
Abstract
Aim: To demonstrate the importance of critical reagent characterization for immunogenicity assay development for multi-specific drugs using two case studies. Methods: Bridging anti-drug antibody (ADA) assay with acid-dissociated samples were used for both cases. Results: In the first case study, the unexpected interference in an ADA assay from clinical samples was identified; a model was created to replicate the issue, and an anti-target antibody was identified to mitigate the target interference. In the second case study, an issue due to non-specific binding of a domain-specific confirmatory reagent was identified, and various mitigation techniques were evaluated. Conclusion: A thorough characterization of the critical reagents helped identify the issues with these ADA case studies and provided strategies for resolving them.
Case study 1: multimeric target interference in anti-drug antibody (ADA) assay
When drugs involve heavily glycosylated targets, an in-study cut point may be more prudent. Geographical differences, stage of progression of disease may affect the target glycosylation impacting its affinity for the drug, eventually impacting the cut point.
Given the unknowns of sample purification impact, patient-derived purified heterogenous targets may not always be the gold standard for target interference assessments in an ADA assay. Homogenous recombinant targets whose sequences have been verified may perform better in replicating target interference issues observed with patient samples.
Case study 2: interference by domain-specific confirmatory reagent in ADA assay
Bispecific biologics require additional critical reagents to support immunogenicity assessments like domain-specific confirmatory reagents which may require additional characterization at the bioanalytical laboratory.
Utilizing an optimal concentration of the sample serum could be an alternative to adding serum for blocking non-specific binding in an ADA assay. Assay sensitivity and drug tolerance are key factors to test when utilizing this method.
1. Background
Bioanalytical strategies for biologics are a critical part of drug discovery and development because they allow the deduction of optimized dosage to maximize the benefit-to-risk ratio, as well as determine the reaction of the immune system against the drug. The success of a bioanalytical assay relies heavily on its critical reagents. With increasing complexity of drug modalities like bispecific antibodies, multi-specific antibodies and antibody–drug conjugates, there is an increase in the complexity of the bioanalytical assays needed to support drug development [Citation1,Citation2]. This results in the increased demand for complex critical reagents needed for the continuum of assay development, validation and sample analysis. Bispecific modalities are of special interest due to their broad range of clinical applications [Citation3], however specific guidance and white papers do not currently discuss the challenges with bioanalysis of bispecific molecules, especially regarding generation of critical reagents for long-term ligand binding assay (LBA) support.
Many articles detail reagent generation, characterization, monitoring and life cycle management of LBA critical reagents [Citation4–8]. Bispecific antibody therapeutics often require additional protein engineering for the generation of anti-idiotypic (anti-ID) antibodies for pharmacokinetic (PK) assay development or for surrogate positive controls for immunogenicity assays against different domains of the molecule. Anti-ID generation can produce a ‘broad toolbox’ of reagents for assay development [Citation9] and engineered protein domains of the bispecific may also serve as critical reagents for immunogenicity domain specificity.
Along with the increased complexity in reagent generation for bispecific drugs, interference from one or both targets may impact bioanalytical assays. Target interference can present as either false positive or false negative in immunogenicity assays [Citation10,Citation11], therefore it is important to have a relevant target during assay development as the target biology under the influence of the drug might not be well known. If the target is soluble, having disease-state individuals with known target levels can reduce the risk of developing a non-target tolerant assay. However, this comes with the challenges of being able to measure the relevant target in the matrix for bioanalytical assay development. Bispecific antibody therapeutics can double the workload of this type of characterization. The matrix lots should also be diverse in terms of disease progression and geographic location to cover all possible stages of clinical development. Bioanalytical challenges abound for soluble target interference. Many papers have been published on mitigation strategies, but few detail strategies for bispecific drugs [Citation12–14].
In this article, we report a couple of case studies that reinforce the requirement for appropriate characterization of critical reagents to support immunogenicity assays for these complex modalities. Both these case studies involve bispecific molecules and their clinical anti-drug antibody (ADA) assays had issues attributed to different critical reagents. The first case study discusses an issue arising from multimeric targets, and the second case study discusses an issue arising from a domain-fragment in the domain-specific confirmatory assay, which is not a concern with single target antibodies. The case study involving the multimeric target interference chronicles how despite prior knowledge of having a multimeric target, target interference was not identified during assay development, only to be identified during validation. It further elaborates on mitigation techniques and how a masking reagent that binds to the soluble target was eventually used to mitigate the interference. The second case study delves into a critical reagent-related assay interference in a domain-specific confirmatory clinical ADA assay. We discuss the various approaches that were utilized to identify and mitigate this domain-specific interference.
We hope these case studies reaffirm the importance of appropriate critical reagent characterization, especially ones associated exclusively with bispecific molecules. These critical reagents exclusive to bispecific molecules potentially do not go through the same level of scrutiny as these are utilized primarily for bioanalysis and are not meant for human use and might require additional scrutiny on the part of the bioanalytical laboratory.
2. Materials & methods
2.1. Case study 1: multimeric target interference in ADA assay
2.1.1. Equipment
The following instruments were used: Meso Scale Discovery (MSD) Sector Imager SQ120 (SN # 13001909171389), Biotek Microplate Washer 405 TS (Biotek, SN # 160913C), Orion Star A211 pH meter (Thermo Scientific, SN # X35584).
2.1.2. Reagents & materials
Affinity-purified rabbit anti-drug polyclonal antibody (pAb) from Biogenes (Berlin, Germany) was used for the positive controls (PCs), while pooled healthy human plasma from BioIVT was used for negative control (NC). The PC was generated by immunizing rabbits with the drug and affinity purified using the drug to purify anti-drug antibodies. An irrelevant human IgG column was then utilized to remove antibodies specific to the Fc region of the drug from the affinity-purified anti-drug antibody mix. Individual disease-state human plasma from BioIVT and Cureline were utilized for cut point determination. Biotinylation and ruthenium conjugation of drug were carried out at EMD Serono using EZ Link Sulfo-NHS-LC Biotin (Thermo Fisher # 39257) and MSD Sulfo-tag™ NHS-Ester (MSD # R91AO-2) kits respectively according to manufacturer's instructions with challenge ratios of 20:1 and 10:1, respectively. Additionally, acetic acid (Fisher Chemical # A35-500), MSD 4X read buffer (MSD # R92TC-1), 1.5M Tris-HCl, pH 10 (Teknova # R92TC-1), SuperBlock™ blocking buffer (Thermo Scientific # 37515), MSD 96-well streptavidin-coated plate (MSD # L15SA-1) and PBST 0.05 (wash buffer, prepared at EMD Serono) were used for the assay development.
For assessments of target A and B in the plasma of the patient samples, commercial research kits from Abcam and R&D systems were utilized respectively and the protocol provided with the kits were utilized to measure the concentrations of the respective targets.
2.1.3. Electrochemiluminescence bridging ADA assay
A bridging ADA assay was developed on the MSD electrochemiluminescence (ECL) platform. Samples were acid dissociated (300 mM acetic acid) for approximately 25 min (with shaking at 450 rpm) by adding approximately 10 μl of samples to 290 μl of acid (volume can be proportionally increased).
Biotinylated and ruthenylated drug (0.25 μg/ml each) were added to the neutralization master mix which included 10% (final volume) of 1.5M Tris-HCl, pH 10 in Superblock™ blocking buffer. Next, 50 μl of the acidified sample was mixed with 100 μl of neutralization master mix. This mixture was incubated at room temperature on a shaker set at 450 rpm for 120 ± 10 min. In parallel, a streptavidin coated MSD plate was blocked with 200 μl/well of superblock for at least an hour at room temperature without shaking. ADA in samples or controls form a bridge with the biotinylated and ruthenylated drug. This bridge complex was then captured on the blocked streptavidin coated MSD plate by adding 50 μl/well of the neutralized sample in duplicate wells and incubating for approx. 60 ± 10 min at room temperature while shaking at 450 rpm, followed by 6X washes which included adding 300 μl/well of PBST 0.05 using an automated plate washer. Following the wash step, 150 μl/well of 2X MSD Read Buffer was added to each well and the plate was read using MSD Sector Imager SQ120. The intensity of the ECL signal was directly proportional to the amount of ADA present in the sample. The overall minimum required dilution (MRD) of the samples from the acid dissociation and neutralization step was 1:90.
2.1.4. Electrochemiluminescence bridging ADA assay with target masking
For the mitigation of the target interference, various anti-target A proteins at different concentrations were added to this neutralization master mix along with the labeled drug, before the acidified samples were neutralized with the neutralization master mix. The remaining assay steps, including the MRD of the assay, were not impacted.
2.1.5. Preparation of control samples
Affinity purified rabbit anti-drug pAb was used to prepare the PC samples at 500, 100 and 10 ng/ml in pooled healthy human plasma. The same pooled human plasma was used as the NC for the assay.
For target tolerance assessment, 2X PC samples and 2X target samples were prepared separately in the pooled human plasma. They were then combined in a 1:1 ratio to achieve 1X concentration and incubated for at least an hour at room temperature before being frozen. The samples were thawed on the day of use and assayed as described above.
2.1.6. Screening cut point calculation
During assay development, the signals from 30 individual disease-state matrices run twice on different days were normalized to the plate NC. This was followed by Tukey's outlier test to remove the analytical outliers, followed by the removal of biological outliers. After a normality and skewness assessment using Shapiro-Wilks test, the parametric method of screening cut point calculation, where cut point = (mean + 1.645 *standard deviation) was used [Citation15].
The cut point determination at the contract research organization (CRO) was done according to the CRO's standard operating procedure utilizing 50 individual disease-state matrices that were run six-times each by multiple analysts on multiple days.
2.2. Case study 2: interference by domain-specific confirmatory reagent in ADA assay
2.2.1. Equipment
The same equipment as listed for case study 1 were used.
2.2.2. Reagents & materials
Affinity-purified rabbit anti-drug pAb from Green Mountain Antibodies (Burlington, Vermont) was used for the PCs, while pooled healthy and individual disease-state human serum were acquired from BioIVT. The PC was generated by immunizing rabbits with the drug and affinity purified using the drug to purify anti-drug antibodies. An irrelevant human IgG column was then utilized to remove antibodies specific to the Fc region of the drug from the affinity-purified anti-drug antibody mix. Biotinylation and ruthenium conjugation of drug were carried out at EMD Serono using EZ Link Sulfo- NHS-LC Biotin (Thermo Fisher # 39257) and MSD Sulfo-tag™ NHS-Ester (MSD # R91AO-2) kits respectively according to manufacturer's instructions with challenge ratios of 10:1 for both labels. Additionally, acetic acid (Fisher Chemical # A35-500), MSD 4X read buffer (MSD # R92TC-1), 1.5M Tris-HCl, pH 10 (Teknova # R92TC-1), SuperBlock™ blocking buffer (Thermo Fisher Scientific # 37515), MSD 96-well streptavidin-coated plate (MSD # L15SA-1) and PBST 0.05 (wash buffer, prepared at EMD Serono) were used for the assay development. Domain-specific confirmatory reagents were prepared at EMD Serono.
2.2.3. Electrochemiluminescence bridging ADA assay
The assay format and conditions were similar to those described above for case study 1 with the same MRD. For confirmatory assessments, 10 µg/ml of the confirmatory reagent was added to the neutralization master mix along with the labeled drugs.
2.2.4. Preparation of control samples
Affinity purified rabbit anti-drug pAb was used to prepare PC samples at 500, 100 and 10 ng/ml in pooled human serum. The same pooled human serum was used as the NC for the assay.
3. Results
3.1. Case study 1: multimeric target interference in ADA assay
The first case deals with an ADA assay against a bispecific molecule (identified as anti-targets A and B henceforth) where target A is known to be a multimer. Multimeric targets are known to be problematic with ADA assessments as they can form a bridge between the biotinylated and ruthenylated drugs, resulting in a false positive signal in a bridging ADA assay. The clinical ADA assay for this molecule was tested for interference from targets A and B during its development. Between the two targets, target B is better understood with molecules against it already in the market and a variety of literature supporting its biology. However, target A is known to be present in a multimeric form, with several repeating units and high levels of glycosylation, increasing the complexity of this target. Hence, a couple of different sources of target were used for interference assessment, the first included the peptide which was utilized for the design of the anti-A arm of the drug, while the other target was purified from a relevant patient population. However, no target interference (up to 1000 ng/ml) from either target A and B was observed during assay development, a level significantly above the shed targets observed in the majority of the patients. Additionally, during assay development, the distribution of signals from commercial treatment naive patients tested did not show any significant variability (CPF was calculated to be 1.25 using 30 samples) which would hint toward any significant interference. The assay was then evaluated for performance in the same disease-state matrix containing the same anti-coagulant but a different counter ion, before being transferred to the CRO for qualification and validation.
During the assay validation at the CRO, the sample-to-sample variability between the commercial treatment naive disease-state samples was found to be significantly higher than observed during the assay development (using a different set of commercial matrices) which resulted in a cut point factor of 3.58 (A). When healthy matrices were tested in the assay, the variability was not observed, suggesting the assay did not have any robustness issue and suggesting interference from the disease-state matrix. Without any known interferent, altering the pH of the assay and affinity capture elution (ACE) were utilized to reduce the interference without much success [Citation16,Citation17].
Figure 1. Root cause analysis for Case Study 1. (A) Spread of instrument responses from individual plasma samples from healthy and diseased individuals from cut point factor (CPF) calculations. Enhanced scatter of signals from diseased matrices caused a high CPF of 3.58. (B) Positive correlation between target A concentrations and signals in the ADA assay suggesting interference in the ADA assay by target A.
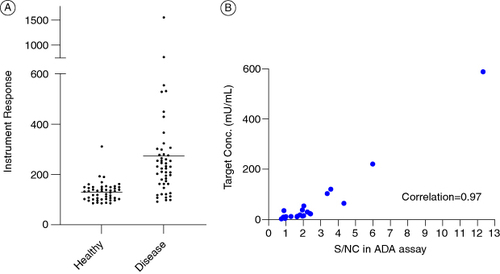
To identify the reason for this high variability, commercially available kits were utilized to measure the levels of targets A and B from a subset of the samples that were used for the cut point calculations at the CRO. The protocols mentioned in the kits were utilized without any further qualification. A positive correlation was observed between the levels of target A and the signal to noise ratio (S/N) from the ADA assay, suggesting potential target interference (B). The target A purified from patient samples was successfully detected by the kit for target A, but no interference could be introduced to the ADA assay by the introduction of the said target up to 2790 U/ml. Targets acquired from purifying patient samples are potentially the most natural representation of targets in patient populations, however the effect of the purification process on the targets is unknown. Hence, rather than proceeding with a heterogeneous mix of natural targets, it was decided to move-forward with well-defined recombinant targets to try and mimic the issue observed with actual samples. The core binding sequence of target A, which can have multiple repeats, was identified, and a couple of commercially available recombinant proteins containing 2 and 12 repeats of the core binding sequence were ordered. It was determined that the 12-repeat target can bridge the labeled drug molecules in the ADA assay setup and provide false positive ADA signals akin to the clinical samples. A concentration dependent increase in ADA signals was observed with the 12-repeat target (Supplementary Figure S1). This finally provided an experimental system to work with and test mitigation efforts. To confirm that the increased diversity of ADA signals from disease-state matrix was only from target A interference, various proteins that bind to target A were tested to demonstrate a signal reduction in the clinical samples. An antibody (shown as anti-target A reagent 3 in the figure) that binds to the target was found to have the best effect in normalizing the signals from the multiple individual lots found to have high signals during validation (A). An anti-target B reagent did not have a significant normalizing impact on the signals, confirming target A as the primary interferent (Supplementary Figure S2). Additionally, the addition of the anti-target A antibody did not have any significant effect on the performance of the positive controls and did not affect the sensitivity of the assay. The concentration of the antibody was titered to provide the best signal normalization without significantly affecting the biological differences between samples irrespective of the target. The antibody was able to reduce the bridging caused by the addition of the recombinant target suggesting the specificity of the signal reduction (B).
Figure 2. Testing masking reagents as mitigation for multimeric target interference. (A) Effect of various anti-target A masking reagents on the S/N ratio of a subset of individual human plasma lots used for the determination of the CPF. No significant impact on positive controls shown on top right. (B) Performance of the ADA assay with and without target masking antibody addition. Samples in blue on the left represent assay performed without any target A mitigation. The middle panel shows the same samples as the left but with the target A mitigation in place. The right-most graph shows the signals from all diseased individual samples tested during method development using the target A mitigation.
The assay was re-validated with the addition of the target A binding antibody as mitigation for target interference and a new cut point factor of 1.37 was established at the CRO, which significantly reduced the chances of false negatives and underestimating the presence of ADAs. The remaining assay parameters remained very similar or improved as compared with the original validation ().
Table 1. Comparison of validation results from the original assay and the target A tolerant assay for Case study 1.
It was investigated why the target-mediated difference in individual matrices was not identified during the initial assay development. It was found that the disease-state matrices used initially during assay development were procured from the US population, whereas the samples procured by the CRO during validation were procured from Europe. The differences between these two groups were not related to the levels of the target though. Since target A is heavily glycosylated, there is potential for glycosylation differences between these populations which could be attributed to various parameters, including the geographical difference and stage of progression at the time of the detection of the disease. Hence, in cases where heavily glycosylated proteins are part of the equation, it may be prudent to look closely at the target populations, the progression of the disease and closely follow the false positive rate to corroborate the cut point determined during validation.
3.2. Case study 2: interference by domain-specific confirmatory reagent in ADA assay
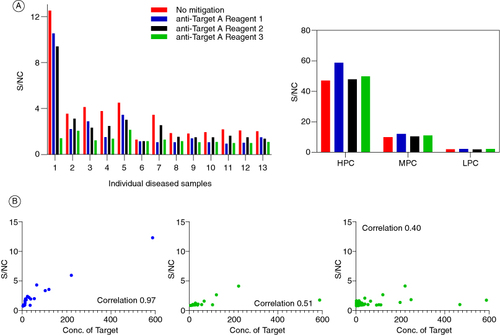
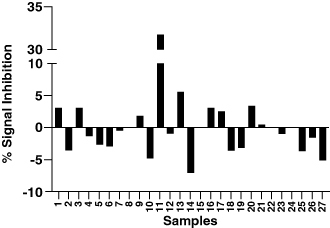
The second case study also deals with the clinical ADA assay for a bispecific molecule. During the development of the assay, all parameters including the whole drug confirmatory assay were within acceptable limits [Citation2,Citation15,Citation18–20]. However, an issue was identified with one of the domain-specific confirmatory assays. For the purposes of this case study, the two domains of the drug are referred to as domain A (binding target X) and domain B (binding target Y). Domain A did not have any issues in the confirmatory assay. However, when domain B was added to the confirmatory assay, the raw signals of the confirmatory assay increased compared with the screening assay (). To identify the source of the issue, various forms of domain B were tested in the confirmatory assay.
Figure 3. Signal inhibition percentages for whole drug and domain-specific confirmatory assays obtained from a set of 32 diseased human subjects. Addition of the drug or domain A to the confirmatory assay causes a signal decrease relative to the screening assay raw signals. On the contrary, addition of domain B causes a signal increase.
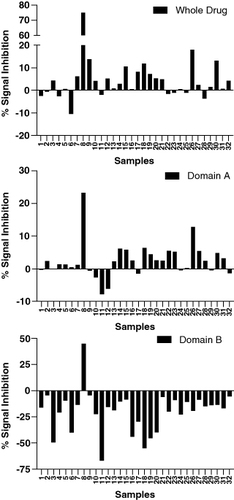
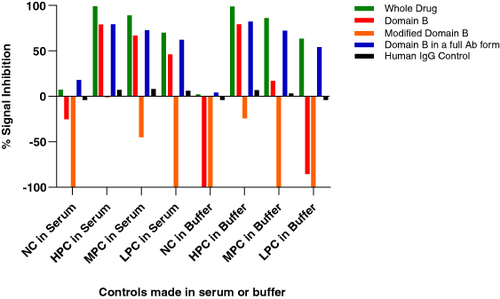
The different reagents used for troubleshooting included the whole drug (full-length antibody consisting of domains A and B), domain B consisting of only the anti-Y fragment, a modified domain B where only the antigen binding residues have been modified to bind to an irrelevant target Z, retaining the same fragment backbone as the original domain B; and finally a modified full length antibody consisting of the original domain B binding to target Y, while domain A was replaced by domain C which binds to target D, an irrelevant target (). In summary, two full length antibodies and two fragments were used for troubleshooting. It was observed that the raw signals of the NC increased upon the addition of both the fragment molecules, but not the full-length molecules. Since both the fragments were against different antigens, it suggested an issue with the backbone of the molecule and not the antigen-binding region itself. These assessments were performed in both serum and buffer conditions, and it was identified that the presence of serum reduced the non-specific signals upon addition of domain B confirmatory reagent ().
Figure 4. Different reagents used for troubleshooting the confirmatory assay. The standard confirmatory reagents shown in the top panel from left to right include the drug (full-length antibody containing domains A and B), domain A (fragment of the drug binding to target X), and domain B (fragment of the drug binding to target Y). The additional confirmatory reagents being utilized include a full-length antibody (bottom left) with domain B (same as the drug) and domain C (binding to an irrelevant target D); and a modified domain B (bottom right) where the paratope has been modified to bind to an irrelevant target Z. The polyclonal positive control (PC) has been purified to remove anti-drug antibodies against the humanized IgG backbone, resulting in the PC containing antibodies specifically targeting domains A and B. When the domain A+B (drug) is added, it binds to all the PCs, confirming the presence of antibodies against all domains of the drug. In contrast, the domain B+C molecule would only bind to the antibodies raised against domain B, with no signal reduction from the antibodies against domain A. The polyclonal PC mix does not contain antibodies against domain C.
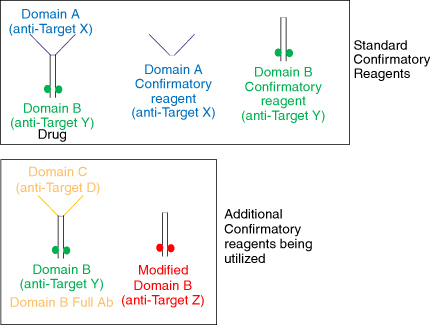
Figure 5. Performance of different domain B containing reagents in the anti-drug antibody confirmatory assay. Both the fragments cause signal increase compared with the screening assay, whereas the full-length antibodies show a signal decrease. Additionally, the signal increase caused by the fragments is higher in the absence of serum (positive controls made in buffer).
To test this hypothesis further, the domain B fragment was labeled with biotin and ruthenium and added to a streptavidin MSD plate in various combinations. It was found that when only the ruthenylated domain B was added to a well, the raw signals were higher than that of biotinylated domain B suggesting non-specific binding of the ruthenylated-domain B to the blocked streptavidin MSD plate. Additionally, the presence of both labeled domains in the well caused a further increase in raw signals compared with just the ruthenylated domain B suggesting non-specific binding within domain B, in addition to the non-specific binding of the domain B to the blocked plate (Supplementary Figure S3)
To explore the possibility of serum blocking the non-specific binding of the domain B fragment, horse serum was introduced into the assay. After titration, it was determined that 5% horse serum added to the assay buffer reduced the increase in assay signals upon addition of the confirmatory reagent (domain B) and led to a positive % inhibition (Supplementary Figure S4). When applied to a set of 32 naive individual matrix lots used for cut point calculations, all samples had a positive % inhibition including that of the plate controls. However, it was noticed that the addition of the horse serum was causing an overall increase in assay signals. An issue arising from the higher signals was an almost tenfold increase in the average % inhibition in the whole drug confirmatory assay (Supplementary Figure S5). This increase in % inhibition was also observed with the domain A confirmatory assay.
The data showed that serum reduces the non-specific binding, but addition of external horse serum, seemed to affect the assay by increasing the overall signals of the assay. One potential solution was to use the serum from the samples itself to reduce domain B bridging. To achieve this, the assay setup was slightly modified and split into three components: the first component was the acid-dissociated samples, the second was the neutralizing buffer containing the labeled drugs, and the final component was the neutralizing buffer containing the confirmatory reagent. After the acid dissociation of the sample, the neutralization buffer containing the labeled drugs were added, directly followed by the addition of the neutralization buffer containing the confirmatory reagent (Supplementary Figure S6). This splitting of the neutralizing buffer/mastermix into two separate components prevented the confirmatory reagent, specifically domain B from having direct contact with the labeled drugs in the absence of the serum from the sample, thereby reducing the opportunity for the domain B to bridge the biotinylated and ruthenylated drugs. The assay had been built at an MRD of 1:90, and a lower MRD was tested to increase the amount of serum in the assay to reduce the non-specific binding. At an MRD of 30, with human serum component at around the 3% mark, the best results were obtained on a training set of 6 samples, where 4 samples were found to have a signal inhibition upon domain B introduction while 2 other samples had a minimal signal increase (Supplementary Figure S7). However, it was observed that the drug tolerance was reduced significantly at the lower MRD which prevented us from pursuing this path forward (data not shown). Though this process was not feasible for this project, we believe this provides a unique alternative to utilize the serum from a sample itself to improve blocking capacity of an ADA assay.
The other option was to identify the sticky residues from domain B and reengineer it to prevent non-specific binding. Due to the restrictive project timelines, re-engineering the molecule was not a feasible option and a workaround was identified in the form of an already available full-length antibody molecule which contained domain B but instead of domain A, contained a domain C which binds to an irrelevant target D as described above. This assay setup assumes that ADAs against the constant region of the drug will not be produced due to the drug backbone being fully humanized. Using this full-length antibody, the domain B confirmatory assay was successfully developed (). Normal variability of signal inhibitions around the no change mark were obtained by using this method which finally allowed the calculation of a confirmatory cut point for domain B which is not possible if signal inhibition is predominantly negative. The PCs, including the LPC (set at 10 ng/ml) consistently had higher levels of inhibition than the confirmatory cut point for domain B. Additionally, the confirmatory assays of the whole drug, or domain A, are not impacted in this strategy suggesting a path forward for this issue.
Figure 6. Domain B confirmatory results when using the Domain B full antibody which includes domain B as well as domain C which is against a non-specific target D. A confirmatory cut point was successfully calculated using this method.
4. Conclusion
Immunogenicity assessments are a critical part of the drug development process, required for the safety evaluation as well as for better understanding of a biotherapeutic's pharmacokinetics, pharmacodynamics and efficacy. The quality of critical reagents often determines the quality of the assay that is available for bioanalysis. The case studies presented here highlight the need for proper characterization of critical reagents during the development of clinical ADA assays. With bispecific molecules, the number of critical reagents is increased significantly, with novel engineering of domain specific reagents also required which are generally not needed for traditional monoclonal antibodies. Since some of these reagents are only needed for in vitro assays and are not meant for human use, the onus is on the bioanalytical laboratory to ensure proper characterization of these reagents to avoid issues in the future. The experiments to identify the issues as described in this paper; including confirmation of target binding in an LBA, as well as enhanced characterization of drug domains for bispecific molecules were made standard practice in the laboratory after these issues were identified. In one of the case studies, even though we were anticipating target interference, we were not able to establish it till late in the validation due to the critical reagents not being appropriate, requiring re-development and validation of the assay and hampering the program timeline. The other case study is potentially a novel issue not described previously in literature, with one of the domains of the bispecific molecule showing non-specific binding and derailing the domain-specific confirmatory assay. Once these reagent related issues were identified, the resolutions described include previously described techniques of using anti-target reagents to reduce target interference, while some included potentially novel approaches of using the sample's own serum to improve blocking activity and reduce non-specific binding. In both cases, a renewed study of the critical reagents helped us identify the issue and guided us in providing resolutions to develop a robust ADA assay which meets regulatory assay performance requirements.
Author contributions
Conceptualization: A Sinha, S Rinker, S Kumar, K Cowan. Data acquisition and/or interpretation: A Sinha, MR Souliotis, R Kumar, S Rinker, S Kumar. Writing original draft: A Sinha, S Rinker, S Kumar, K Cowan. Review & editing: A Sinha, S Rinker, MR Souliotis, R Kumar, S Kumar, K Cowan.
Financial disclosure
The work was supported entirely by EMD Serono Research and Development Institute/Merck KGaA. Some of the authors are owners of Merck KGaA shares. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Writing disclosure
No writing assistance was utilized in the production of this manuscript.
Supplementary Figures S1-S7
Download MS Word (995.6 KB)Acknowledgments
The authors thank C Carter, presently at Bioagilytix who was involved in the initial method development of one of the case studies, and L Chung who was involved in the validation of one of the studies at the CRO.
Supplemental material
Supplementary data for this article can be accessed at https://doi.org/10.1080/17576180.2024.2366091
Competing interests disclosure
The authors have no competing interests or relevant affiliations with any organization or entity with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, stock ownership or options and expert testimony.
Additional information
Funding
References
- Mu R, Yuan J, Huang Y, et al. Bioanalytical methods and strategic perspectives addressing the rising complexity of novel bioconjugates and delivery routes for biotherapeutics. BioDrugs. 2022;36(2):181–196. doi:10.1007/s40259-022-00518-w
- Gorovits B, Wakshull E, Pillutla R, et al. Recommendations for the characterization of immunogenicity response to multiple domain biotherapeutics. J Immunol Methods. 2014;408:1–12. doi:10.1016/j.jim.2014.05.010
- Li H, Er Saw P, Song E. Challenges and strategies for next-generation bispecific antibody-based antitumor therapeutics. Cell Mol Immunol. 2020;17(5):451–461. doi:10.1038/s41423-020-0417-8
- Rup B, O'Hara D. Critical ligand binding reagent preparation/selection: when specificity depends on reagents. AAPS J. 2007;9(2):E148–155. doi:10.1208/aapsj0902016
- O'Hara DM, Theobald V, Egan AC, et al. Ligand binding assays in the 21st century laboratory: recommendations for characterization and supply of critical reagents. AAPS J. 2012;14(2):316–328. doi:10.1208/s12248-012-9334-9
- Nowatzke W, Woolf E. Best practices during bioanalytical method validation for the characterization of assay reagents and the evaluation of analyte stability in assay standards, quality controls, and study samples. AAPS J. 2007;9(2):E117–122. doi:10.1208/aapsj0902013
- O'Hara DM, Theobald V. Life cycle management of critical ligand-binding reagents. Bioanalysis. 2013;5(21):2679–2696. doi:10.4155/bio.13.241
- Pihl S, van der Strate BW, Golob M, et al. EBF recommendation on practical management of critical reagents for antidrug antibody ligand-binding assays. Bioanalysis. 2019;11(19):1787–1798. doi:10.4155/bio-2019-0248
- Oquendo E, Savoie J, Swenson JM, et al. Critical reagent generation, characterization, handling and storage workflows: impact on ligand binding assays. Bioanalysis. 2021;13(10):847–860. doi:10.4155/bio-2020-0252
- Zhong ZD, Clements-Egan A, Gorovits B, et al. Drug target interference in immunogenicity assays: recommendations and mitigation strategies. AAPS J. 2017;19(6):1564–1575. doi:10.1208/s12248-017-0148-7
- Dai S, Schantz A, Clements-Egan A, et al. Development of a method that eliminates false-positive results due to nerve growth factor interference in the assessment of fulranumab immunogenicity. AAPS J. 2014;16(3):464–477. doi:10.1208/s12248-014-9581-z
- Jordan G, Moheysen-Zadeh M, Dahl U, et al. Novel isoelectric target depletion (ITaD) protocol reduces the need for specific reagents for immunogenicity testing. Bioanalysis. 2022;14(10):725–735. doi:10.4155/bio-2022-0060
- Pohler A, Jany C, Butzer J, et al. High ionic strength dissociation assay reduces dimeric target interference in immunogenicity testing. Bioanalysis. 2023;15(14):823–832. doi:10.4155/bio-2023-0082
- Carrasco-Triguero M, Mahood C, Milojic-Blair M, et al. Overcoming soluble target interference in an anti-therapeutic antibody screening assay for an antibody-drug conjugate therapeutic. Bioanalysis. 2012;4(16):2013–2026. doi:10.4155/bio.12.165
- Devanarayan V, Smith WC, Brunelle RL, et al. Recommendations for systematic statistical computation of immunogenicity cut points. AAPS J. 2017;19(5):1487–1498. doi:10.1208/s12248-017-0107-3
- Bourdage JS, Cook CA, Farrington DL, et al. An Affinity Capture Elution (ACE) assay for detection of anti-drug antibody to monoclonal antibody therapeutics in the presence of high levels of drug. J Immunol Methods. 2007;327(1-2):10–17. doi:10.1016/j.jim.2007.07.004
- Kavita U, Duo J, Crawford SM, et al. A systematic study of the effect of low pH acid treatment on anti-drug antibodies specific for a domain antibody therapeutic: impact on drug tolerance, assay sensitivity and post-validation method assessment of ADA in clinical serum samples. J Immunol Methods. 2017;448:91–104. doi:10.1016/j.jim.2017.06.002
- Shankar G, Devanarayan V, Amaravadi L, et al. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J Pharm Biomed Anal. 2008;48(5):1267–1281. doi:10.1016/j.jpba.2008.09.020
- Myler H, Pedras-Vasconcelos J, Phillips K, et al. Anti-drug antibody validation testing and reporting harmonization. AAPS J. 2021;24(1):4. doi:10.1208/s12248-021-00649-y
- Services USDoHaH. Guidance for Industry: immunogenicity testing of therapeutic protein products-developing and validating assays for anti-drug antibody detection. In: Services USDoHaH, editor. 2019. https://www.fda.gov/media/119788/download