
Abstract
Aim: Endogenous interferents can cause nonselectivity in ligand binding pharmacokinetic assays, leading to inaccurate quantification of drug concentrations. We describe the development of a Gyrolab immunoassay to quantify a new modality, CB307 and discuss strategies implemented to overcome matrix effects and achieve selectivity at the desired sensitivity. Results: Matrix effects were mitigated using strategies including increasing minimum required dilution (MRD) and lower limit of quantification, optimization of antibody orientation, assay buffer and solid phase. Conclusion: The strategies described resulted in a selective method for CB307 in disease state matrix that met bioanalytical method validation (BMV) guidance and is currently used to support clinical pharmacokinetic sample analysis in the first-in-human POTENTIA clinical study (NCT04839991) as a secondary clinical end point.
Introduction
Selectivity is the ability of the ligand-binding assay (LBA) to quantify accurate levels of drug in the presence of nonspecific matrix components and is a requirement within the scope of the ICH M10 bioanalytical method validation guidelines adopted by regulatory authorities.
This paper will present the development of a Gyrolab-based LBA for the determination of a new biological modality, CB307, in human serum from patients with prostate-specific membrane antigen (PSMA) positive tumors, as a case study of approaches for the successful resolution of selectivity issues in a LBA pharmacokinetic method development.
CB307 is a novel half-life extended bispecific Humabody® therapeutic molecule that binds to CD137, PSMA and human serum albumin. CB307 is designed to deliver CD137 agonism in a tumor-specific context by targeting PSMA expressing cells in the tumor microenvironment.
Experimental
A three-step sequential Gyrolab method was developed for determination of CB307 in individuals with PSMA positive tumors using two anti-idiotype antibodies targeting the αCD137 VH and αPSMA VH domains on CB307.
The original assay format was developed on the Bioaffy 1000 CD using a tenfold minimum required dilution (MRD) in Rexxip HX, however nonselectivity was observed.
Results & discussion
Nonselectivity of a method can be caused by various endogenous matrix components such as circulating drug ligand, closely related endogenous compounds, human antispecies antibodies and more, which can bind to or block epitopes on the capture and detection antibodies or drug.
Method development variables for the Gyrolab format can include standard parameters for an immunoassay, such as buffer choice, MRD and reagent concentrations. In addition, there can also be instrument-specific parameters, such as a variety of different CD types (varying capture volume and affinity) plus spin speed, number of wash cycles and incubation times.
Strategies were implemented to overcome the matrix effects including increasing the MRD, raising the assay LLOQ, testing an alternative antibody orientation, optimizing assay buffer and selecting the most suitable solid phase for the assay.
Use of the Gyrolab Bioaffy high-capacity CDs can be particularly helpful for lower affinity capture and detection reagents and can result in improved binding within the affinity column.
Alongside selectivity in healthy individuals, assessing selectivity in disease state matrix is recommended as early as possible in method development to ensure the assay is fit for measuring drug in the target population.
Conclusion
Use of an 80-fold MRD in Rexxip HX with a 100 ng/ml LLOQ on the Bioaffy 1000 HC CD eliminated matrix effects and led to a robust method for determination of CB307. The method was validated to local BMV guidelines and supports clinical study POTENTIA (NCT04839991).
The strategies described within this research article can be used for all ligand-binding assays to overcome undesirable matrix effects.
1. Background
Understanding the pharmacokinetic (PK) properties of a drug in participants’ samples is a key feature of early-stage clinical trials. Drug exposure and the associated absorption, distribution, metabolism and excretion properties, inferred from PK, allow clinicians to better understand the toxicity and efficacy of drugs [Citation1]. This aids determination of the optimal dosing strategy for patients, informing the recommended phase 2 dose and schedule determination for future clinical development.
The quantification of large molecule drugs is typically performed using ligand-binding assay (LBA) techniques, where antibodies or ligands specific to the drug are used to capture and detect free or total drug in samples [Citation2–4]. However, the presence of nonspecific matrix components can make the accurate quantification of biologics more challenging. Factors such as soluble drug target, degrading enzymes, heterophilic antibodies and other interfering components can all impact the selectivity of a method [Citation5–6].
Selectivity is the ability of the LBA to quantify accurate levels of drug in the presence of nonspecific matrix components and is a requirement within the scope of the ICH M10 bioanalytical method validation (BMV) guidelines adopted by regulatory authorities. Assessment of selectivity is performed by exogenous addition of drug into at least 10 individual sources of blank matrices at two concentrations within the analytical range; the lower limit of quantification (LLOQ) and high-quality control (HQC) concentrations. For an assay to meet validation acceptance criteria, accuracy of the spiked selectivity samples must be within ±25.0% relative error (RE%) at the assay LLOQ and ±20.0% at the HQC in a minimum of 80.0% of the individual matrix sources evaluated. In situations where a clinical trial is being conducted in a specific target indication or disease population, selectivity must also be confirmed in individual matrix reflective of the trial participants, as endogenous matrix components may differ to that of healthy individuals. The importance of a selectivity assessment in the regulatory validation of LBA format PK assays is well established, as such regular assessment is crucial in the method development phases to achieve a reliable method validation [Citation7]. Practical approaches for resolving a lack of selectivity in method development are less well documented.
This paper will present the development of a Gyrolab-based LBA for the determination of a new biological modality, CB307, in human serum from patients with prostate-specific membrane antigen (PSMA) positive tumors, as a case study of approaches for the successful resolution of selectivity issues in a LBA PK method development. This paper will focus on resolution of selectivity at the assay LLOQ, as this is the concentration where matrix effects typically impact assay performance the most; it is however still important to confirm selectivity at both the LLOQ and HQC when final assay conditions are established. Many of the approaches discussed within this paper can be applied to mitigate selectivity and matrix effects in all LBA assays, across multiple platforms at both the LLOQ and HQC concentrations.
CB307 is a novel half-life extended bispecific Humabody® therapeutic molecule that binds to CD137 (HGNC:TNFRSF9 - also known as 4-1BB), PSMA (HGNC: FOLH1) and human serum albumin (HSA) (see ). CB307 is designed to deliver high-potency CD137 agonism in a tumor-specific context by targeting PSMA expressing cells in the tumor microenvironment [Citation8].
Figure 1. Diagram of CB307 format and structure. CB307 is made up of 3 Humabody® VH units connected with flexible peptide linkers that targets PSMA (HGNC: FOLH1), CD137 (HGNC:TNFRSF9 - also known as 4-1BB) HSA.
HSA: Human serum albumin; PSMA: Prostate-specific membrane antigen.
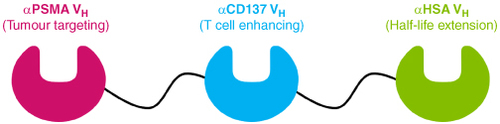
Humabody® VH units are antibody fragments known as heavy chain variable domains and are human antibody protein sequences derived from the Crescendo transgenic mouse [Citation9]. Multiple Humabody® VH domains can be joined together with peptide linkers and expressed as single polypeptide chains with scope to include different target binding valencies and/or specificities. Furthermore, the reduced molecular weight of Humabody VH with respect to monoclonal antibodies may also aid tumor penetration [Citation10].
The Gyrolab immunoassay platform was developed by Gyros Protein Technologies, Uppsala, Sweden. It is designed to automate and streamline the analysis of proteins and biomolecules in biological samples and it employs microfluidic technology to perform immunoassays in a miniaturized format, requiring only small sample volumes. This platform was selected to improve assay performance with respect to plate-based technologies used in earlier iterations of the CB307 PK assay. The Gyrolab allows for higher throughput and reduced hands-on time compared with traditional immunoassay methods [Citation11–12], but importantly the environment within the microfluidic assay column requires binding events to occur with rapid kinetics and equilibria to those which take place with a microtitre plate format. The small contact time between drug and critical reagents can reduce the impact of endogenous interferents causing method nonselectivity [Citation13].
2. Experimental
2.1. Instrument & software
Relative fluorescence units (instrument response) were measured on a Gyrolab® xP using Gyrolab® software v7.1. The raw data generated was processed using Watson LIMS v7.2, GraphPad Prism 9.4.1 and Microsoft Excel.
Anti-idiotype antibody binding to CB307 was evaluated on the Biacore 8K (Cytiva) using Biacore 8K Control Software (version 3.0.11.15423) and Biacore Insight Evaluation Software (version 3.0.11.15423).
2.2. Anti-idiotype antibodies
Two monoclonal anti-idiotype antibodies were generated against CB307, one targeting the αCD137 VH and one the αPSMA VH, from Fabs identified with Bio-rad's HuCAL platinum® phage display platform [Citation14]. The identified Anti-VH Fabs were converted to full antibodies by Crescendo Biologics with the addition of human IgG1 constant regions and provided to Resolian for use in method development. The anti-idiotype antibodies were identified as type 1 anti-idiotypes (binding only to CB307 not complexed with their respective target VH‘s analyte) during Fab generation and confirmed through assessment of target interference in the final assay format. Each anti-idiotype antibody was biotinylated with EZ-Link™ NHS-PEG4-Biotin, No-Weigh™ Format (Thermo Fisher) and labelled with Alexa Fluor™ 647 using Alexa Fluor™ 647 Antibody Labelling Kit (Thermo Fisher, MA, USA), allowing assessment of each anti-idiotype as capture and/or detection on the Gyrolab® instrument. For biotinylation reactions, antibodies were challenged at a 20-fold molar excess of label. All unconjugated material was removed by buffer exchange into phosphate-buffered saline (PBS) using Zeba desalting columns (Thermo Fisher). For Alexa Fluor™ 647 labelling reactions, 100 μg of antibody was added to an Alexa Fluor vial as per kit instructions.
2.3. Reference standard
Clinical grade CB307 was supplied at 5.0 mg/ml, in the clinical formulation buffer, by Crescendo Biologics for use in method development.
2.4. Gyrolab reagents
RexxipTM H, HN, HX and F buffers were purchased from Gyros Protein Technologies, Uppsala, Sweden. Two wash solutions were used; PBS-T (0.01%) prepared from phosphate buffered saline tablets (Oxoid, UK) with Tween-20 (Sigma-Aldrich, UK) and pH 11 Buffer (Gyros Protein Technologies AB, Uppsala, Sweden). The assay was developed using Gyrolab BioaffyTM 1000 and 1000 HC compact discs (CDs) (Gyros Protein Technologies).
2.5. Human matrices
Pooled and individual human sera were purchased from BioIVT (Burgess Hill, UK).
Human sera from individuals with identified target tumor types were provided by Crescendo Biologics for use as disease state matrix.
2.6. PK assay method (final assay format)
A three-step sequential sandwich immunoassay was developed on the Gyrolab xP using Bioaffy 1000 and 1000 HC CDs using two wash solutions (PBS-T (0.01%) and pH 11 buffer). Calibration curves were prepared by exogenous addition of CB307 into pooled human serum. Quality controls and other samples (such as selectivity and dilutional linearity samples) were also prepared by adding CB307 into human serum. Unspiked pooled human serum acted as a control blank. Following preparation, the calibration curve and all samples underwent a minimum required dilution (MRD) in Rexxip HX (except where specified in the method development).
Biotinylated capture antibody was prepared at a concentration of 100 μg/ml in Rexxip F and detection antibody was prepared at a concentration between 5 nM and 25 nM in Rexxip F and loaded onto the instrument. PBS-T (0.01%) was also loaded onto the instrument for CD microstructure washing steps.
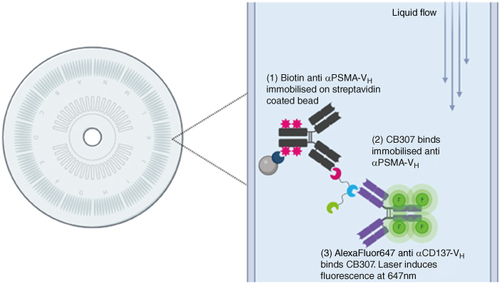
Following these steps, the run was executed on the Gyrolab xP instrument. Transfer needles within the Gyrolab instrument dispensed capture solution onto the CD microstructures where the solution is held in a volume defined chamber by a hydrophobic barrier. Following addition, the CD is spun at high speeds to push the capture solution through a column, packed with streptavidin-coated resin, by centrifugal force. As the capture is biotinylated, it binds the streptavidin coated solid phase. The microstructure is then washed with PBS-T (0.01%) to remove any unbound reagent. This process is repeated for sample and detection addition. After all assay steps are performed, laser induced fluorescence is measured from each CD microstructure; the fluorescent intensity is relative to the amount of CB307 present in the microstructure. Standards, QC’s and samples were analysed in duplicate measurements. A schematic of the assay format is presented in . Method development variables for this format can include standard parameters for an immunoassay, such as buffer choice and reagent concentration. In addition, there can also be instrument-specific parameters, such as a variety of different CD types (varying capture volume and affinity) plus spin speed, number of wash cycles and incubation times.
Figure 2. Assay format for the determination of CB307 in human serum on the Gyrolab. (1) Biotinylated Anti-αPSMA VH Anti-id binds to streptavidin beads within the 15 nl affinity column of the Gyrolab CD. Following a wash step (2) samples containing CB307 pass through the column. Any CB307 present binds to the immobilised Anti-40C5 anti-idiotype. (3) Detection antibody AlexaFluor647 conjugated Anti-αCD137 VH Anti-id binds to CB307. A laser within the Gyrolab instrument induces fluorescence from the AlexaFluor at 647 nm which is measured and is relative to the amount of CB307 present in the sample. Figure generated using BioRender.com.
2.7. Binding kinetic method
Anti-Id binding kinetics to CB307 was assessed on the Biacore 8K (Cytiva). Anti-Id surfaces were prepared via amine-coupling of Series S CM5 chips (29149603, Cytiva). CM5 surfaces were conditioned with 1:1 ratio of N-hydroxysuccinimide and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride for 6 min. Each anti-Id molecule was diluted to 2.50 μg/ml in 10 mM acetate buffer pH 5.5 (Cytiva) and injected for 1.5 min, excess reactive groups were deactivated by a 7 min injection of 1 M ethanolamine-HCl, pH 8.50. All amine-coupling reagents were sourced from Cytiva (BR100050). CB307 affinity for the generated anti-Id surfaces was assessed using the single-cycle kinetics method with association time of 2 min and dissociation time of 20 min. Four successive injections of the CB307 analyte at 12.5, 50.0, 200 and 1000 nM, were tested. The resulting data was fitted using a 1:1 binding stoichiometry model.
3. Results & discussion
Antibody orientation for both anti-idiotypes (each either acting as the capture or detection) was assessed using the Gyrolab Bioaffy 1000 CD (this CD type was selected as the suitable starting point based on the expected sensitivity and anticipated sample range). Pilot studies demonstrated that both orientations could produce acceptable curves. However, the use of the biotinylated-anti-αCD137 VH anti-id Ab acting as the capture antibody (100 μg/ml in PBS-T 0.01%), with the AlexaFluor-647 conjugated anti-αPSMA VH anti-id Ab serving as the detection antibody (25 nM in Rexxip F) provided the best sensitivity and dynamic range when a tenfold MRD was performed on serum samples and calibrators using Rexxip HX. An assay was established with a range of 30–2,500 ng/ml using a 4PL regression model with a 1/y weighting factor.
Prior to progression to a regulated validation phase, pre-validation experiments were performed to assess precision and accuracy across three occasions, dilutional linearity, prozone, carryover and selectivity of the method. The performance of these assessments in a non-regulated development study is recommended to ensure the method will meet ICH M10 BMV guidelines prior to progression to a regulated validation study [Citation15].
Results generated for precision and accuracy, dilutional linearity, prozone and carryover assessments were within acceptance limits set out in the ICH M10 validation guidelines. However, data generated in the selectivity assessment showed variable recovery of CB307 across ten healthy serum individuals spiked with drug at the anticipated LLOQ of 30.0 ng/ml, shown in (light gray bars). The accuracy (RE%) of recovered concentration from individual samples when compared with the nominal spiked concentration ranged between -30.3% and 145.0%. A high degree of over-recovery was seen in individual 1 with a back-calculated concentration of 73.5 ng/ml, nearly 2.5x the nominal spiked concentration of CB307. Under-recovery was seen in several other individuals, including 2, 4 and 6; however, only individual 1 and 6 were outside of validation acceptance criteria.
Figure 3. Selectivity at LLOQ using ten- and 20-fold MRDs.10 healthy human serum individuals were spiked with CB307 at the assay LLOQ of 30.0 ng/ml (tenfold MRD) or 60.0 ng/ml (20-fold MRD) and back-calculated recovery was compared against the theoretical concentration.
Ind: Individual; LLOQ: Lower limit of quantification; MRD: Minimum required dilution.
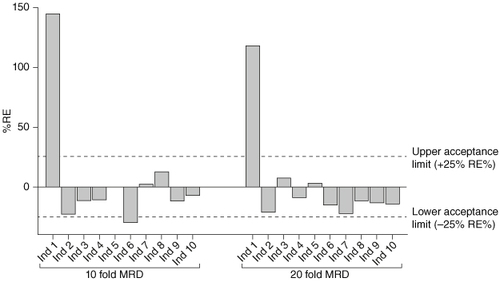
Although these results theoretically meet BMV guidelines with ≥80.0% of individuals meeting acceptance criteria of within ±25.0% RE%, the results clearly indicate the presence of matrix effects leading to inaccurate quantification of CB307 across individuals. This is of importance, as although PK assays have clear acceptance criteria, it is still critical to interpret the data scientifically and where significant over- or under-recovery is seen in any individuals, consideration must be given as to whether the assay is ‘fit for purpose’. Where an assay is destined to support decision making on dose, efficacy or toxicity within a clinical study (as is the case with CB307), evidence of matrix effects causing shifts in back-calculated concentrations must be investigated or mitigated, even when it is present in less than 20.0% of the population.
3.1. Potential causes of assay nonselectivity for detection of CB307
Where selectivity data shows variability between individuals, the cause is typically due to endogenous matrix components either specifically or nonspecifically binding to the analyte of interest or capture and detection antibodies within the assay. Determination of the specific cause of nonselectivity can be challenging due to the complexity of the matrix used and typically the focus for method development has to be on overcoming the matrix effects without fully understanding the cause.
Despite this, selectivity data can give clues to the type of interference present. Where over-recovery is seen in an individual, it may indicate nonspecificity of capture and/or detection antibodies allowing an endogenous molecule to form a successful bridge between the two, causing a false positive result. This can be caused by factors such as heterophilic antibodies, endogenous antispecies antibodies, or endogenous molecules that are closely related to the drug [Citation16].
Where under-recovery is seen, this may indicate binding of an endogenous compound to either the capture antibody or the drug itself, leading to masking of epitopes required to form a successful bridge, leading to a false negative result. Where anti-idiotypes are type 1 and therefore compete with the drug's ligand for binding, under-recovery can be caused by varying levels of soluble target within patient samples. This can be of particular importance where disease states show elevated levels of the drug's ligand or where the therapeutic can cause upregulation or cell lysis leading to shedding of target (in the case of membrane bound receptors) into the blood [Citation17]. In the case of CB307, reported serum concentrations of PSMA can be between 100 and 360 ng/ml in healthy individuals and rise to over 600 ng/ml in individuals with malignant prostate cancer which may explain some of the variation in CB307 recovery across different matrix lots [Citation18].
For the specific example shown in , where both under-recovery and over-recovery of CB307 concentrations were seen in different individuals, this can also suggest an assay issue that is not related specifically to a matrix impacting factor and that the issue may be assay reagent and/or platform related. Although strategies such as heterophilic blockers or antitarget antibodies can be used to mitigate specific interferences such as antispecies antibodies or soluble ligand, in cases where both under- and over-recovery are observed, general strategies can be employed to overcome such matrix effects [Citation19–20].
3.2. Resolving matrix effects – increasing MRD & LLOQ
Often, the simplest and most effective way to reduce matrix effects is to increase the MRD, reducing the percentage of matrix in the final sample thus diluting any endogenous interferents present. When increasing MRD, it is often necessary to also increase the concentration of drug in the whole matrix sample to maintain the same concentration post-dilution as with the previous MRD. This ensures that the ratio of interfering factors to specific analyte is lower than the previous MRD and the drug signal will not be rendered undetectable at higher dilution, increasing the chance of success.
This strategy was implemented to try and improve selectivity in detection of CB307 in human serum. The assay MRD was increased from tenfold to 20-fold and the whole matrix range was increased twofold (range of 60.0–5000 ng/ml) for the 20-fold diluted samples to maintain the same in assay concentrations of the previous MRD. Selectivity was reassessed with the same 10 individuals with both MRDs; a comparison of recovery when using a tenfold MRD and 20-fold MRD can be seen in .
Increasing the MRD to 20-fold improved recovery at the new assay LLOQ of 60.0 ng/ml, where 9/10 healthy individuals met validation acceptance criteria. However, individuals still showed poor accuracy in the measurement of CB307. Significant over-recovery of analyte remained apparent with individual 1, having a back-calculated concentration of CB307 over 2x the nominal spiked concentration and individuals 2 and 6 still showing significant under-recovery.
As the assay for CB307 was destined for use in patients with PSMA positive tumors, selectivity in disease state patients was tested with the increased 20-fold MRD to further assess the selectivity in samples from a clinically relevant population. Selectivity testing of disease state matrix within method development is often difficult to complete due to the availability of relevant matrix. Often it is performed within regulated sample analysis studies using pre-dose clinical samples as they become available. Should relevant drug naive samples be available, it is recommended to test selectivity in disease state individuals as early as possible as endogenous components and concentrations within disease state individuals can differ from healthy populations. As such, it is important to confirm the absence of matrix effects within the disease population. Samples of six patients from target tumor populations were spiked with analyte at the assay LLOQ (60.0 ng/ml) and assessed after applying a 20-fold MRD (shown in Supplementary Figure S1).
Four out of six disease state individuals showed good recovery for CB307 however significant under-recovery (individual 3) and over-recovery (individual 4) were observed in 2/6 (33.3%) of the individuals tested, indicating the selectivity results observed in the healthy populations remained present within the disease state population. This emphasized the importance of resolving the selectivity issues in this method as it was clear that the developed assay was not fit for the intended purpose.
3.3. Resolving matrix effects – reverse antibody orientation
With the current assay design, further increases to MRD was not possible as this would result in poor signal/noise at the desired whole matrix assay sensitivity. An alternative strategy to overcoming matrix effects can be to reverse the orientation of the antibody pairing to ensure the most specific and sensitive anti-idiotype is the capture antibody; this can improve assay selectivity as only the capture antibody is exposed to interfering matrix components in a sequential assay format.
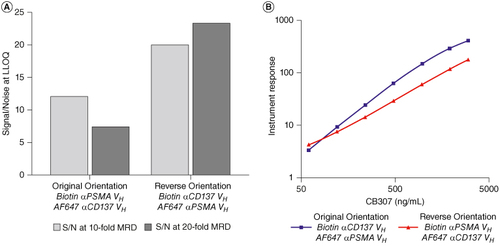
Initial orientation assessments, carried out with a tenfold MRD, demonstrated that the original assay orientation (Biotinylated- Anti-αCD137 VH Anti-id Ab as capture and Alexa647 conjugated anti-αPSMA VH anti-id Ab as detection) demonstrated a twofold improvement in assay sensitivity compared with the reverse orientation (Biotinylated- anti-αPSMA VH anti-id Ab as capture and Alexa647 conjugated anti-αCD137 VH anti-id Ab as detection). However, further analysis of the reverse orientation with a 20-fold MRD demonstrated a significant decrease in blank signal compared with the original orientation, leading to a 4–fivefold improvement in signal/noise (S/N) ratio at the assay LLOQ (A). Interestingly, greater sensitivity was also achieved with a tenfold MRD with the reverse orientation, differing from the results initially obtained. Higher S/N ratio allows for further increases in MRD, if required, without reducing whole matrix sensitivity. Furthermore, the slope of the calibration curve with the reverse orientation was less steep providing a wider dynamic range (B).
Figure 4. Comparison of original and reverse antibody orientations. (A) S/N of CB307 at LLOQ of 60.0 ng/ml using a tenfold and 20-fold MRD with original antibody orientation (Capture: Biotin anti-αCD137 VH Anti-id, Detection: AlexaFluor647 anti-αPSMA VH anti-id) and reverse antibody orientation (Capture: biotin anti-αPSMA VH anti-id, Detection: AlexaFluor647 anti-αCD137 VH anti-id). (B) Calibration curve of CB307 (between 60.0 and 4000 ng/ml) with a 20-fold MRD using the original and reverse antibody orientation.
S/N: Signal/noise; MRD: Minimum required dilution.
A selection of individuals showing poor and acceptable accuracy from previous experiments (disease state individuals 2, 3, 4 and healthy individual 4) were used to perform a selectivity assessment using both the original and reverse assay orientation. CB307 was spiked at a LLOQ of 60.0 ng/ml and a 20-fold MRD was applied as in previous experiments. Results are presented in .
Figure 5. Selectivity in individuals showing matrix effects in previous experiments using the original and new antibody orientation. CB307 added into four individuals at 60.0 ng/ml using a 20-fold MRD and back-calculated recovery was compared against the theoretical concentration.
DS: Disease State; Ind: Individual; MRD: Minimum required dilution; RE: Relative error.
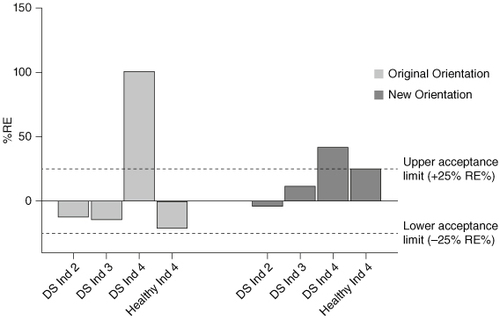
The accuracy of spiked drug recovery from the four individuals tested showed overall improvement with the new assay orientation compared with the original orientation. Disease state individual 4 still failed to meet validation acceptance criteria with both assay formats and other individuals still demonstrated poor recovery.
The improved accuracy with the new assay orientation likely stems from several factors. Firstly, the increased S/N at the LLOQ alone can be helpful in improving selectivity results as variation in background signals of individuals is less likely to impact recovery of analyte. Furthermore, it is likely the anti-αPSMA VH anti-id Ab shows higher specificity for CB307 than anti-αCD137 VH anti-id Ab. As such, use of the anti-αPSMA VH anti-id as capture antibody led to a more selective method as this antibody is more suited to selectively binding CB307 in the presence of interfering matrix components. It is still important to consider the impact of circulating target when reversing the antibody orientation for a multispecific drug such as CB307. Although selectivity improved with use of the anti-αPSMA VH anti-id as capture compared with the anti-αCD137 VH anti-id, soluble PSMA exists at much higher circulating serum concentrations than CD137 and therefore the likelihood of target interference may increase. Where target concentrations differ significantly between individuals and/or between healthy and disease state populations, assay target interference can cause under-recovery of analyte in selectivity assessments, as much of the drug is complexed to the target, preventing binding to the capture antibody. Target interference for determination of CB307 was tested in the eventual method validation by mixing recombinant CD137 and PSMA with CB307 QCs at the assay LLOQ and ULOQ and allowing to incubate for 1 h at 37°C prior to analysis (a summary of the final validation data is presented in Supplementary Tables S1–S4). As expected with type 1 anti-idiotypes, increasing concentrations of both PSMA and CD137 did show evidence of interference in the detection of CB307, particularly at the assay LLOQ. This does not invalidate PK assays but does inform stakeholders that the assay measures free drug only and upstream data interpretation should take this into consideration. It is recommended to review PK profiles alongside PD data to understand if elevated target is impacting PK measurements [Citation21–22].
3.4. Resolving matrix effects – assay buffer
Change in capture and detection antibody orientation alone did not sufficiently resolve the selectivity issues. Different assay buffers and higher MRDs were investigated in both orientations. Different Rexxip buffers, which contain a variety of additives such as salt, detergent and heterophilic blockers; can dissociate or block endogenous molecules from interfering in determination of analyte and therefore optimization can be useful in resolution of matrix effects.
Disease state individuals 2–4 and healthy individual 1 were tested for selectivity at a slightly higher LLOQ of 100 ng/ml with MRDs of tenfold and 20-fold in three different diluents: Rexxip HX, HN and H, in both orientations. The increased S/N, at lower concentrations, provided by the changed orientation allowed for the investigation of higher MRDs in this orientation without the loss of sensitivity. MRDs of 40-fold and 60-fold were tested in Rexxip HX in disease state individual 4 and healthy individual 1, in the reverse orientation only. No improvement or poorer performance were observed with the original orientation with different buffers, this data has not been presented. Supplementary Figure S2 shows recovery of spiked individuals at different conditions in the reverse orientation.
Results presented in Supplementary Figure S2 show the impact assay buffer selection and increasing MRD can have on selectivity results. The use of Rexxip HN yielded poor recovery and large variation between individuals tested at both tenfold and 20-fold MRD. Rexxip H showed better performance at a tenfold MRD with all 4 individuals within validation acceptance criteria; however, when the MRD was increased to 20-fold, individuals fell outside of acceptance criteria. The most accurate results were obtained with Rexxip HX, recovery of CB307 improved with each increase in MRD; all individuals fell within validation acceptance criteria at a 40-fold MRD or greater.
As ingredients of Rexxip buffers are proprietary, it is difficult to gain mechanistic insight into why certain Rexxip buffers can improve selectivity more than others. Rexxip HX, the buffer selected in this assay, is described as having agents to neutralise heterophilic antibodies and suit negatively charged analytes; whereas the other buffers tested suit positively charged analytes. Factors such as the isoelectric point of the analyte, the formulation of heterophilic blockers present and salt and detergent content can all influence assay selectivity [Citation23]. Furthermore, this dataset reiterates the importance of selecting a suitable MRD to dilute out any interfering matrix components. Following this experiment, a further selectivity screen was performed with seven new healthy individuals (individual 11–17) with drug added at the LLOQ of 100 ng/ml and diluted with either a 40-fold or 80-fold MRD in Rexxip HX. Results are presented in Supplementary Figure S3. All individuals tested met validation acceptance criteria at both MRDs, however an 80-fold MRD was selected as this MRD yielded the most accurate recovery of CB307.
3.5. Resolving matrix effects – solid phase selection
Following optimization of the assay buffer detailed above, a pre-validation study was performed where key validation parameters were assessed in a non-regulated study to ensure the method was likely to pass BMV guidance. Selectivity was assessed in a panel of six previously untested healthy individual samples with 80.0% of tests meeting validation acceptance criteria at the assay LLOQ. Despite this, three out of the five acceptable individuals were very close to failing acceptance criteria, with relative error of +25.0%, +19.0% and +21.0% respectively.
Alongside this, failure of duplicate precision, due to %CV >20.0% (or 25.0% at LOQs), was noted to occur more regularly than expected for a Gyrolab assay. Use of the Gyrolab column viewer can provide information on the fluorescence intensity within the 15 nl streptavidin column in the Gyrolab CD microstructure [Citation24]. Upon review of these profiles, it was apparent that abnormally broad peaks were observed (shown in ).
Figure 6. A column profile showing fluorescent intensity within the 15 nl affinity column of the Bioaffy 1000 CD and Bioaffy 1000 HC CD. The profile shows fluorescent intensity, radius of the affinity column and angle of the affinity column on the y-axis, z-axis and x-axis, respectively. High fluorescent intensity is detailed by a red peak, whereas low fluorescent intensity is detailed by a blue peak. The white box is known as the Integration Area and is where the instrument measures the fluorescence. (A) Column profile on the 1000 CD for CB307 determination; a broad, poorly defined peak extending outside the integration area is shown. (B) Column profile on the 1000 HC CD for CB307 determination; a sharp, well-defined peak contained within the integration area is shown.
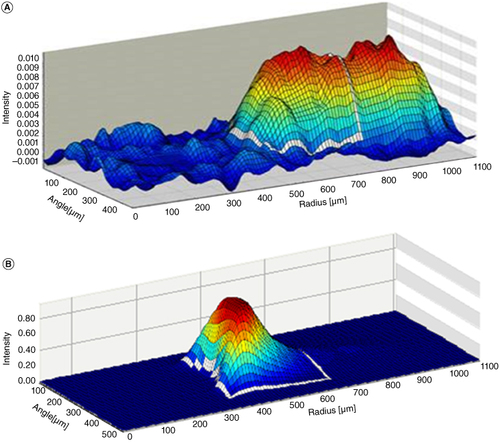
Ideally, peak of fluorescence intensity should be sharp, well defined and sit entirely within the integration area (shown by the superimposed white box in ) indicating high affinity of capture and detection reagents to the analyte of interest. Where a broad peak is observed, this indicates that affinity of the anti-idiotypes to CB307 may be lower than required for a robust assay on the standard CD type. This can lead to the fluorescent peak mainly falling outside of the integration area, toward the back of the column, resulting in variability between data points. This may also impact selectivity data where affinity is not high enough to outcompete interfering endogenous compounds.
The affinity of the anti-αCD137 VH anti-id Ab and anti-αPSMA VH anti-id Ab for CB307 was assessed at Crescendo Biologics using a Biacore 8K (Cytiva). Binding of CB307 to immobilised anti-αCD137 VH anti-id Ab and anti-αPSMA VH anti-id Ab was shown to have an equilibrium binding constant (KD) of 20.0 nM and 68.0 nM, respectively (binding kinetics parameters shown in ). Although these affinities are not low, high affinity antibodies are considered to have KD in the low nanomolar range or even picomolar range [Citation25]. From our experience, standard Gyrolab CDs suit antibodies with higher affinities and with high on-rate constants (kon) due to the shorter contact time required between reagents in the capture column of CDs. Where this is not the case, use of the higher capacity (HC) CDs from Gyrolab can be helpful as these are designed to accommodate antibodies with reduced affinity to its target [Citation26].
Table 1. CB307 binding kinetics to anti-αCD137 VH anti-id and anti-αPSMA VH anti-id (anti-idiotype) Abs.
As a result of these data, the assay was transferred from the standard 1000 CD to the 1000 HC CD with the view that column profiles for added drug control samples could be further improved on this CD type, which in turn may improve selectivity results. Selectivity was retested at the assay LLOQ with an 80-fold MRD in Rexxip HX in seven new healthy individuals on both the 1000 CD and 1000 HC CD within the same analytical run, results of which are shown in .
Figure 7. Comparison of selectivity results in healthy individuals at assay LLOQ using the Bioaffy 1000 CD and Bioaffy 1000 HC CD. Seven healthy individuals were spiked at the LLOQ of 100 ng/ml, with an 80-fold MRD applied and analyzed on the Bioaffy 1000 and Bioaffy 1000 HC CD. Back-calculated recovery was compared against the theoretical concentration.
LLOQ: Lower limit of quantification; MRD: Minimum required dilution; RE: Relative error.
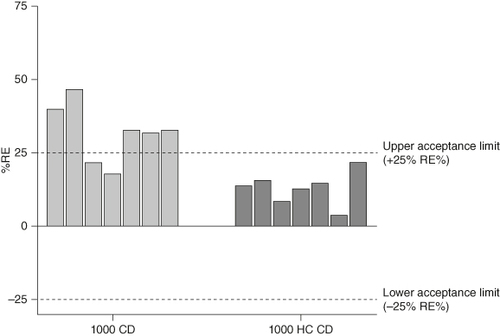
The data generated from this experiment showed a significant improvement in selectivity when using the 1000 HC CD compared with the standard 1000 CD. Only 2/7 individuals met validation acceptance criteria using the standard Bioaffy 1000 CD, whereas all individual test samples were within acceptance criteria when the Bioaffy 1000 HC CD was used. To confirm this change was key to improving selectivity, disease state individuals were reassessed on this CD type, again at the assay LLOQ.
Back-calculated recovery of CB307 in all ten disease state samples at the assay LLOQ showed a high degree of accuracy on the 1000 HC CD, with %RE all within ±5.9% RE% (shown in Supplementary Figure S4). As the method is destined to support dosing in disease state patients only, robust selectivity in this population was of critical importance. Final disease state and healthy selectivity results obtained in the method validation are shown in Supplementary Table S2.
As with Rexxip buffers, the components of the 15 nl affinity columns of the various Gyrolab Bioaffy CDs are proprietary technology which makes it challenging to interpret why use of the HC CD yielded the observed improvements in selectivity for CB307. Gyros describes the HC CDs as having a more porous resin which lends itself to lower affinity reagents [Citation27]; perhaps this change in resin structure alters the flow or contact time of analyte to the critical reagents in the column, allowing lower affinity reagents to bind the analyte more selectively over interfering factors.
The choice of solid phase is not typically considered when resolving selectivity issues, however this data shows that selection of solid phase can be a key component in assay selectivity, particularly on the Gyrolab. In the development of an assay for determination of CB307, transfer of the assay to a more suitable CD was one of the most impactful changes in improving selectivity. Conditions established on the Bioaffy 1000 HC CD provided a fit-for-purpose assay to support clinical sample analysis and as such it was out of scope of the project to test if simply changing the solid phase at the start of the assay development would have resolved selectivity issues without the requirement to raise the LLOQ/MRD and reverse the antibody orientation. In any case, selection of a suitable solid phase and antibody orientation in combination with increasing assay LLOQ and MRD yielded a selective and robust analytical method for a phase 1 clinical study.
Following the implementation of various strategies detailed within this paper, the method for determination of CB307 in human serum from PSMA positive individuals was successfully validated where precision, accuracy, selectivity (in healthy and disease state matrix), dilutional linearity, prozone and stability successfully met acceptance criteria detailed within BMV guidance. A summary of the bioanalytical validation results for precision, accuracy, selectivity, dilutional linearity and target interference is presented in Supplementary Tables S1–S4. The method is currently being used to analyse samples from the first-in-human clinical study POTENTIA (NCT04839991) [Citation28].
4. Conclusion
Accurate quantitation of drug concentrations to understand the PK characteristics of developmental drugs is critical to the drug discovery process. The advancement of new therapeutic modalities such as antibody/protein fragments and multispecific molecules can increase the complexity faced when developing assays for these novel molecules.
The importance of demonstrating selectivity in PK assays has been well documented, however practical approaches to improving nonselectivity of an assay has been less well explored in the literature.
This paper explores strategies used to overcome nonselectivity in the measurement of CB307, a multispecific Ab fragment molecule, in human serum. These included:
Increasing MRD and assay LLOQ.
The importance of testing disease state selectivity, as relevant, as early as possible.
Testing orientations of antibody pairings and revisiting this component as other parameters change.
Testing different assay buffers with varying levels of heterophilic blockers, detergent, salt etc.
Ensuring the optimal solid phase is used for the assay.
Although this case study focused on the Gyrolab immunoassay platforms and a multispecific Ab fragment, these same strategies can be used on alternative platforms and protein therapeutics such as ELISA or Meso Scale Discovery (MSD) technologies. For instance, testing of high bind, standard bind and small spot streptavidin plates on the MSD may provide similar improvements in assay performance as switching CD type on the Gyrolab.
This paper also highlights the importance of fully understanding the affinity of the critical reagents for the analyte. This information can help guide method developers in selecting the most optimal antibody orientation and solid phase. In our case the characteristics of the relationship between assay reagents and test item were such that plate based methods had proved unsuitable and an acceptable path forward was achieved with a Gyrolab method, with lower affinity antibodies. In other circumstances where affinity for the analyte is low, use of plate-based methodologies may be more suitable owing to the increased contact time between critical reagents, during longer incubation steps, allowing binding reactions to reach equilibrium. Where affinity is high, the Gyrolab may be a preferable option as the shorter contact time between critical reagents will not impact the critical reagents binding the analyte but may be low enough to overcome interference from endogenous molecules in the matrix.
Finally, this paper demonstrates the requirement for using the BMV guidance alongside scientific interpretation of data. Throughout this paper, many of the selectivity assessments theoretically met validation acceptance criteria; however, there were clearly matrix effects present in the failing 10.0–20.0% of individuals tested. Had this been taken forward to method validation, this may have resulted in failure of the selectivity assessment in validation of a new set of individuals. More importantly however, had this progressed to analysis of clinical samples, it is possible that 10.0–20.0% of patient results may be inaccurate, potentially impacting PK characteristics and clinical decision making around dosage and safety. For this reason, we recommend a strict adherence to performing a pre-validation piece of work to fully understand method performance prior to entrance into method validation. A regulated validation should contain no surprises and should only serve to confirm what you already know. Performing a comprehensive pre-validation is crucial along with setting tighter acceptance limits for parameters assessed during the pre-validation phase of method development to ensure assay robustness and suitability moving into the next stage of the method's lifecycle.
Author contributions
TW did the experimental design, generated the experimental data and is the main contributor to the writing of the manuscript. RH directed, reviewed and gave input into experimental design; reviewed the experimental data and manuscript; edited the manuscript. PB reviewed and gave input into the experimental design; reviewed the experimental data and manuscript; edited the manuscript. AP reviewed and edited the manuscript and funded and directed the work. AS and LJ did the generation of antibody affinity data and writing of affinity data section; reviewed the experimental data and manuscript. HP was the principle investigator on the human trial that provided the disease state material used for the selectivity assessment and reviewed the experimental data and manuscript.
Financial disclosure
Financial support was provided by Crescendo Biologics. All authors have financial relationships in as much as they are employees of Resolian, Crescendo Biologics or the University of Surrey. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Writing disclosure
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
The full name of the institutional review board which approved the study: Surrey Research Ethics Committee, Senate House, University of Surrey, Guildford, Surrey, GU2 7XH, UK.
Supplementary Materials
Download Zip (299.1 KB)Supplemental material
Supplemental data for this article can be accessed at https://doi.org/10.1080/17576180.2024.2365545
Competing interests disclosure
The authors have no competing interests or relevant affiliations with any organization or entity with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, stock ownership or options and expert testimony.
References
- Guttendorf R. Meeting the demands of regulatory requirements: the significance of ADME. Bioanalysis. 2012;4(12):1395–1397. doi:10.4155/bio.12.135
- Lee JW, Kelley M, King LE, et al. Bioanalytical approaches to quantify “total” and “free” therapeutic antibodies and their targets: technical challenges and PK/PD applications over the course of drug development. AAPS J. 2011;13(1):99–110. doi:10.1208/s12248-011-9251-3
- Lequin RM. Enzyme Immunoassay (EIA)/Enzyme-Linked Immunosorbent Assay (ELISA). Clin Chem. 2005;51(12):2415–2418. doi:10.1373/clinchem.2005.051532
- Tu J, Bennett P. Parallelism experiments to evaluate matrix effects, selectivity and sensitivity in ligand-binding assay method development: pros and cons. Bioanalysis. 2017;9(14):1107–1122. doi:10.4155/bio-2017-0084
- Wang Q, Ma M. Are matrix effects the Achilles heel in the bioanalysis of biotherapeutics by ligand-binding assays? Bioanalysis. 2014;6(8):1041–1044. doi:10.4155/bio.14.62
- White A. Ligand-binding assays vs chromatographic platforms for oligonucleotide quantification. Bioanalysis. 2023;15(2):53–55. doi:10.4155/bio-2023-0032
- EMA/CHMP/ICH/172948/2019. ICH guideline M10 on bioanalytical method validation. European Medicines Agency [Internet]. [ cited 2023 Aug 25]. Available from: https://www.ema.europa.eu/en/ich-m10-bioanalytical-method-validation-scientific-guideline
- Archer S, Brailey PM, Song M, et al. CB307: a dual targeting costimulatory Humabody VH therapeutic for treating PSMA-positive tumors. Clin Cancer Res. 2024;30(8):1595–1606. doi:10.1158/1078-0432.CCR-23-3052
- Teng Y, Young JL, Edwards B, et al. Diverse human VH antibody fragments with bio-therapeutic properties from the Crescendo Mouse. New Biotechnology. 2020;55:65–76. doi:10.1016/j.nbt.2019.10.003
- Nessler I, Khera E, Vance S, et al. Increased tumor penetration of single-domain antibody–drug conjugates improves in vivo efficacy in prostate cancer models. Cancer Res. 2020;80(6):1268–1278. doi:10.1158/0008-5472.CAN-19-2295
- Mora JR, Obenauer-Kutner L, Vimal Patel V. Application of the GyrolabTM platform to ligand-binding assays: a user's perspective. Bioanalysis. 2010;2(10):1711–1715. doi:10.4155/bio.10.122
- Liu R, Hoffpauir B, Chilewski SD. Accelerating regulated bioanalysis for biotherapeutics: case examples using a microfluidic ligand-binding assay platform. AAPS J. 2017;19(1):82–91. doi:10.1208/s12248-016-0006-z
- Yang T-Y, Uhlinger DJ, Ayers SA, et al. Challenges in selectivity, specificity and quantitation range of ligand-binding assays: case studies using a microfluidics platform. Bioanalysis. 2014;6(8):1049–1057. doi:10.4155/bio.14.60
- Prassler J, Thiel S, Pracht C, et al. HuCAL PLATINUM, a synthetic Fab library optimized for sequence diversity and superior performance in mammalian expression systems. J Mol Biol. 2011;413(1):261–278. doi:10.1016/j.jmb.2011.08.012
- Meng M, Wang L, Voelker T, et al. A systematic approach for developing a robust LC-MS/MS method for bioanalysis. Bioanalysis. 2013;5(1):91–115. doi:10.4155/bio.12.295
- Selby C. Interference in immunoassay. Ann Clin Biochem. 1999;36(6):704–721. doi:10.1177/000456329903600603
- Xiao Z, Adam BL, Cazares LH, et al. Quantitation of serum prostate-specific membrane antigen by a novel protein biochip immunoassay discriminates benign from malignant prostate disease. Cancer Res. 2001;61(16):6029–6033.
- Spriggs FP, Duriga N, Rathi A, et al. Resolution of matrix interference: quantitative and quasi-quantitative ligand-binding assays case studies. Bioanalysis. 2014;6(8):1093–1101. doi:10.4155/bio.14.74
- DeForge LE, Loyet KM, Delarosa D, et al. Evaluation of heterophilic antibody blocking agents in reducing false positive interference in immunoassays for IL-17AA, IL-17FF and IL-17AF. J Immunol Methods. 2010;362(1–2):70–81. doi:10.1016/j.jim.2010.09.004
- Chen J, Kendra K, Torri A, et al. Overcoming multimeric target interference in a bridging immunogenicity assay with soluble target receptor, target immunodepletion and mild acidic assay pH. Bioanalysis. 2020;12(15):1071–1085. doi:10.4155/bio-2020-0110
- Roskos LK, Schneider A, Vainshtein I, et al. PK–PD modeling of protein drugs: implications in assay development. Bioanalysis. 2011;3(6):659–675. doi:10.4155/bio.11.28
- Stevenson LF. Biomarkers, PK and immunogenicity: are we ready for integration? Bioanalysis. 2016;8(22):2287–2289. doi:10.4155/bio-2016-0226
- Schuurmans Stekhoven FMAH, Gorissen MHAG, Flik G. The isoelectric point, a key to understanding a variety of biochemical problems: a minireview. Fish Physiol Biochem. 2008;34(1):1–8. doi:10.1007/s10695-007-9145-6
- Engström J. Gyrolab immunoassays: miniaturization, automation and integration into a rapid workflow. Methods Mol Biol. 2023;2612:109–127. doi:10.1007/978-1-0716-2903-1_9
- Drake AW, Myszka DG, Klakamp SL. Characterizing high-affinity antigen/antibody complexes by kinetic- and equilibrium-based methods. Anal Biochem. 2004;328(1):35–43. doi:10.1016/j.ab.2003.12.025
- Gyros Protein Technologies. D0012493 PIS-Gyrolab-Bioaffy-CDs.pdf. (2022) [ Internet] [ cited 2023 Aug 31]. Available from: https://www.gyrosproteintechnologies.com/hubfs/GPT/pdfs/3-Gyros/Marketing%20material/PIS/D0012493%20PIS-Gyrolab-Bioaffy-CDs.pdf
- Gyros Protein Technologies. D0026196 Gyrolab-assay-developement-guide.pdf. (2020) [ Internet] [ cited 2024 Mar 8]. Available from: https://www.gyrosproteintechnologies.com/hubfs/GPT/pdfs/3-Gyros/User%20Zone/D0026196%20Gyrolab-assay-developement-guide.pdf
- Crescendo Biologics Ltd. A phase 1 open-label, dose escalation and expansion trial to investigate the safety, pharmacokinetics and pharmacodynamics of CB307, a trispecific Humabody® T-cell enhancer, in patients with PSMA+ advanced and/or metastatic solid tumours. [ Internet] [ cited 2024 Jan 1]. Available from: https://clinicaltrials.gov/study/NCT04839991