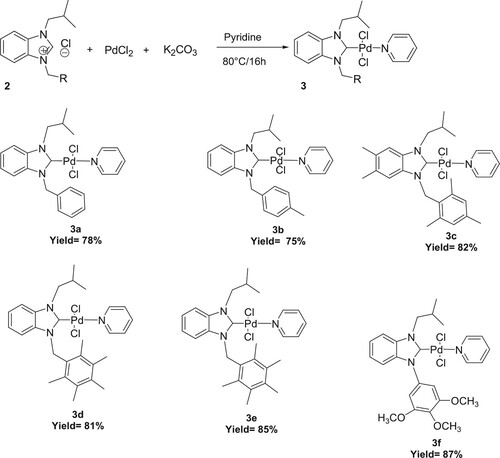
2.3. General procedure for the preparation of the palladium-PEPPSI complexes, (3a-f)
A solution of benzimidazolium salt 2 (1 mmol), palladium(II) chloride (1.05 mmol, 0.18 g), potassium carbonate (5 mmol, 0.69 g) in pyridine (1 mmol, 3 mL) was prepared at 80 °C Stir at 4pm. Thin layer chromatography (eluent dichloromethane) showed formation of a new, more polar product 3 after 16 h. After the reaction was complete, the mixture was cooled to room temperature and filtered through celite. The filtrate solvent was removed in vacuo. The resulting product was washed with n-pentane (3×5 mL) and dried in vacuo. Pure complex 3 was isolated by column chromatography on silica gel (eluting with dichloromethane) or by recrystallization from DCM/DE (1:3).
2.3.1. Dichloro[1-(methoxyethyl)-3-(2,3,4,5,6-pentamethylbenzyl)benzimidazole-2-ylidene]pyridinepalladium(II), 3a
Yield: 78% (0.4 g); m.p. = 243.9°C.ν(CN) = 1447.16 cm−1. 1H NMR (CDCl3, 400 MHz, δ, ppm): 2.25 [s, 6H, CH2C6(CH3)5-2,6], 2.32 [s, 6H, CH2C6(CH3)5-3,5], 2.34 [s, 3H, CH2C6(CH3)5-4], 3.38 [s, 3H, CH2CH2OCH3], 4.23 [t, J = 4 Hz, 2H, CH2CH2OCH3], 5.13 [t, J = 4 Hz, 2H, CH2CH2OCH3], 6.26 [s, 2H, CH2C6(CH3)5-2,3,4,5,6], 6.44, 6.97 and 7.18 [t, 3H, NC6H4N], 7.54 [d, J = 8 Hz, 1H, NC6H4N], 7.39 [t, J = 8 Hz, 2H,, NC5H5-H3,5], 7.81 [t, J = 8 Hz, 1H, NC5H5-H4], 8.98 [d, J = 8 Hz, 2H, NC5H5-H2,6]. 13C NMR (CDCl3, 100 MHz, δ, ppm): 16.9, 17.3 and 17.5 [CH2C6(CH3)5-2,3,4,5,6], 48.6 [CH2CH2OCH3], 51.2 [CH2C6(CH3)5-2,3,4,5,6], 59.2, 72.0 [CH2CH2OCH3], 111.3 [NC6H4N-o], 122.7 [NC5H5, C3,5], 123.0 [NC6H4N-m], 124.5 [CH2C6(CH3)5-2,3,4,5,6;C3,5], 127.8 [CH2C6(CH3)5-2,3,4,5,6;C4], 133.1 [NC6H4N-i], 134.7 [CH2C6(CH3)5-2,3,4,5,6;C2,6], 135.9 [NC5H5, C4], 138.1 [CH2C6(CH3)5-2,3,4,5,6;C1], 151.2 [NC5H5, C2,6], 163.27 [Pd-Ccarb]. Anal. Calc. for C27H33N3OCl2Pd (%): C, 54.70; H, 5.61; N, 7.09. Found (%): C, 54.78; H, 5.73; N, 7.21. HR-MS(ESI), m/z = 468,1243 [M+Na+H]+ (calcd for C22H28N2OPdNa:468,1217).
2.3.2. Dichloro[1-(methoxyethyl)-3-(2,4,6-trimethylbenzyl)benzimidazole-2-ylidene]pyridinepalladium(II), 3b
Yield: 75% (0.35 g); m.p. = 215.5°C. ν(CN) = 1448.80 cm−1. 1H NMR (CDCl3, 400 MHz, δ, ppm): 2.26 [s, 9H, CH2C6H2(CH3)3-2,4,6], 3.28 [s, 3H, CH2CH2OCH3], 4.14 [t, J = 4 Hz, 2H, CH2CH2OCH3], 5.04 [t, J = 4 Hz, 2H, CH2CH2OCH3], 6.11 [s, 2H, CH2C6H2(CH3)3-2,4,6], 6.39 and 7.47 [d, J = 8 Hz, 2H, NC6H4N], 6.86 [s, 2H, CH2C6H2(CH3)3-2,4,6], 6.89 and 7.10 [t, J = 8 Hz, 2H, NC6H4N], 7.31 [t, J = 8 Hz, 2H,, NC5H5-H3,5], 7.72 [t, J = 8 Hz, 1H, NC5H5-H4], 8.91 [d, J = 8 Hz, 2H, NC5H5-H2,6]. 13C NMR (CDCl3, 100 MHz, δ, ppm): = 20.8 and 21.1 [CH2C6H2(CH3)3-2,4,6], 48.6 [CH2CH2OCH3], 50.0 [CH2C6H2(CH3)3-2,4,6], 59.2, 72.0 [CH2CH2OCH3], 111.1 and 111.4 [NC6H4N-o], 122.8 [NC5H5, C3,5], 123.1 [NC6H4N-m], 124.5 [CH2C6H2(CH3)3-2,4,6;C3,5], 127.5 [CH2C6H2(CH3)3-2,4,6;C4], 134.7 [CH2C6H2(CH3)3-2,4,6;C2,6], 134.4 and 135.6 [NC6H4N-i], 138.2 [NC5H5, C4], 138.8 [CH2C6H2(CH3)3-2,4,6;C1], 151.3 [NC5H5, C2,6], 163.5 [Pd-Ccarb]. Anal. Calc. for C25H29N3OCl2Pd (%): C, 53.16; H, 5.17; N, 7.44. Found (%): C, 53.22; H, 5.24; N, 7.56. HR-MS(ESI), m/z = 440,0914 [M+Na+H]+ (calcd for C20H24N2OPdNa:440,0904); m/z = 413,0680 [M-H]+ (calcd for C20H24N2OPd:413,0845).
2.2.3. Dichloro[1-(methoxyethyl)-3-(3,5-dimethylbenzyl)benzimidazole-2-ylidene]pyridinepalladium(II), 3c
Yield: 82% (0.32); m.p. = 205.5°C. ν(CN) = 1445.58 cm−1. 1H NMR (CDCl3, 400 MHz, δ, ppm):2.21 [s, 6H, CH2C6H3(CH3)2-3,5], 3.30 [s, 3H, CH2CH2OCH3], 4.23 [m, 2H, CH2CH2OCH3], 5.10 [d, J = 6 Hz, 2H, CH2CH2OCH3], 6.07 [m, 2H, CH2C6H3(CH3)2-3,5], 6.86 [s, 1H, NC6H4N], 7.05[m, 2H, NC6H4N], 7.48 [d, J = 8 Hz, 1H, NC6H4N], 7.15 and 7.19 [s, 3H, CH2C6H3(CH3)2-3,5], 7.28 [m, 2H, NC5H5-H3,5], 7.48 [d, J = 8 Hz, 1H, C6H4−H4], 7.67–6.72 [m, 1H, NC5H5-H4], 8.95 [m, 2H, NC5H5-H2,6]. 13C NMR (CDCl3, 100 MHz, δ, ppm): 21.3 [CH2C6H3(CH3)2-3,5], 48.8 [CH2CH2OCH3], 53.3 [CH2C6H3(CH3)2-3,5], 59.2[CH2CH2OCH3], 71.7 [CH2CH2OCH3], 111.2 and 111.5 [NC6H4N-o], 123.1 [NC5H5, C3,5], 124.6 [NC6H4N-m], 125.9 [CH2C6H3(CH3)2-3,5;C2,6], 129.8 [CH2C6H3(CH3)2-3,5;C4], 134.8 and 134.3 [NC6H4N-i], 135.9 [CH2C6H3(CH3)2-3,5;C1], 138.1 [NC5H5, C4], 138.4 [CH2C6H3(CH3)2-3,5;C3,5], 152.7 [NC5H5, C2,6], 163.4 [Pd-Ccarb]. Anal. Calc. for C24H29N3OCl2Pd (%): C, 52.33; H, 4.94; N, 7.63. Found (%): C, 52.39; H, 5.01; N, 7.95. HR-MS(ESI), m/z = 571,6105 [M+Na-H]+ (calcd for C24H27N3OCl2PdNa:571,0385); m/z = 426,0798 [M+Na+H]+ (calcd for C19H22N2OPdNa:426,0786).
2.2.4. Dichloro-[1-(isobutyl)-3-(methoxyethyl)-5,6-dimethylbenzimidazole-2-ylidene] (pyridine)palladium(II)3d
Yield: 81%, m.p. = 242.5°C, C21H29Cl2N3OPd, M = 516,81. gmol−1, IR νCN, cm−1: 1400, 1H NMR (CDCl3, 400 MHz, δ, ppm): 0.91 (d, 6H, CH3(a,b), J = 4 Hz), 1.28 (sept, 1H, H2′, J = 8 Hz), 1.57 (s, 3H, CH3(e)), 2.20 (s, 6H, CH3(c,d)), 3.97 (m, 2H, H2″), 5.07 (m, 2H, H1″), 6.57 (s, 2H, H1′), 6.88 (s, 2H, H4,7), 7.68–7.72(m, 2H, H3′″, 5′″), 8.84–8.86 (m, 3H, H2′″,6′″, 4′″). 13C NMR (CDCl3, 100 MHz, δ, ppm): 20.0 (Cc,d), 20.3(Ca,b), 20.6(C2′), 21.1(C1″), 29.1 (Cc), 53.6 (C1′,2″), 109.9 (C4), 114.3(C7), 124.8 (C3′″;5′″), 129.2 (C5), 129.4 (C6), 133.8 (C8), 137.9 (C9), 140.3 (C4′″), 151.5 (C2′″;6′″), 161.7 (C2). Anal.Calcd. pour C19H25Cl2N3OPd (%):C, 48.806; H, 5.656; N, 8.131; Trouvé (%):C, 48.9; H, 5.7; N, 8.3. DART-TOF-MS (m/z) = 516,81
2.2.5. Dichloro-[1-(isobutyl)-3-(2.4.6 trimethylbenzyl)benzimidazol-2-ylidene](pyridine)
palladium(II)3e
Yield: 85%, m.p. = 220.2°C, C26H31Cl2N3Pd, M = 562.87. gmol−1, IR νCN, cm−1: 1458, 1H NMR (CDCl3, 400 MHz, δ, ppm): 1.15 (d, 6H, CH3 (a,b), J = 8 Hz), 2.33 (s, 9H, CH3(c,d,e)), 3.11 (sept, 1H, H2′, J = 8 Hz), 4.65 (d, 2H, H1′, J = 8 Hz), 6.24 (s, 2H, H1″), 6.93 (s, 2H, H4″,6″) 6.95–7.36 (m, 4H, H4, 5, 6, 7), 7.38 (m, 2H, H3′″,4′″), 7.79 (t, 1H, H4′″, J = 8 Hz), 8.99 (d, 2H, H2′″,6′″, J = 4 Hz). 13C NMR (CDCl3, 100 MHz, δ, ppm): 20.9 (C(c,e)), 21.0 (C(a,b)), 21.2 (Cd), 29.7 (C2′), 50.4 (C1″), 55.5 (C1′), 110.6 (C4), 111.6 (C7), 122.8 (C3′″), 123.1 (C5′″), 124.6 (C5;6), 129.7 (C4″;6″), 134.4 (C8), 135.5 (C9), 138.2 (C5″;4′″), 138.6 (C2″), 138.8 (C3″;7″), 151.4 (C2′″;6′″), 163.6 (C2). Anal.Calcd. pour C26H31Cl2N3Pd (%): C, 55.48; H, 5.55; N, 7.47; Trouvé (%):C, 55.60; H, 5.40; N, 7.60.
2.2.6. Dichloro-[1-(isobutyl)-3-(3.4.5 trimethoxylbenzyl)benzimidazol-2-ylidene](pyridine) palladium(II) 3f
Yield: 87%, m.p. = 158.4°C, C26H31Cl2N3O3Pd, M = 610.86 gmol−1, IR νCN, cm−1: 1431, 1H NMR (CDCl3, 400 MHz, δ, ppm): 1.14 (d, 6H, CH3 (a,b), J = 8 Hz), 3.09 (sept, 1H, H2′, J = 8 Hz), 3.81 (s, 3H, CH3(d)), 3.82 (s, 6H, CH3(c,e)), 4.68 (d, 2H, H1′, J = 8 Hz), 6.14(s, 2H, H1″), 6.86 (s, 2H, H3″,7″), 7.14–7.23 (m, 2H, H3′″,5′″), 7.32–7.38 (m, 4H, H4, 5, 6, 7), 7.78 (t, 1H, H4′″, J = 8 Hz), 9.00(d, 2H, H2′″,6′″, J = 4 Hz), 13C NMR (CDCl3, 100 MHz, δ, ppm): 20.8 (Ca,b), 29.5 (C2′), 53.4 (C1″), 55.7 (C1′), 56.6 (Cc;e), 60.9 (Cd), 105.1 (C3″;7″), 110.8 (C4), 111.5 (C7), 123.3 (C3′″), 124.7 (C5′″), 125.1 (C5;6), 130.89, (C2″), 131.0 (C8), 134.0 (C9), 135.6 (C5″), 138.7 (C4′″), 151.3 (C2′″;6′″), 153.5 (C4″,6″), 163.9 (C2). Anal.Calcd. pour C28H35Cl2N3Pd (%): C, 51.12; H, 5.12; N, 6.88; Trouvé (%):C, 51.20; H, 5.10; N, 3.90.
2.4. General procedure for Suzuki–Miyaura reactions
To initiate the reaction, a small round bottom flask was used to introduce air, Pd-PEPPSI complex (0.1 mol %), aryl bromide (1.0 mmol), phenylboronic acid (1.2 mmol), K2CO3 (1 mmol), and 4 mL of water/i-PrOH (3:1 v/v). The mixture was then stirred for an appropriate amount of time. Following this, the reaction mixture was extracted with Et2O, and the extract was filtered through a pad of silica with copious washing of diethyl ether. To ensure the purity of the compounds, GC was used to check yields based on aryl chloride and calibrated internal standard (undecane).
3. Results and discussion
3.1. Preparation of benzimidazolium salts 2
New benzimidazolium salts 2, which are carbene precursors, were synthesized firstly by N-alkylation reaction using benzimidazole (1 mmol) with isobutyl bromide (1 mmol) in the presence of KOH for 8h at reflux to prepare the starting material 1-Isobutyl-1H-benzoimidazole (1). The precursor (2) was then synthesized by the quaternization of the intermediate (1) with same benzyl chloride in DMF for 24h in degassed dimethylformamide at 80°C (Scheme 1). The new benzmidazolium salts were obtained and isolated as air- and moisture-stable white solid in high yield (>87%). The structures of the salts were determined by their characteristic spectroscopic data and elemental analyses. The benzimidazolium salts include an acidic NCHN proton, which can be deprotonated easily to form an NHC, at the C2 position of the benzimidazole ring. The sharp salt peak indicating the synthesis of a benzimidazolium salt came quite downfield at δ 12.07, 11.81, 11.61,11.44,11.29 and 12 ppm, in the 1H NMR spectra for 2, respectively. The NCHN peaks of the carbene precursors were observed at δ 144.05, 143.96, 144.2, 143.9, 143.87 and 143.7ppm in the 13C{1H} NMR spectra for 2, respectively. The formation of the benzimidazolium salts was also evident through their IR spectra, which showed peaks at 1650.9,1550,1469,1660.1,1546 and 1542cm−1 for the ν(CN) bond of 2, respectively.
Scheme 1. Synthesis of NHC precursors 2a-f.
The one-bond CH J-coupling value (1 JCH) is related to the hybridization of the carbon atom involved (Citation48). There is an empirical correlation between the carbon s-fraction ‘s’ and the coupling constant J as follows: 1 JCH = 500 s, with ‘s’ ranging from 0.25 to 0.33 to 0.5 as the hybridization changes from sp3 to sp2 to sp, respectively (Citation49). Higher one-bond CH J-coupling constants correspond to poorer r-donating properties of NHC ligands due to increased s-orbital character of the CAH bond. The one-bond CH J-coupling value can in principle be obtained either from the 1H-coupled 13C NMR spectrum or from the 13C satellites of the 1 H NMR spectrum. If the main 1 H peaks are well resolved, the latter represents a much more efficient approach given the higher sensitivity of 1H vs 13C NMR spectroscopy (Citation50). The one-bond JCH coupling constant value can be obtained either from the 1Hcoupled 13C-NMR spectrum or from the 13C satellites of the 1H NMR spectrum. In a simple 13C-NMR spectrum, the carbene carbon appears as a singlet. But it displays two peaks in proton-coupled 13C-NMR. From this, we can calculate the 1JCH coupling constant which is related to the sigma donating properties of the ligands. The value of one-bond JCH coupling is inversely related to the sigma-donating properties. The JCH values of all ligands were listed in .
Table 1. 1JCH coupling values of NHC Ligands 2a-f in Hz.
The 1JCH coupling constant values for 2b and 2c were 165.44 and 162.29 Hz, respectively. On the other hand, the 1JCH coupling constant values of 2d and 2e were 225.19 and 220.16 Hz, respectively. The ligands 2b and 2c exhibited strong sigma donating properties, and 2d-f exhibited weak sigma donating properties.
3.2. Synthesis of PEPPSI Pd–NHC complexes 3
Our aim was to synthesize novel Pd–NHC complexes containing a PEPPSI skeleton. The target PEPPSI Pd–NHC complexes 3 were successfully synthesized by using carbene precursors 2, PdCl2 and K2CO3 as a base in pyridine at 60 °C in the presence of excess K2CO3 (Scheme 2). The colors of the obtained solid complexes were either yellow or white. These complexes, which are stable both in solution and in solid states against air, light and moisture, were obtained in low yields of 75–85%. All complexes with a benzimidazolium moiety display characteristic signals for the pyridine ligand in the 1H and 13C{1H} NMR spectra. For the 1H NMR spectra of the metal complexes, sharp peaks in the lower field belonging to the benzimidazolium salts (NCHN) were not observed between δ 10 and 12 ppm. The FTIR data clearly indicated the presence of ν(CN) at 1447.16, 1448.80, 1445.58,1400,14459 and 1431. for the PEPPSI Pd–NHC complexes 3, respectively. Unfortunately, we were unable to yield a proper single crystal from these complexes for X-ray diffraction. The signal of benzylic proton (N–CH2–Ar) belonging to Pd-NHC complexes 3a-f complexes was observed as a sharp singlet signal at around δ 6.26 ppm, 6.11 ppm,6.07 ppm,6.57 and 6.24 ppm respectively. While signals for the pyridine ring, protons are found downfield. The signals for aromatic protons appear between 7.10 and 7.68 ppm. The presence of pyridine in the coordination sphere was confirmed by characteristics 1 H NMR signals at 8.98, 7.71 and 7.29 ppm. These signals suggest that the pyridine ring coordinated to the palladium center to form a PEPPSI-type palladium complexes 3. In 13C NMR the signals of the aromatic carbons of the pyridine ring were detected at δ = 152.8, 152.2, 128.1 ppm. The characteristic Pd-C2-carbene signals of Pd-complex (3i) appear as a singlet at δ = 163.5 ppm in 13C NMR spectra. The aromatic carbons of the benzene ring resonate between 112.03 and 152.6 ppm. These results are in agreement with the data of other such complexes (Citation51–53). The contents of C, H, and N in palladium PEPPSI-type complexes 3a–f were determined by elemental analysis. The results agreed well with the theoretical formula of the complexes. The obtained fragments are typical for each palladium PEPPSI-type complexes 3a–f and can provide further evidence for the characterization of the examined compounds.
Scheme 2. Synthesis of Pd-PEPPSI complexes 3.
After the characterization of these complexes, their catalytic behavior was studied in the Suzuki coupling reaction of aryl boronic acids with aryl halides in water
3.3. Suzuki–Miyaura cross-coupling reaction
Compared to conventional phosphine-based catalysts, the NHC-PdPEPPSI complexes typically show very good activity for a diverse range of cross-coupling reactions including Suzuki–Miyaura reactions. However, the activity and selectivity of such PEPPSI complexes largely depends on the combined effects of backbone and N-substituents of the NHC (Citation54). To check the catalytic performance of our PEPPSI complex, the Suzuki–Miyaura cross coupling reaction of aryl bromides / chlorides with arylboronic acid was explored. The initial optimization study was conducted using p-bromoanisole (1mmol) and phenyl boronic acid (1.1 mol) as model substrates in presence of K2CO3 (1mmol) as base taking i-PrOH/H2O (1:3) (4 mL) as solvent at room temperature with 0.1 mol% catalyst. When a single type of solvent, such as water, EtOH, MeOH, DMF, or i-PrOH, was used, low coupling product was observed (, entries 1–10). However, increasing the volume ratio of water to any solvent, such as EtOH, DMF and i-PrOH, from 1:1 to 3:1 (v/v) and changing the solvent type, led to a significant increase in coupling product (, entries 1–14). The control experiments showed that without the Pd-PEPPSI complex 3a, only a trace amount of the product was obtained (, entry 15). As shown in , the heterocoupling of boronic acid derivatives with various aryl bromides gave the corresponding biaryl compounds in good to excellent yields, and only o-methyl bromobenzene gave a low yield (entries 4, 9, and 14). We think this is due to steric hinderance. Through the experiments performed, we were able to achieve a quantitative yield (97%) using the i-PrOH-water solvent system at ratios of 1:3 and 1:1 v/v, as indicated in , entry 11. However, when the ratio of i-PrOH or water was increased, it resulted in a decrease in the coupled product due to the solubility of the base in i-PrOH or the solubility of the complex in water. Therefore, we opted for a 1:3 ratio to achieve quantitative yields and also implemented a green chemistry procedure that utilizes more water. By screening different bases, in i-PrOH-water (1:3 and 1:1 v/v) during the Suzuki reaction including K2CO3, Na2CO3, Cs2CO3, K3PO4, KOH and K2CO3 (), Results showed that different bases produced varying levels of activity in the reaction (, entries 1–14), we found that K2CO3 was the best when using the Pd-PEPPSI complex 3a (0.1 mol %) for 1 h in water giving a 97% yield when isolated within an hour. For KOH, the yield was not satisfactory. We are quite satisfied to report that trace amount of undesired homo-coupling product was observed in optimization of Suzuki coupling reaction proceed at room temperature in air.
Table 2. The optimization for the Suzuki-Miyaura reaction of 4-bromoanisoleTable Footnotea.
Under these optimum conditions, a range of aryl bromides/chlorides containing different types of electrons donating or withdrawing substituents were screened as coupling partners with phenylboronic acid (). As shown in , the either electron-donating or electron-withdrawing groups bearing aryl bromides effectively to afford the corresponding products in good yields (51–96%) in i-PrOH/H2O mixture in 1h at rt (, entries 1–6). For example, 4-bromoacetophenone afforded a good yield in 10 min, resulting in a TOF of 1000 h−1 (, entry 2). However, -CH3 and -OCH3 substituted aryl bromides showed slightly lower activity than –NO2, -COCH3 and -CHO substituted aryl bromides due to the high nuclophilicity of the electron donating aryl bromides (, entries 1,2,5,6). Trace amount homo-coupled by-product was observed although cross-coupling reactions were carried out on air. Overall, these results direct that this PEPPSI-Pd-NHC complex efficient for the coupling of aryl bromides/chlorides with phenylboronic acid. Based on literature support we can conclude that our catalyst follow Organ-type (Citation55–Citation59) mechanism where the Pd-NHC-PEPPSI is reduced to Pd (0) followed by pyridine dissociation. The dissociation of pyridine generate the Pd(0) active species which subsequently undergo oxidative addition of aryl halide, transmetallation of phenyl boronic acid and reductive elimination of cross-coupling product complete the reaction cycle.
Table 3. Catalytic activities of 3a-f complexes in the Suzuki coupling reactions under optimized conditions.Table Footnotea
When the efficiency of catalysts is compared, the complex 3a has performed slightly higher catalytic activity then rest of complexes. Also, this catalytic system is almost equally effective for electronically rich and poor aryl bromides. For the coupling reaction of 1-chloro aryl derivatives with phenylboronic acid (, entries 1–4), the catalytic activity of the Pd-PEPPSI complex 3a at 0.01 mol % loading was not satisfactory and only 71% of the coupling product was obtained using p-nitrochlorobenzene (, entry 3) as the substrate.
Table 4. Catalytic activities of 3a complex in the Suzuki coupling reactions under optimized conditions.Table Footnotea
The yields with other substrates were very low under the same conditions. Thus, our study is pretty significant for produce coupled product at room temperature in very short time using green solvents with low catalyst loading (0.1 mol%) without need to any additives.
4. Conclusion
In this work, we have demonstrated synthesis of benzimidazolium salts (2) and PEPPSI Pd–NHC complexes and explored its potential as catalyst for the SuzukiMiyaura Cross coupling reactions of aryl halides with phenyl boronic acid. Good-to-excellent yields of cross-coupling products were obtained with a range of substrates in aqueous-isopropanol under mild reaction conditions. The structures of the obtained compounds were confirmed via 1H and 13C{1H} NMR and elemental analysis. Among these complexes, complexes 3a and 3b showed good +--catalytic performance towards various types of aryl bromides, while the other complexes showed lower catalytic activity. Ultimately, the catalyst system described here has several advantages: low catalyst loading, substrate tolerance, quantitative yield, room temperature, environmentally friendly solvent, short reaction time, and very simple procedure. The use of green solvents such as water and i-PrOH, the low catalyst loading and the fact that the reaction takes place in air at room temperature are the main advantages of this catalytic system.
Supplemental Material
Download PDF (1.4 MB)Acknowledgements
The authors extended their appreciation to the Researchers Supporting Project number (RSP2024R75), King Saud University, Riyadh, Saudi Arabia.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- (a) Li, C.J. Chem. Rev. 2005, 105, 3095–3165; (b) Simon, M.O.; Li, C.J. Chem. Soc. Rev. 2012, 41, 1415–1427.
- (a) Aravinda, R.P.; Babul, R.A.; Ramachandra, R.G.; Subbarami, R.N. J. Heterocycl. Chem. 2013, 50, 1451–1456; (b) Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483; (c) Cao, Y. Adv. Mater. Res. 2011, 284–286, 2404–2408; (d) Organ, M.G.; Chass, G.A.; Fang, D.C.; Hopkinson, A.C.; Valente, C. Synthesis. (Mass), 2008, 2776–2797; (e) Botella, L.; Nájera, C. Angew. Chem., Int. Ed. 2002, 41, 179–181; (f) Lei, Z.; Yao, J.; Xiao, Y.; Liu, W. H.; Yu, L.; Duan, W.; Li, C.-J. Chem. Sci. 2024, 15, 3552–3561; (g) Yao, J.; Yu, L.; Duan, W.; Li. C.-J. Org. Chem. Front. 2023, 10, 524–530; (h) Feng, L.; Yao, J.; Yu, L.; Duan, W. Org. Chem. Front. 2022, 9, 2351–2356; (i) Hu, S.; He, X.; Lei, Z.; Yu, L.; Duan, W. J. Mol. Struct. 2023, 1286, 135565; (j) X. He, S. Hu, Y. Xiao, L. Yu, W. Duan. Eur. J. Org. Chem. 2022, 2022, e202200731.
- Shaughnessy, K.H.; DeVasher, R.B. Curr.: Org. Chem. 2005, 9, 585–604. (b) Zhao, D.; Fei, Z.; Geldbach, T.J.; Scopelliti, R.; Dyson, P.J. J. Am. Chem. Soc. 2004, 126, 15876–15882.
- Casalnuovo, A.L.; Calabrese, J.C. J. Am. Chem. Soc. 1990, 112, 4324–4330.
- (a) Çetinkaya, B.; Gurbuz, N.; Seckin, T.; Ozdemir, I. J. Mol. Catal. A: Chem. 2002, 184, 31–38; (b) Cauzzi, D.; Lanfranchi, M.; Marzolini, G.; Predieri, G.; Tiripicchio, A.; Costa, M.; Zanoni, R. J. Organomet. Chem. 1995, 488, 115–125; (c) Cazin, C.S.J.; Veith, M.; Braunstein, P.; Bedford, R.B. Synthesis 2005, 622–626; (d) Cazin, C.S.J.; C.R. Chim. 2009, 12, 1173–1180; (e) Cazin, C.S.J.; Veith, M.; Braunstein, P.; Bedford, R.B. Synthesis 2005, 622–626; (f) Shaughnessy, K.H. Eur. J. Org. Chem. 2006, 1827–1835.
- Herrmann, W.A. Angew. Chem., Int. Ed. 2002, 41, 1290–1309. (b) Mata, J.A.; Poyatos, M.; Peris, E. Coord. Chem. Rev. 2007, 251, 841–859; (c) Azua, A.; Sanz, S.; Peris, E. Organometallics. 2010, 29, 3661–3664.
- Valentine, D.H.; Hillhouse, J.H. Synthesis. (Mass) 2003, 2437–2460.
- Alberico, D.; Scott, M.E.; Lautens, M. Chem., Rev 2007, 107, 174–238.
- Li, B.J.; Yang, S.D.; Shi, Z.-J. Synlett. 2008, 7, 949–957.
- (a) Bellina, F.; Rossi, R. Tetrahedron 2009, 65, 10269–10310; (b) Shi, S.; Nolan, S.P.; Szostak, M. Well-Defined Palladium(II)–NHC Precatalysts for Cross-Coupling Reactions of Amides and Esters by Selective N–C/O–C Cleavage. Acc. Chem. Res. 2018, 51 (10), 2589–2599.
- (a) Ackermann, L.; Vincente, R.; Kapdi, A.R. Angew. Chem. Int. Ed. 2009, 48, 9792–9826; (b) Zhao, Q.; Meng, G.; Nolan S.P.; Szostak, M. N-Heterocyclic Carbene Complexes in C–H Activation Reactions. Chem. Rev. 2020, 120 (4), 1981–2048.
- Roger, J.; Gottumukkala, A.L.; Doucet, H. Chem.: Cat. Chem 2010, 2, 20–40.
- Wu, X.F.; Anbarasan, P.; Neumann, H.; Beller, M. Angew. Chem. Int. Ed. 2010, 49, 7316–7319.
- Kuhl, N.; Hopkinson, M.N.; Wencel-Delord, J.; Glorius, F. Angew.: Chem. Int. E.d 2012, 51, 10236–10254.
- Wencel-Delord, J.; Glorius, F. Nat. Chem 2013, 5, 369–375.
- Rossi, R.; Bellina, F.; Lessi, M.; Manzini, C. Adv. Synth. Catal. 2014, 356, 17–117.
- Yuan, K.; Soule, J.-F.; Doucet, H. ACS. Catal 2015, 5, 978–991.
- Zhao, L.; Bruneau, C.; Doucet, H. Chem. Commun 2013, 49, 5598–5600.
- Negishi, E., Ed. Handbook of organopalladium chemistry for organic synthesis, John Wiley & Sons: New York, 2002; pp 3424.
- Schnurch, M.; Flasik, R.; Khan, A.F.; Spina, M.; Mihovilovic, M.D.; Stanetty, P. Eur. J. Org.Chem. 2006, 15, 3283–3307.
- Ackermann, L., Ed. Arylation Reactions of Alkynes: The Sonogashira Reaction, School of Chemistry, EaStCHEM: Weinheim, 2009.
- (a) Kaloğlu, N.; Özdemir, İ. Tetrahedron 2019, 75, 2306; (b) Slimani, I.; Boubakri, L.; Özdemir, N.; Mansour, L.; Özdemir, I.; Gürbüz, N.; Yaşar, S.; Sauthier, M.; Hamdi, N. Inorganica Chimica Acta. 2022, 532, 120747.
- Jacobsen, H.; Correa, A.; Poater, A.; Costabile, C.; Cavallo, L. Coord. Chem. Rev. 2009, 253, 687.
- Scott, N.M.; Dorta, R.; Stevens, E.D.; Correa, A.; Cavallo, L.; Nolan, S.P. J. Am. Chem. Soc. 2005, 127, 3516.
- Herrmann, W.A.; Elison, M.; Fischer, J.; Kocher, C.; Artus, G.R.J. Angew. Chem. Int. Ed. 1995, 34, 2371–2374. doi:10.1002/anie.199523711.
- Li, G.; Lei, P.; Szostak, M.; Casals Cruañas, E.; Poater, A.; Cavallo, L.; Nolan, S.P. Chem. Cat. Chem 2018, 10, 3096–3106. doi:10.1002/cctc.201800511.
- Diebolt, O.; Braunstein, P.; Nolan, S.P.; Cazin, C.S. Chem. Commun 2008, 27, 3190–3192. doi:10.1039/B804695F.
- Magi Meconi, G.; Vikrama Chaitanya Vummaleti, S.; Antonio Luque-Urrutia, J.; Belanzoni, P.; Nolan, S.P.; Jacobsen, H.; Cavallo, L.; Solà, M.; Poater, A. Organometallics 2017, 36, 2088–2095. doi:10.1021/acs.organomet.7b00114.
- Organ, M.G.; Çalimsiz, S.; Sayah, M.; Hoi, K.H.; Lough, A.J. Angew. Chem. Int. Ed. 2009, 48, 2383–2387. doi:10.1002/anie.200805661.
- Froese, R.D.J.; Lombardi, C.; Pompeo, M.; Rucker, R.P.; Organ, M.G. Acc.: Chem. Res 2017, 50, 2244–2253. doi:10.1021/acs.accounts.7b00249.
- Valente, C.; Baglione, S.; Candito, D.; O’Brien, C.J.; Organ, M.G. Chem. Commun 2008, 6, 735–737. doi:10.1039/B715081D.
- Wang, C.A.; Rahman, M.M.; Bisz, E.; Dziuk, B.; Szostak, R.; Szostak, M. ACS Catal. 2022, 12, 2426–2433. doi:10.1021/acscatal.1c05738.
- Zhou, T.; Li, G.; Nolan, S.P.; Szostak, M. Org. Lett 2019, 21, 3304–3309. doi:10.1021/acs.orglett.9b01053.
- Popp, B.V.; Wendlandt, J.E.; Andis, C.R.L.; Stahl, S.S. Angew. Chem 2007, 119, 607–610. doi:10.1002/ange.200603667.
- Valente, C.; Çalimsiz, S.; Hoi, K.H.; Mallik, D.; Sayah, M.; Organ, M.G. Angew. Chem. Int. Ed. 2012, 51, 3314–3332. doi:10.1002/anie.201106131.
- Reddy, P.V.G.; Reddy, M.V.K.; Kakarla, R.R.; Ranganath, K.V.; Aminabhavi, T.M. Environ.: Res 2023, 225, 115515. doi:10.1016/j.envres.2023.115515.
- Zhong, R.; Pöthig, A.; Feng, Y.; Riener, K.; Herrmann, W.A.; Kühn, F.E. Green Chem. 2014, 16, 4955–4962. doi:10.1039/C4GC00986J.
- Reeves, E.K.; Humke, J.N.; Neufeldt, S.R. J. Organomet. Chem 2019, 84, 11799–11812. doi:10.1021/acs.joc.9b01692.
- Mazars, F.; Zaragoza, G.; Delaude, L. Inorg. Chim. Acta 2023, 556, 121676. doi:10.1016/j.ica.2023.121676.
- O’Brien, C.J.; Kantchev, E.A.B.; Valente, C.; Hadei, N.; Chass, G.A.; Lough, A.; Hopkinson, A.C.; Organ, M.G. Chem. Eur. J 2006, 12, 4743–4748. doi:10.1002/chem.200600251.
- Karataş, M.O.; Alıcı, B. Inorg. Chem. Commun 2020, 116, 107890. doi:10.1016/j.inoche.2020.10789.
- Kaloglu, N.; Ozdemir, İ. Tetrahedron 2019, 75, 2306–2313. doi:10.1016/j.tet.2019.02.062.
- Bhattacharyya, B.; Kalita, A.J.; Guha, A.K.; Gogoi, N. J. Organomet. Chem 2021, 953, 122067. doi:10.1016/j.jorganchem.2021.122067.
- Organ, M.G.; Chass, G.A.; Fang, D.C.; Hopkinson, A.C.; Valente, C. Synthesis. (Mass) 2008, 17, 2776–2797. doi:10.1055/s-2008-1067225. (Mass).
- Touj, N.; Gürbüz, N.; Hamdi, N.; Yaşar, S.; Özdemir, İ. Inorg. Chim. Acta 2018, 478, 187–194. doi:10.1016/j.ica.2018.04.018.
- Jose, D.E.; Kanchana, U.S.; Mathew, T.V.; Anilkumar, G. J. Organomet. Chem 2020, 927, 121538. doi:10.1016/j.jorganchem.2020.121538.
- Mazars, F.; Zaragoza, G.; Delaude, L. J. Organomet. Chem 2022, 978, 122489. doi:10.1016/j.jorganchem.2022.122489.
- Casalnuovo, A.L.; Calabrese, J.C. J. Am. Chem. Soc 1990, 112, 4324–4330.
- (a) Leadbeater, N.E. Chem. Commun. 2005, 2881–2902; (b) Li, C.J. Chem. Rev. 2005, 105, 3095–3166; (c) Polshettiwar, V.; Decottignies, A.; Len, C.; Fihri, A. ChemSusChem. 2010, 3, 502–522; (d) Shaughnessy, K.H. Chapter 1: Metal-Catalyzed Cross-Couplings of Aryl Halides to Form C–C Bonds in Aqueous Media. In Metal-Catalyzed Reactions in Water; Dixneuf, P.H.; Cadierno, V.; Ed.; Wiley-VCH: Weinheim, 2013; pp. 1–46.
- (a) Chatterjee, A.; Ward, T.R. Catal. Lett. 2016, 146, 820–840; (b) Dong, D.Q.; Yang, H.; Zhou, M.Y.; Wei, Z.H.; Wu, P.; Wang, Z.L. Curr. Opin. Green Sustain. Chem. 2023, 40, 100778.
- Mazars, F.; Zaragoza, G.; Delaude, L. J. Organomet. Chem 2022, 978, 122489.
- Boubakri, L.; Dridi, K.; AlAyed, A.A.; Özdemir, İ; Yaşar, S.; Hamdi, N. J. Coord. Chem 2019, 72, 516.
- Bent, H.A. Chem. Rev. 1961, 61, 276.
- Tapu, D.; Dixon, D.A.; Roe, C. Chem. Rev. 2009, 109, 3385.
- Nakano, M.; Tsurugi, H.; Satoh, T.; Miura, M. Org. Lett 2008, 10, 1851–1854.
- Chen, L.; Roger, J.; Bruneau, C.; Dixneuf, P.H.; Doucet, H. Chem. Commun 2011, 6, 1872–1874.
- Toure, B.B.; Lane, B.S.; Sames, D. Org. Lett 2006, 8, 1979–1982.
- Kaloğlu, N.; Özdemir, İ; Günal, S.; Özdemir, İ. Appl. Organometal. Chem. 2017, 31, e3803.
- Muñoz, S.B. III; Fleischauer, V.E.; Brennessel, W.W.; Neidig, M.L. Organometallics 2018, 37, 3093–3101. doi:10.1021/acs.organomet.8b00466.