
ABSTRACT
These Lipid Association of India (LAI) recommendations refer to specific patient populations. They follow the previously published LAI part 1 recommendations. These part 2 LAI recommendations focus on specific patient groups. These include patients with heart failure, chronic kidney disease, non-alcoholic fatty liver disease, cerebrovascular disease, thyroid disorders, inflammatory joint diseases, familial hypercholesterolaemia and human immunodeficiency virus infection. We also consider women, the elderly and post-transplantation patients. The current recommendations are based, as much as possible, on available data from Indian populations.
Abbreviations:
- 4D: Die Deutsche Diabetes Dialyses Studie
- ABCD: Amsterdam Born Children and Their Development
- ACC: American College of Cardiology
- ACCF: American College of Cardiology Foundation
- ACCORD: Action to Control Cardiovascular Risk in Diabetes
- AFCAPS/TexCAPS: Air Force/Texas Coronary Atherosclerosis Prevention study
- AHA: American Heart Association
- AIM-HIGH: Atherothrombosis Intervention in Metabolic Syndrome with low HDL/High Triglyceride
- ALA: Alpha Linolenic Acid
- ALERT: Assessment of Lescol in Renal Transplantation
- ALT: Alanine Aminotransferase
- AST: Aspartate Aminotransferase
- Apo A: Apolipoprotein A
- Apo B: Apolipoprotein B
- Apo C: Apolipoprotein C
- Apo E: Apolipoprotein E
- AS: Ankylosing Spondylitis
- ASCOT-LLA: Anglo-Scandinavian Cardiac Outcomes Trial-Lipid-Lowering Arm
- ASCVD: Atherosclerotic Cardiovascular Disease
- AURORA: A Study to Evaluate the Use of Rosuvastatin in Subjects on Regular Hemodialysis: An Assessment of Survival and Cardiovascular Events
- bDMARDs: Biological Disease Modifying Anti-Rheumatic Drugs
- BNP: Brain Natriuretic Peptide
- CAD: Coronary Artery Disease
- CARDS: Collaborative Atorvastatin Diabetes Study
- CARE: Cholesterol and Recurrent Events
- CAV: Cardiac Allograft Vasculopathy
- cDMARDs: Conventional Disease Modifying Anti-Rheumatic Drugs
- CETP: Cholesteryl Ester Transfer Protein
- cIMT: Carotid Intima Media Thickness
- CK: Creatine Kinase
- CKD: Chronic Kidney Disease
- Co-Q10: Coenzyme Q10
- CORONA: Controlled Rosuvastatin Multinational Study in Heart Failure
- COX-2: Cyclooxygenase-2
- CRP: C-Reactive Protein
- csDMARDs: Conventional Synthetic Disease Modifying Anti-Rheumatic Drugs
- CTT: Cholesterol Treatment Trialists’
- CV: Cardiovascular
- CVD: Cardiovascular Disease
- DHA: Docosahexaenoic Acid
- DM: Diabetes Mellitus
- DMARDs: Disease Modifying Anti-Rheumatic Drugs
- DOPPS: Dialysis Outcome and Practice Patterns Study
- eGFR: Estimated Glomerular Filtration Rate
- EPA: Eicosapentaenoic Acid
- ESRD: End Stage Renal Disease
- FH: Familial Hypercholesterolaemia
- FDA: Food and Drug Administration
- FOURIER: Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk
- FIELD: Fenofibrate Intervention and Event Lowering in Diabetes
- GISSI-HF: GruppoItaliano per lo Studio dellaSopravvivenzanell’InfartoMiocardico Heart Failure
- GPIHBP: Glycosylphosphatidyl Inositol Anchoring High-Density Lipoprotein Binding Protein
- GREACE: Greek Atorvastatin and Coronary Heart Disease Evaluation
- HDL: High-Density Lipoprotein
- HDL-C: High-Density Lipoprotein Cholesterol
- HeFH: Heterozygous FH
- HoFH: Homozygous FH
- HF: Heart Failure
- HIV: Human Immunodeficiency Virus
- HMG CoA reductase: 3-Hydroxy-3-Methyl-Glutaryl-Coenzyme a Reductase
- HPS: Heart Protection Study
- HPS2-THRIVE: Heart Protection Study 2-Treatment of HDL to Reduce the Incidence of Vascular Events
- HR: Hazard Ratio
- HRT: Hormone Replacement Therapy
- hsCRP: High sensitivity C-Reactive Protein
- ICH: Intracerebral Haemorrhage
- IDL: Intermediate Density Lipoprotein
- IJD: Inflammatory joint disorder
- IMPROVE-IT: Improved Reduction of Outcomes: Vytorin Efficacy International Trial
- JBS: Joint British Society
- JELIS: Japan EPA Lipid Intervention Study
- JUPITER: Justification for the use of Statin in Prevention: An Intervention Trial Evaluating Rosuvastatin
- KDIGO: Kidney Disease: Improving Global Outcomes
- LAI: Lipid Association of India
- LMF: Lipase Maturation factor
- LPL: Lipoprotein Lipase
- LDL: Low-Density Lipoprotein
- LDL-C: Low-Density Lipoprotein Cholesterol
- LDLR: Low-Density Lipoprotein Receptor
- LIPID: Long-Term Intervention with Pravastatin in Ischemic Disease
- Lp(a): Lipoprotein (a)
- LVEF: Left Ventricular Ejection Fraction
- MEDPED: Make Early Diagnosis to Prevent Early Deaths
- MEGA: Management of Elevated Cholesterol in the Primary Prevention Group of Adult Japanese
- MetS: Metabolic Syndrome
- MHC: Major Histocompatibility Complex
- MI: Myocardial Infarction
- mTOR: Mammalian Target Of Rapamycin
- NAFLD: Non-alcoholic Fatty Liver Disease
- NASH: Non-alcoholic Steatohepatitis
- NICE: National Institute for Health and Care Excellence
- NLA: National Lipid Association
- NNT: Number Needed to Treat
- Non-HDL-C: Non-High-density Lipoprotein Cholesterol
- NSAID: Non-Steroidal Anti-Inflammatory Drug
- NT-proBNP: N-Terminal pro-Brain Natriuretic Peptide
- NYHA: New York Heart Association
- OCP: Oral Contraceptive Pill
- ODYSSEY Outcomes: Evaluation of Cardiovascular Outcomes after an Acute Coronary Syndrome During Treatment With Alirocumab
- PCOS: Polycystic Ovary Syndrome
- PCSK9: Proprotein Convertase Subtilisin Kexin Type 9
- PPAR: Peroxisome Proliferator-Activated Receptor
- PRECISION: Prospective Randomised Evaluation of Celecoxib Integrated Safety VS Ibuprofen or Naproxen
- PROSPER: Prospective Study of Pravastatin in the Elderly at Risk
- PsA: Psoriatic Arthritis
- RA: Rheumatoid Arthritis
- RCT: Randomised Controlled Trial
- RR: Relative Risk
- SAGE: Study Assessing Goals in the Elderly
- SHEP: Systolic Hypertension in the Elderly Program
- T2DM: Type 2 DiabetesMellitus
- TG: Triglyceride
- THRaST: THRombolysis and STatins
- TNT: Treating to New Targets
- TRACE-RA: Trial of Atorvastatin for the Primary Prevention of Cardiovascular Events in Patients with RA
- tsDMARDs: Targeted Synthetic Disease Modifying Anti-Rheumatic Drugs
- TSH: Thyroid Stimulating Hormone
- ULN: Upper Limit of Normal
- VLDL: Very Low-density Lipoprotein
- VLDL-C: Very Low-density Lipoprotein cholesterol
- WHO: World Health Organization
- WOSCOPS: West of Scotland Coronary Prevention Study
1. Introduction
Dyslipidaemia is widely prevalent and is one of the most important risk factors for atherosclerotic cardiovascular disease (ASCVD). Accordingly, effective management of dyslipidaemia is an important goal for the prevention of ASCVD.
Numerous leading international societies have published guidelines for the management of dyslipidaemia but these guidelines are not directly applicable to Indians because of various reasons. Indians have an unusually high prevalence of dyslipidaemia comprising of low high-density lipoprotein cholesterol (HDL-C), high triglycerides (TG), moderately elevated low-density lipoprotein cholesterol (LDL-C) and high lipoprotein (a), particularly at a younger age; this pattern of dyslipidaemia is also distinct compared with Western populations. The distribution and interplay of various other ASCVD risk factors and genetic susceptibility may also be different. In addition, population awareness about prevention of ASCVD, cultural beliefs, socio-economic conditions, diet and other factors contribute to their adverse risk profile. For these reasons, it is important to formulate policies and guidelines that accommodate these differences and propose recommendations that are specific to the Indian population. To meet this objective, the Lipid Association of India (LAI) has produced a consensus document on the management of dyslipidaemia in Indians. Part 1 of the document dealt with all the common issues related to lipid management encountered during routine clinical practice and was published last year [Citation1, 2]. The present document – part 2 of the consensus document – outlines the management of dyslipidaemia in specific patient populations.
2. Methodology
The LAI initiated the process of developing this consensus document by inviting leading experts from various specialities including Internal Medicine, Cardiology, Endocrinology, Nephrology, Neurology, Pharmacology and Vascular Surgery to participate in this process. A series of regional meetings were held with these experts in different parts of the country. In all, there were 8 meetings held at Delhi, Patna, Mumbai, Chennai and Jaipur with 213 experts from 17 states, 2 union territories and 33 cities of India. In addition, two experts were from the UK and 1 from the USA.
These meetings followed a standard format. First, the key issues related to lipid management in specific patient populations were presented to the entire group in the form of lectures, followed by extensive discussion among experts on these topics. The discussions held in the meetings were recorded. Subsequently, the experts of the core committee reviewed the discussions of all the meetings and prepared a consensus draft. This draft was circulated to the remaining experts for their suggestions and approval. Suggestions were re-evaluated by the core committee and were incorporated in the recommendations. After the final round of discussion, the expert panel prepared the final document, considering the following main objectives:
To ensure that the document remained simple, yet scientifically robust.
To be informative rather than prescriptive.
To maximally utilise the available Indian data for evidence-based practice.
To ensure practical applicability of the recommendations to the Indian population.
3. Dyslipidaemia in heart failure (HF)
3.1. Cholesterol levels in HF
Low total cholesterol levels have been shown to be associated with increased cardiovascular (CV) events and mortality in HF patients in several studies [Citation3, 4]. In a Korean HF Registry, patients (n = 2797; 44% with ischemic HF) were stratified into quartiles of serum total cholesterol [Citation5]. Patients with lower serum total cholesterol had lower blood pressure, lower haemoglobin, low serum sodium and increased brain natriuretic peptide (BNP) levels, suggesting that low total cholesterol coexists with other markers of poor risk. Low total cholesterol was associated with increased risk for death and readmission due to HF. However, propensity score matching analysis revealed that low cholesterol itself did not affect outcome [hazard ratio, HR, 1.12]. Hence, low cholesterol may merely be a consequence of advanced HF and has no pathophysiologic role. In vitro studies have implicated cytokines, such as tumour necrosis factor, as a causative factor in the hypocholesterolaemia of inflammatory conditions through mechanisms such as decreased hepatic lipoprotein production and increased low-density lipoprotein (LDL) receptor activity.3 Further, low cholesterol is a marker of cachexia related to HF [Citation6, 7].
3.2. Statins in HF
Several randomised trials have shown that statins reduce major adverse CV events in patients with risk factors for, or clinically apparent, ASCVD. However, these studies have excluded HF patients. The relative risk reduction (RRR) of cardiac events was seen regardless of baseline LDL-C levels [Citation8].
The beneficial effects of statins in HF were demonstrated in various registries, observational studies and post hoc analyses of randomised clinical trials [Citation9]. However, two large randomised controlled trials failed to confirm the benefit of statins in HF. The Controlled Rosuvastatin Multinational Trial in Heart Failure (CORONA) [Citation10] and GruppoItaliano per lo Studio dellaSopravvivenzanell’InfartoMiocardico Heart Failure (GISSI-HF) [Citation11] trials evaluated the use of statins in patients with symptomatic HF and reduced left ventricular ejection fraction (LVEF).
The CORONA trial [Citation10] randomised 5011 patients aged ≥ 60 years (mean age 73 years) with ischemic HF, LVEF ≤ 40% and New York Heart Association (NYHA) class II-IV symptoms to 10 mg/day rosuvastatin vs. placebo. The GISSI-HF trial [Citation11] randomised 4574 patients aged ≥ 18 years (mean age 68 years) with NYHA class II-IV symptoms; HF aetiology being ischemic (40%), non-ischemic (35%) or other causes (25%) with LVEF ≤ 40% (or >40% if hospitalised within the past year) also to 10 mg/day rosuvastatin vs. placebo. There was no statistically significant difference in the primary endpoint of death from CV causes, nonfatal MI and nonfatal stroke in CORONA and co-primary endpoints of time to death, or admission to hospital for CV reasons in GISSI-HF between the rosuvastatin and placebo groups. It is noteworthy that both trials included older and sicker patients with high mortality rates in both the treatment and placebo groups signifying advanced HF. Further, most of the deaths in the two trials were arrhythmic or due to progressive HF. There were only few deaths attributed to myocardial infarction (MI) or stroke. Since statin therapy is likely to be beneficial for plaque stabilisation or pleiotropic effects, perhaps it could not affect mortality due to the low incidence of MI or stroke. Nevertheless, pleiotropic effects also failed to show any effect on primary endpoint in this population with advanced HF [Citation10, 11]. However, an individual-level pooled data re-analysis of both trials demonstrated a significant 19% reduction in risk of MI among patients with ischemic HF [Citation12].
As per the 2013 American College of Cardiology Foundation (ACCF)/American Heart Association (AHA) guidelines [Citation13], statins have class IA recommendation in stage B ischemic HF (post-MI without symptoms of HF) to prevent progression of HF. In symptomatic HF patients, statins have class III recommendation meaning that statins are not useful as adjunctive therapy in the absence of other indications for their use [Citation13]. However, the recent 2016 American College of Cardiology (ACC) expert consensus document recommends that patients with HF in NYHA class II to III due to ischemic aetiology may be given statins [Citation14]. The role of statins in non-ischemic or dilated cardiomyopathy is more controversial especially in advanced HF as in the negative GISSI-HF trial where 34.5% of the participants had non-ischemic cardiomyopathy [Citation11].
The low uptake of hydrophilic rosuvastatin by the heart (approximately 1% of that by liver) compared with the lipophilic simvastatin (approximately 80%) has been postulated to explain the lack of benefit with rosuvastatin in the CORONA and GISSI-HF trials. A meta-analysis of 17 studies showed that atorvastatin but not rosuvastatin increased LVEF and decreased BNP levels in HF patients [Citation15]. However, no randomised trial has been carried out with atorvastatin in HF.
Coenzyme Q10 (CoQ10) plays an essential role in the mitochondrial respiratory chain through oxidative phosphorylation with the concomitant production of adenosine triphosphate. Statins block production of farnesyl pyrophosphate, an intermediate in the synthesis of CoQ10 or ubiquinone. A meta-analysis showed that statins significantly reduced plasma CoQ10 concentrations [Citation16]. Whether depletion of CoQ10 by statins could actually worsen HF is highly debated. In the CORONA study, lower CoQ10 levels were associated with several other markers of greater disease severity like older age, higher N-terminal pro-BNP (NT-proBNP) levels, lower lipid levels, lower LVEF and lower glomerular filtration rate. Rosuvastatin 10 mg/day significantly reduced plasma CoQ10 by 39% [Citation17]. Multivariable models demonstrated no independent relationship between CoQ10 levels and muscle pain or CV outcomes [Citation17]. Review of the data on coenzyme Q10 depletion and statin myopathy did not find definitive evidence for either a causative role or a therapeutic benefit from coenzyme Q10 supplements [Citation18, 19].
There is evidence suggesting that cumulative exposure to lifelong low LDL C is likely to confer more protection than when treatment is delayed and the cumulative exposure to LDL C remains high [Citation20, 21]. Similarly, it is likely that in the HF trials, sicker patients with advanced HF failed to derive benefit from statins because of delayed treatment. In fact, in a post hoc analysis of the CORONA study, NT-proBNP was measured in 3664 (73% of total cohort) patients. The mid-tertile included values between 868 and 2348 pg/ml. Patients in the lowest tertile of NT-proBNP had the best prognosis and if assigned to rosuvastatin rather than placebo had a greater reduction in primary endpoint (HR0.65, p = 0.019) than patients in higher tertiles. Patients with ischemic HF who have NT-proBNP values < 868 pg/ml may benefit from rosuvastatin resulting in fewer atherothrombotic events and sudden deaths. This occurred despite similar effects of rosuvastatin on the lipid profile and high-sensitivity C-reactive protein (hsCRP) in each tertile of NT-proBNP [Citation22]. These findings suggest that rosuvastatin was effective in patients with less advanced disease.
Galectin-3 is a biomarker of myocardial fibrosis and may play a role in the pathophysiology of HF through promotion of myocardial fibrosis and inflammation thereby influencing ventricular remodelling [Citation23]. Galectin-3 levels correlate with severity of HF [Citation24]. In the CORONA trial, patients with plasma galectin-3 levels less than the median (≤19.0 ng/ml) benefited more from statin therapy and had a lower primary event rate (HR 0.65; p = 0.014), lower total mortality (HR 0.70; p = 0.038), lower all-cause mortality and HF hospitalisations (HR 0.72; p = 0.017) compared with placebo, raising the possibility that it might be used to define HF subtypes that respond differently to rosuvastatin. The combination of concurrently low concentrations of galectin-3 and NT-proBNP (<868 pg/ml) identified patients with a greater benefit from rosuvastatin (HR 0.33; p = 0.002) [Citation25]. There is insufficient evidence to support that heart failure improves with use of ezetimibe.
3.3. LAI recommendations
Besides lifestyle measures, statins may be administered to patients with ischemic HF with NYHA Class II-III symptoms.
Patients with advanced symptomatic HF require individualised care, e.g. if a coronary artery disease (CAD) patient who was on statins develops symptomatic HF, statins may be continued. In these groups, intensive statin therapy with a goal to achieve 50% reduction in LDL-C levels is justified.
Statins are also recommended for ischemic HF patients awaiting heart transplantation.
Statins are not recommended in NYHA Class IV HF. However, three points can be considered in patients with advanced HF. Firstly, no excess of side effects with rosuvastatin 10 mg/day was observed in CORONA and GISSI-HF trial compared with placebo, proving the safety of rosuvastatin at this dose. Secondly, the degree of LDL-C lowering did not correlate with event rate. Thirdly, there were fewer hospitalisations of any type in the rosuvastatin group in the CORONA study. Therefore, if statin therapy is administered to advanced HF patients, statins with potency equivalent to rosuvastatin 10 mg is recommended. The above two trials included patients with NYHA III and IV.
Statin therapy is not recommended in advanced HF patients who have a short life expectancy (e.g. because of comorbidities like malignancy).
In the absence of trials of non-statin therapies, including Proprotein Convertase Subtilisin Kexin Type 9 (PCSK9) inhibitors in HF, such therapies are not recommended on a general basis. However, they may be considered in an individual patient based on clinical need.
Statins are not recommended in non-ischemic HF or dilated cardiomyopathy just for the management of HF since no benefit was shown as stated in a recent meta-analysis [Citation26].
4. Dyslipidaemia in chronic kidney disease
Chronic kidney disease (CKD) is defined as structural or functional kidney abnormalities persisting for > 3 months with or without a decrease in estimated glomerular filtration rate (eGFR) [Citation27]. CKD is divided into five stages, with stage 1 indicating relatively preserved renal function with eGFR > 90 ml/min/1.73 m2, with steady decline in renal function on progression to stage 5 defined as an eGFR < 15 ml/min/1.73 m2. Several studies have conclusively demonstrated progressive increase in CV morbidity and mortality on progression from stage 1 to stage 5 CKD [Citation28]. CKD stage 5D or end stage renal disease (ESRD) increases the risk of CV disease (CVD) several fold, irrespective of age. In this context, young people with ESRD are at 10–30-fold higher risk for CV mortality than age-matched healthy counterparts [Citation29]. The Indian subcontinent faces a higher burden of CKD [Citation30], which is often considered a CAD equivalent.
CKD is a risk factor for atherogenic dyslipidaemia [Citation31] and hence, management of dyslipidaemia can potentially reduce mortality in these patients. CKD is associated with high TG, high very low-density lipoprotein cholesterol (VLDL-C) levels, low HDL-C levels and modestly elevated LDL-C levels. However, on progression to ESRD, LDL-C and total cholesterol levels remain low to normal in the majority of patients [Citation31, 32]. In advanced CKD patients with decreased LDL-C levels, there are increased levels of small dense LDL particles, so that the small dense LDL particle to normal size LDL particle ratio is significantly elevated [Citation33]. There is some evidence that statins can improve the LDL subclass distribution in patients on haemodialysis [Citation34].
Several randomised, large-scale trials in CKD patients such as the Die Deutsche Diabetes Dialyses Study (4D) [Citation35], A Study to Evaluate the Use of Rosuvastatin in Subjects on Regular Haemodialysis: An Assessment of Survival and Cardiovascular Events (AURORA) [Citation36], Study of Heart and Renal Protection (SHARP) [Citation37] and Assessment of Lescol in Renal Transplantation (ALERT) trial [Citation38] have provided the basis for guidelines on the management of dyslipidaemia in CKD by organisations such as Kidney Disease: Improving Global Outcomes (KDIGO) [Citation39], the Joint British Society (JBS3) [Citation40] and Canadian Society of Nephrology [Citation41]. It is also important to note that there is paucity of data on dyslipidaemia management in Indian patients with CKD. Of the above, the KDIGO guidelines include a detailed analysis and discussion on relevance and management of dyslipidaemia in CKD. Hence, the executive summary of these guidelines is mentioned below. The salient features of KDIGO guidelines on management of dyslipidaemia [Citation39] include:
A full evaluation of lipid profile in all subjects with CKD at the time of diagnosis. This includes all subjects receiving dialysis and kidney transplant recipients. | |||||
Routine follow-up measurement of lipid levels is not recommended, except to establish adherence to treatment and documentation of >30% reduction in LDL-C. | |||||
All adults with CKD should receive therapeutic lifestyle advice. | |||||
Treatment with statins or statins/ezetimibe combination is recommended for all adults equal to or greater than 50 years with an eGFR < 60 mL/min/1.73 m². | |||||
In adults between the ages of 18 and 49 with CKD should be treated with statins if they have one of the following-
| |||||
| • | ESRD patients who are on lipid-lowering therapy at the time of starting dialysis should continue to receive these agents. | ||||
| • | In statin-naive adult dialysis patients, initiation of statins is not recommended. | ||||
| • | All adult kidney transplant recipients should receive statin therapy. | ||||
| • | Statins should be started at a lower dose in subjects with eGFR < 30 ml/min with gradual escalation to the recommended dose along with monitoring for any adverse events. | ||||
4.1. LAI recommendations
A full evaluation of lipid profile (total cholesterol, LDL-C, HDL-C and TG) is recommended in all CKD patients (including patients with ESRD and renal transplant recipients) at the time of diagnosis.
Follow-up measurement of lipid levels should be performed routinely (at treating physicians’ discretion) in subjects with CKD until the LDL-C and non-HDL-C target is achieved. | |||||
Therapeutic lifestyle modification should be recommended to all patients. | |||||
A combination of statins/statin plus ezetimibe is recommended for all adults over the age of 40 years with eGFR < 60 mL/min/1.73 m². | |||||
Adults between the ages of 18 and 39, with CKD should be treated with statins if they have one of the following:
| |||||
| • | Patients who are already receiving lipid-lowering therapy at the time of starting dialysis should continue to receive these agents. | ||||
| • | In adults with dialysis-dependent CKD who are not on statins, statins could be considered at the lowest dose possible and should be titrated up carefully to achieve the LDL-C level for very high risk patients (see ). | ||||
| • | All adult kidney transplant recipients should receive statin therapy. | ||||
Table 1. Renal dosing for commonly available statins [Citation50–Citation53].
Statins should be started at a lower dose in all subjects with eGFR <30 ml/min with gradual escalation to the recommended dose along with monitoring for any adverse events (see ).
4.2. Rationale for LAI recommendations
Experts at LAI are largely in agreement with the KDIGO regarding lipid-lowering therapy in CKD patients. The expert opinion differs in the following aspects:
Recommendations for pursuing target LDL-C and non-HDL-C levels
Age cut-off for initiation of statin therapy has been lowered to 40 years
Initiation of statin therapy in dialysis patients
CKD patients remain at high risk for CVD; hence, it is prudent to check the lipid profile at the time of diagnosis of CKD, whatever the stage of renal failure, including patients who have undergone renal transplantation. Secondary causes of dyslipidaemia should be ruled out. Follow-up lipid profile should be checked in order to ensure compliance with medications and to achieve target LDL-C and non-HDL-C levels. South Asians have an atherogenic lipid profile at baseline and are at risk for premature and severe CVD compared with Caucasians [Citation42, 43]. This underlying risk multiplies with development of CKD. Hence, it is prudent to mitigate the risk associated with dyslipidaemia as early and as effectively as possible.
The use of statins in early stages of CKD has modest cardio-protective benefit, though less than what has been noted in general population and the magnitude of benefit decreases as the CKD stage increases [Citation44]. The SHARP trial, a large randomised trial evaluating the use of simvastatin (20 mg)/ezetimibe (10 mg) combination vs. placebo in both dialysis and non-dialysis patients, reported a 17% proportional reduction in major atherosclerotic events, significant reductions in non-haemorrhagic stroke and arterial revascularisation procedures in CKD-ND (non-dialysis) patients [Citation37]. In contrast, large randomised trials in ESRD patients on dialysis like 4D (with atorvastatin) [Citation35] and AURORA (with rosuvastatin) [Citation36] failed to show cardio-protective or mortality benefit. Hence, international recommendations do not favour initiation of statins in dialysis patients. We differ from the KDIGO guidelines [Citation39] in this regard, the reasons being that the Indian population has a higher risk for both CAD and ESRD compared with the Western population which may have a genetic basis and an associated accelerated CVD and CAD [Citation42]. In addition, it is to be noted that the dialysis population in India is younger (mean age 44 years) compared with the Western population [Citation30]; hence, therapies to mitigate CV risk instituted earlier have the potential to reduce mortality in this population of Indian origin. In such a situation, where Indian data are limited, we encourage informed and shared decision-making. We emphasise that other reversible and modifiable risk factors for ESRD and CVD need to be strictly controlled as well.
KDIGO states an age cut-off of 50 years or greater for initiation of statins in CKD patients. This age cut-off was based on a Canadian cohort study consisting predominantly of Caucasians. The age cut-off of 40 years in our expert opinion is based on the risk factor profile of our population, as discussed above [Citation42, 43].
Management of dyslipidaemia in kidney transplant recipients is discussed in the solid organ transplantation section.
In patients with nephrotic syndrome (NS), the concentration of VLDL, IDL and lipoprotein (a) is higher with increasing content of TG within the lipoprotein particles. They exhibit exaggerated post-prandial hyperlipidaemia. Reduced lipoprotein lipase activity, increased PCSK9 activity, increased Acetyl CoA Acyl Transferase 2(ACAT-2) activity and increased 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMG Co-A reductase) activity have all been implicated in the generation of dyslipidaemia of NS [Citation45, 46]. Treatment of dyslipidaemia has not been shown to reduce the risk of renal failure; however, there is some evidence that statins reduce proteinuria and the reduction in, as well as deterioration of, eGFR in these patients [Citation47, 48]. However, caution should be exercised in using rosuvastatin in this group of patients due to reported increase in proteinuria and deterioration of renal function [Citation47, 48].
Consideration has to be given to the dose of statins with respect to an individual’s renal function in order to avoid adverse reactions. shows the dose adjustment of commonly available statins. Major trials mentioned earlier such as 4D [Citation35], AURORA [Citation36], SHARP [Citation37] and ALERT [Citation49] utilised renal dosing for atorvastatin, rosuvastatin, simvastatin/ezetimibe and fluvastatin, respectively. In all these trials, there was no statistically significant increase in adverse reactions related to statins. For clinical practice, statin monotherapy can be used since trials in the general population have shown desirable effects of monotherapy with statins. Combination with ezetimibe can be used if a desirable LDL-C level has not been achieved.
Currently, there is no good evidence to support the routine use of fibric acid derivatives for management of hypertriglyceridaemia in CKD patients due to high toxicity of these drugs (rhabdomyolysis and acute kidney injury) in patients with renal dysfunction [Citation52, 54]. In cases with serum TG > 500 mg/dL despite lifestyle modification and statin therapy, a fibric acid derivative may therefore be considered but due care should be taken in patients with renal impairment as fibrates are renaly excreted [Citation52]. The National Kidney Foundation recommends that the dose of fenofibrate should be reduced by 50% in patients with GFR of 60–90 ml/min/1.73 m2 and about 75% if GFR is between 15 and 59 ml/min 1.73 m2 and avoid using fibrates if GFR is <15 ml/min (https://www2.kidney.org/professionals/kdoqi/guidelines_lipids/iii.htm).
5. Dyslipidaemia and non-alcoholic fatty liver disease (NAFLD)
NAFLD is associated with metabolic factors and evidence suggests that CVD is the most common cause of death in patients with NAFLD. Statins are used to treat atherogenic dyslipidaemia to reduce atherosclerotic cardiac events. However, statins are known to elevate liver enzymes and hence there is significant concern among physicians to prescribe statins in NAFLD. There is evidence that suggests that statins may improve liver enzymes and histology in NAFLD [Citation55–Citation57].
NAFLD is an important health issue in India and is closely associated with marked metabolic derangements such as insulin resistance and the metabolic syndrome, which are important determinants of T2DM and CVD [Citation58].
Hence, there are two important issues that need to be addressed: Should statins be used to treat dyslipidaemia in NAFLD and can statins be used for NAFLD/non-alcoholic steatohepatitis (NASH) for primary prevention?
5.1. Evidence from meta-analyses
The Prospective Pravastatin Pooling (PPP) project [Citation59] was a prospective analysis of three major large, placebo-controlled trials of pravastatin: West of Scotland Coronary Prevention Study (WOSCOPS) [Citation60], Cholesterol and Recurrent Events (CARE) study [Citation61] and the Long-term Intervention with Pravastatin in Ischemic Disease (LIPID) study [Citation62] which included 19,768 patients treated for 5 years (). Its aim was to assess potential safety issues and concluded that 40 mg of pravastatin was well tolerated, with no serious adverse events, including liver function abnormalities.
Table 2. Meta-analyses of statin trials reporting on risk of liver toxicity with statins.
One meta-analysis of randomised control trials (RCTs) of statins for primary or secondary prevention included a total of 49,275 patients from 13 trials. The duration of the included trials varied from 48 weeks to 6 years. The results showed that low to moderate doses of pravastatin, lovastatin and simvastatin were not associated with a significant risk of liver test abnormalities and that monitoring was not warranted in patients taking a low-to-moderate dose other than at the onset of therapy [Citation63].
Another meta-analysis of 75,317 subjects addressed the issue of effect of higher vs. lower intensity statin therapy on liver toxicity [Citation64]. It included 23 statin arms for 0.9–6 years of follow-up. Liver enzymes increased significantly with higher statin dose while there was no significant relationship between LDL-C reduction and liver damage. It concluded that the risk of statin-associated rise in liver enzymes is not related to the magnitude of LDL-C lowering but is more likely determined by drug and dose-specific effects.
5.2. Should statins be used to treat dyslipidaemia in patients with NAFLD?
NAFLD is characterised by the presence of hepatic steatosis by imaging or histology and absence of secondary causes of fat accumulation in the liver. It is divided into two types on the basis of histological findings: NAFLD and NASH. NAFLD is generally associated with metabolic disorders like obesity, insulin resistance, diabetes mellitus and dyslipidaemia [Citation65]. Aggressive treatment of dyslipidaemia is essential in the management of patients with NAFLD. However, there is concern that patients with NAFLD and dyslipidaemia who are treated with statins may develop abnormal liver tests [Citation66].
A post hoc analysis of the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) study evaluated the risk-to-benefit ratio of using a statin to treat dyslipidaemia in patients with NAFLD [Citation67]. The GREACE study randomised 1600 patients with CAD to receive statin or usual care, which could include statins. Participants were randomised to receive either usual care or structured care with atorvastatin (10–80 mg) to achieve an LDL-C below 100 mg/dL. The primary endpoints were the first occurrence of any CV event and the secondary endpoints were elevation of liver enzymes or liver-related adverse effects. Post hoc analysis of 437 out of total cohort of 1600 patients with baseline elevation in the liver enzymes [<3 times upper limit of normal (ULN)] showed that 227 subjects treated with a statin had significant improvement in liver tests, whereas 210 participants who were not treated with a statin had further increases of liver enzymes. Other causes of elevated liver enzymes like alcohol, hepatitis B and C and autoimmune hepatitis were ruled out. CV events occurred in 22 (10%) of 227 patients with deranged liver enzymes who received statins (3.2 events/100 patient-years) and 63 (30%) of 210 patients with deranged liver enzymes who did not receive statins (10 events/100 patient-years). There was a 68% RRR in patients with abnormal liver tests on statins vs. 39% RRR in patients with normal liver tests on statins [Citation67, 68]. Importantly, patients with increase in liver enzymes up to 3 times the ULN showed improvement in liver tests with statins compared with a further rise in liver tests in patients not treated with statins. The relationship between statins, hepatic TG content and serum alanine aminotransferase (ALT) levels was examined in 2264 Dallas Heart Study participants and it concluded that there was no association between statins and hepatic steatosis or serum ALT abnormalities, even among those with hepatic steatosis [Citation69].
In another study, 342 hyperlipidaemic patients with elevated baseline liver enzymes were prescribed a statin and the incidence of mild-moderate and severe elevations in liver enzymes was 4.7 and 0.6%, respectively. It concluded that individuals with deranged liver enzymes did not have higher risk for hepatic adverse effects due to statins [Citation70].
High-dose pravastatin (80 mg/day) when used for the treatment of dyslipidaemia in patients with chronic liver disease significantly lowered LDL-C, total cholesterol and TGs compared with placebo and it was well tolerated [Citation71]. Similar findings were noted in a small prospective study involving 23 dyslipidaemic patients with biochemical evidence of NAFLD, where rosuvastatin 10 mg/day for 8 months normalised liver function tests in all patients [Citation72].
In a prospective study of 25 dyslipidaemic and NAFLD patients with elevated baseline liver enzymes, the ALT levels returned to normal in 36.3% of patients after 6 months and 20% after 12 months of treatment with atorvastatin [Citation73]. In another randomised study, pitavastatin and atorvastatin equally reduced LDL-C and significantly reduced ALT in patients with hypercholesterolaemia and elevated ALT concentration [Citation74].
5.3. Should statins be used to specifically treat NAFLD?
Statins have pleiotropic effects besides lipid lowering which include anti-thrombotic, anti-inflammatory and antioxidant effects. Hence, they have been considered as therapeutic agents in NAFLD because inflammation and oxidative stress play a major role in the pathogenesis of NAFLD [Citation75].
An additional benefit seen in the post hoc analysis of GREACE study was the reduction in ALT and aspartate aminotransferase (AST) levels during treatment in patients with NAFLD on statins. Nearly 47% reduction in AST levels and 35% reduction in ALT levels were noted. In addition, 89% of the participants on statins had normal liver enzymes and gamma-glutamyl transferase levels at the end of the three-year period. A 12% increase in AST and ALT was noted in patients with NAFLD who did not receive statins at the end of the three-year period. A rise in liver enzymes more than 3 × ULN was observed in only 10 of 880 patients taking statins. In three of these patients, the liver enzymes normalised after dose adjustment of the statin. Only seven (<1%) patients on statins had to discontinue therapy because of liver-related side effects [Citation68].
While the GREACE study included patients with CAD and NAFLD, another study – ATTEMPT [Citation76] – was carried out in patients with MetS (some with NAFLD) but without CVD or DM. The ATTEMPT was a prospective, randomised trial in 1123 MetS patients (aged 45–65 years), followed up for 42 months. Patients received intensive lifestyle intervention and pharmacotherapy: atorvastatin in all patients (LDL-C targets of <100 mg/dL or <130 mg/dL), inhibitors of the renin–angiotensin–aldosterone axis for hypertension, metformin for dysglycaemia and for obesity [Citation77]. A post hoc analysis was carried out to assess the effects of multifactorial intervention on liver tests and their association with CAD in subjects with MetS but without CVD and DM. Among the whole population, 326 patients had elevated liver tests and evidence of NAFLD on imaging studies. Of these, 165 patients received an average of 34 mg/day of atorvastatin to attain the LDL-C < 100 mg/dL target (Group A). The remaining 161 patients received an average of 24 mg/day of atorvastatin to attain the LDL-C < 130 mg/dLtarget (Group B). NAFLD resolved during the 42-month treatment period in 86% of patients in the high atorvastatin group and in 74% of patients in the low atorvastatin group. Nearly 90% of patients attained their target lipid goals. There were no CVD events in group A, whereas five non-fatal events occurred in group B. No major adverse events were reported [Citation78].
A Cochrane database review of 653 records assessed the effects of statins on all-cause and liver-related mortality, adverse events, histological, biochemical and imaging responses in patients with NAFLD or NASH. It found only two RCTs eligible but neither reported on histological changes, liver-related morbidity or mortality. It concluded that statins may improve liver enzymes as well as ultrasound findings but that larger trials are needed [Citation79].
Dongiovanni et al. studied the relationship between statins, genetic risk factors and liver damage in 1201 Europeans who underwent a liver biopsy for suspected NASH [Citation80]; 107 of these patients received simvastatin or rosuvastatin. There was a significant reduction in steatosis by 48–91% and NASH by 38–76% independent of steatosis, while significant fibrosis was almost halved.
An open-label study evaluated the efficacy of pitavastatin for the treatment of NASH with dyslipidaemia. Follow-up biopsies available for 13 NASH patients showed a slight reduction of NASH activity score although there was no significant change in the fibrosis stage [Citation55]. In another open-label study by the same authors in patients with NASH and dyslipidaemia [Citation56], atorvastatin was administered to the patients and follow-up biopsies were carried out after 24 months. There was significant improvement in liver macrovascular steatosis and NASH activity score in both studies but some patients had increased fibrosis stage. The population-based Rotterdam Study was carried out in 2578 subjects who underwent liver ultrasonography. A lower prevalence of liver steatosis in the patients treated with statins for >2 years was reported [Citation57].
In another study, patients with NASH were randomised to receive simvastatin or placebo. There was no statistically significant improvement in serum ALT, hepatic steatosis, necro-inflammatory activity or stage of fibrosis [Citation81, 82].
A post hoc analysis of the IDEAL trial (n = 8863) suggested that the CV benefit of intensive lipid lowering with atorvastatin compared with a more moderate regimen with simvastatin was generally greater in patients with mild-to-moderately elevated baseline ALT than patients with normal baseline ALT [Citation83]. The statin treatment was safe.
A recent review concluded that the prevalence of NAFLD is increasing in South Asians with male predominance. South Asians develop NAFLD at an earlier age as compared with the Western population. BMI does not fully appreciate the risk of developing NAFLD due to variation in ethnic distribution of fat. This increasing prevalence contributes to an increased incidence of hepatocellular carcinoma, requiring population based studies [Citation84].
5.4. LAI recommendations
The available evidence suggests that the risk for serious liver injury from statins is quite rare and patients with NAFLD and dyslipidaemia are not at an increased risk for statin-induced hepatotoxicity. Hence, statins could be used by physicians to treat dyslipidaemia in patients with NAFLD.
There are few data to suggest usage of statins as a treatment option for NAFLD. However, it continues to be a matter of debate and treatment of NAFLD with statins cannot be recommended at present. Larger trials with histological evidence or use of follow-up Fibroscan are required to investigate the efficacy of statins in NAFLD.
6. Statins and cerebrovascular disease
The crude prevalence of stroke in India is estimated as 1.27–2.20 per 1000 and a higher rate is recorded in the Parsi community. A large number of these are ischemic strokes compared with haemorrhagic strokes. Of the total number of strokes, 10–15% are in the young; hypertension, smoking and hyperlipidaemia contribute to stroke risk, especially in the young. There is also a higher prevalence of haemorrhagic stroke in the young [Citation85, 86].
Statins are the lipid-lowering agents most commonly prescribed to reduce the risk of cerebrovascular events. But what is the evidence supporting their use in different cerebrovascular conditions? Can they cause any harm? We briefly discuss the current evidence for and against the use of statins in various cerebrovascular diseases.
SPARCL is a prospective double-blind placebo-controlled trial in 4731 patients with stroke or TIA randomised either to placebo or atorvastatin 80 mg (4.9 years of follow up). Those with haemorrhagic stroke without risk factors for ischemic stroke were excluded from the trial. Several subgroup analyses from this important multicentre study have been published [Citation87–Citation91].
The primary endpoint – fatal or non-fatal stroke – was less frequent in the atorvastatin group with a RRR of 16% (p = 0.03 CI 0.71–0.99). This was mostly driven by reduction in acute stroke with RRR of 43% with no effect on non-fatal stroke. Atorvastatin reduced ischemic stroke by 21% and the partial attenuation to 16% was due to increase in haemorrhagic stroke. Those with haemorrhagic stroke did not benefit from treatment (HR 2.82; 95% CI 0.89–0.90).
There was a reduction of TIA and stroke by 23% and TIA alone by 26%.
Major coronary events, revascularisation and any CV event were reduced by 35% (HR 0.65, CI 95% 0.49–0.87), 45% (HR 0.55, 95% CI 0.43–0.72) and 42% (HR 0.58, 95% CI 0.46–0.73), respectively although there was no difference in total mortality. In patients with >50% reduction in LDL-C, a combined risk reduction of fatal and non-fatal stroke was seen and no increase in risk of intracerebral haemorrhage (ICH) was seen in those with the greatest reduction of LDL-C.
Primary endpoint was a 10% RRR in the elderly (over 65 years) group compared with 26% in the younger (HR 0.74, 0.57–0.96, p = 0.02).
The risk reduction in stroke in those with carotid stenosis confirmed by ultrasound was (HR0.67 CI 0.47–0.94; p = 0.02); an annual risk reduction of 2.5% was observed with NNT of 8 comparable to carotid endarterectomy but without the problems associated with surgery. As carotid artery evaluation was not required by the SPARCL protocol, carotid imaging carried out by the local investigator was used in the study.
Studies aimed at patients with coronary heart disease (CHD) showed a decrease in stroke by one-quarter by addition of a statin [Citation8].
6.1. Acute ischemic stroke
An important question in patients with acute ischemic stroke is whether statins should be started in those who are not already on statins. There is a lack of robust clinical evidence supporting this. However, several studies have shown that statins have many pleiotropic effects viz. expression of endothelial nitric oxide synthase which causes vasodilatation and enhancement of cerebral blood flow, enhancement of collateral circulation, angiogenesis, anti-inflammatory action, stabilisation of the endothelial layer and enhancement of tissue plasminogen activator. All these effects are thought to benefit by reducing infarct volume in acute ischemic stroke [Citation92]. In addition, a recent meta-analysis of mainly observational studies has suggested that immediate statin therapy in acute ischemic stroke might improve functional disability and short-term mortality [Citation93]. Although the American Stroke Association (ASA) Guidelines [Citation94] do not provide any specific recommendation as to if and when to start statins in patients of acute ischemic stroke, it seems prudent to initiate statins in acute ischemic stroke.
The contention that statins may be beneficial in acute ischemic stroke was supported in a small randomised study of 89 patients already taking chronic statins at the time of ischemic stroke, where brief withdrawal of statins during the acute period was associated with increased odds of death or dependency at 3 months [Citation95]. Based on these observations, it is recommended to continue statin therapy among patients already taking statins at the time of onset of ischemic stroke (AHA/ASA Classification of Recommendations and Level of Evidence: class IIa; level of evidence B) [Citation94].
6.2. Statins and thrombolysis
A meta-analysis of three studies totalling 4339 ischemic stroke patients treated with thrombolysis showed an association between statins and increased fatality at 90 days. However, this association was no longer present after adjusting for age and stroke severity in the largest study [Citation96]. On the other hand, a more recent thrombolysis and statins (THRaST) study reported that statin use within 72 h after stroke was associated with favourable functional outcome and a reduced risk of mortality at 90 days in 1844 patients treated with intravenous thrombolysis [Citation97].
Therefore, in an acute stroke patient eligible for thrombolysis and already taking statin, thrombolysis is not contraindicated, and statins should not be discontinued. If the patient is not already on a statin, then a statin should be started (as per THRaST study).
6.3. Acute ICH
Several studies have shown that low serum total cholesterol level is associated with increased risk of ICH [Citation87, 98–100].
However, in a meta-analysis of 31 randomised controlled trials, active statin therapy was not associated with significant increase in the risk of ICH. A significant reduction in all strokes and all-cause mortality was observed with statin therapy [Citation101].
A prospective study from Canada in patients of ICH demonstrated that prior use of statins is not associated with worse outcome and patients who discontinue their statin therapy after admission to hospital have higher mortality and bad prognosis [Citation102].
Based on these observations, in patients of acute haemorrhagic stroke, it is best not to start a statin, but if the patient is already on statin, it is prudent not to discontinue it.
6.4. Prevention of ischemic stroke
6.4.1. Primary prevention
The Medical Research Council/British Heart Foundation Heart Protection Study (HPS) found a 24% reduction in strokes (95% CI, 6–39%; p = 0.01) among 5963 diabetic individuals treated with a statin in addition to best medical care [Citation8].
The Collaborative Atorvastatin Diabetes Study (CARDS) reported that in patients with type 2 diabetes with at least one additional risk factor (retinopathy, albuminuria, current smoking or hypertension) and anLDL-C ≥ 160 mg/dL but without a prior history of CVD, treatment with a statin resulted in a 48% reduction in stroke (95% CI, 11–69%) [Citation103].
Based on the above, statin treatment along with lifestyle changes is recommended in adults with diabetes, especially those with additional risk factors, or CAD, to lower the risk of a first stroke (AHA/ASA class I; level of evidence A) [Citation104].
6.4.2. Secondary prevention
A meta-analysis of several studies has shown that statins are beneficial in preventing a second ischemic stroke, but do not prevent fatal stroke [Citation105].
As per ACC/ASA guidelines, therapy with a high intensity statin is recommended if LDL-C level is ≥100 mg/dL (AHA/ASA class I; level of evidence B) [Citation106].
Therapy with high intensity statin is also recommended if LDL-C level is <100 mg/dL, (AHA/ASA class I; level of evidence C) [Citation106].
As per 2013 ACC/AHA cholesterol guidelines, patients with ischemic stroke or transient ischemic attacks should be managed with lifestyle modification, dietary recommendations and medication recommendations [Citation107](AHA/ASA class I; level of evidence A) [Citation106].
However, for the Indian population, treatment goals and statin intervention thresholds are as per LAI part 1 recommendations [Citation1].
6.5. Patients with previous haemorrhagic stroke
Notwithstanding older studies suggesting a higher incidence of ICH in patients on aggressive statin therapy, recent large studies do not indicate that long-term statin treatment confers a higher risk of ICH [Citation108]. Although they may be associated with a non-significant risk of haemorrhage, other benefits may outweigh this risk, and the current recommendation is to use statins, preferably at a lower dose. However, in patients of chronic haemorrhage who have low risk of ischemic stroke or MI but high risk of ICH recurrence, for example, after primary lobar haemorrhage, statins are best avoided [Citation109, 110].
6.6. LAI recommendations
Acute ischemic stroke: Starting a statin is probably beneficial. Continue statin if patient is already receiving it. Statins are not contraindicated if patient is thrombolysed. It may be given by nasogastric tube in patients with dysphagia.
Acute ICH: Do not initiate statin if patient is not already on it. Do not discontinue statin if patient is already on a statin. Optimal blood pressure control is essential in those already on a statin.
Primary prevention of stroke: Statin treatment is recommended in adults with diabetes, or CAD, to lower the risk of a first stroke.
Secondary prevention of stroke: Statins are beneficial in preventing a second ischemic stroke, but do not prevent fatal stroke.
Patients with previous haemorrhagic stroke: Statins are best avoided in patients with previous lobar haemorrhage, but may be beneficial in preventing stroke in patients with previous basal-ganglionic haemorrhage who have a high risk for ischemic events.
7. Dyslipidaemia in thyroid disorders
Thyroid hormones influence glucose, lipid and protein metabolism and are one of the major regulators of energy metabolism. Therefore, most thyroid function disorders are associated with lipid abnormalities. Management of lipid abnormalities in thyroid disorders is challenging because it depends on the treatment of thyroid function abnormalities as well as primary genetically determined abnormal lipid metabolism. Additionally, there are other interfaces of thyroid and lipid metabolism such as the effects of thyroid hormone on glucose metabolism, body weight, adipo-cytokines and turnover of apolipoproteins and lipoprotein receptors, all of which are involved with lipid disorders.
7.1. Lipid abnormalities in thyroid diseases
The relationship between lipid abnormalities and thyroid function/dysfunction was first reported by Epstein and Lande, who in 1922 demonstrated an association between thyroid disorders and cholesterol [Citation111]. Turner [Citation112] observed in 1933 that the whole thyroid gland when administered simultaneously with cholesterol prevented the atheromatous changes produced by the latter in the aorta of rabbits in 17 of 19 animals.
Total cholesterol and LDL-C levels are increased by around 30% in patients with overt hypothyroidism. The magnitude of increase in total cholesterol is to some extent dependent on the severity of hypothyroidism and largely reversible with thyroid hormone replacement [Citation113–Citation116]. The higher the degree of hypercholesterolaemia, the more the reduction in cholesterol level after thyroxinereplacement [Citation117, 118]. However, total cholesterol and LDL-C levels are not completely normalised with thyroxine replacement in all patients, particularly in those with underlying cholesterol metabolic disorders [Citation119]. The primary lipid metabolism defect persists in them even after complete thyroxine replacement [Citation120]. Apart from quantitative changes, there are also reports on qualitative changes in LDL particles in hypothyroidism. Elevation in small-dense LDL particle has been reported in some, but not all studies. The amount of oxidised LDL particle, apolipoprotein A-1 (Apo A1), lipoprotein (a) [Lp(a)] and apolipoprotein B (ApoB), has also been reported to be elevated in hypothyroidism [Citation121, 122].
Overt hypothyroidism has also been reported to be associated with increase in TG in some, but not all patients [Citation123]. The elevation in TG levels is associated with increased levels of VLDL and occasionally fasting chylomicronaemia [Citation124]. The VLDL and intermediate density lipoprotein (IDL) particles in hypothyroidism are rich in cholesterol and apolipoprotein E (Apo E), thus resembling VLDL particles of type III hypolipoproteinaemia [Citation125]. HDL-C has also been reported to be normal or marginally elevated in overt hypothyroidism [Citation124].
Sub-clinical hypothyroidism has also been reported to be associated with increase in total cholesterol and LDL-C level [Citation126, 127]. The other abnormalities mentioned above in overt hypothyroidism are usually not seen in subclinical hypothyroidism [Citation128]. Even within the normal range, there is a positive association between the thyroid stimulating hormone (TSH) level and the cholesterol level [Citation129].
Thyrotoxicosis is associated with not only weight loss, but also lipid changes opposite to those seen in hypothyroidism. LDL-C and TG levels are reduced in thyrotoxicosis [Citation130, 131].
7.2. Mechanism of thyroid-related dyslipidaemia
Thyroid hormone triiodothyronine increases intestinal cholesterol absorption and the activity of 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMG CoA reductase), the rate-limiting enzyme in cholesterol biosynthesis [Citation132]. But it also increases the expression of LDL receptors in liver [Citation133, 134]. Though the former two mechanisms increase cholesterol levels, the latter mechanism dominates and the net result is an increase in cholesterol level in hypothyroidism and the reverse in thyrotoxicosis. Triiodothyronine can influence HDL metabolism by increasing cholesteryl ester transfer protein (CETP) activity, which exchanges cholesteryl esters from HDL2 to VLDL and TG in the opposite direction. Triiodothyonine stimulates the activity of lipoprotein lipase (LPL), which catabolises the TG-rich lipoproteins, and decreases TG concentration. They also activate hepatic lipase, which hydrolyses HDL2 to HDL3. These, mechanisms contribute to the conversion of IDL to LDL and in turn LDL to small-dense LDL [Citation121].
The metabolic effects of thyroid hormone are through both nuclear receptor and non-receptor mechanisms [Citation135, 136]. Its effects on lipid and glucose metabolism are through TR-β subunit activation predominantly [Citation137]. It indirectly activates peroxisome proliferator-activated receptor (PPAR)-α and PPAR-γ receptors as well [Citation138]. It also activates the respiratory chain [Citation139]. Recently, it has been found that thyroid hormone also increases browning of adipose tissue in individuals with severe insulin resistance [Citation140].
7.3. Cardiovascular risk in hypothyroidism
Apart from dyslipidaemia, obesity, increased waist circumference, increased homocysteine level, high-hsCRP, hypercoagulability, hypertension and increased arterial stiffness are other mechanisms through which hypothyroidism contributes to CV risk [Citation141]. Hypothyroidism has been found to be associated with increased carotid intima thickness (cIMT) and impaired endothelial function [Citation142].
7.4. Clinical epidemiology
As hypothyroidism (including subclinical hypothyroidism) is a common condition, lipid abnormalities and thyroid disorders coexist in a substantial number of individuals. The prevalence of dyslipidaemia in subjects with clinically overt significant hypothyroidism is reported as high as 80% [Citation143]. Among individuals with hypercholesterolaemia, the reported prevalence of elevated TSH is around 15% [Citation144–Citation146]. However, the majority of them have sub-clinical hypothyroidism [Citation144]. According to a study, 13% of individuals in the general population had sub-clinical hypothyroidism and 25% of them had lipid abnormalities [Citation147]. Therefore, hypothyroidism is an important cause of secondary dyslipidaemia and all subjects with dyslipidaemia should be screened for hypothyroidism.
7.5. Management of dyslipidaemia in thyroid disorders
All patients with dyslipidaemia and overt hypothyroidism should be treated with thyroxine initially. A period of 4–6 weeks of thyroxine replacement therapy is usually needed to correct the dyslipidaemia [Citation148]. As this treatment may exacerbate myocardial ischaemia in patients with underlying CAD, it is recommended to start treatment at lower doses (25 μg/day) and titrate gradually (12.5–25.0 μg increments) at intervals of 4–6 weeks with monitoring of clinical features, TSH levels and electrocardiogram. If adequate response is not seen with thyroxine treatment alone after 4–6 weeks of therapy, lipid-lowering medications can be then initiated [Citation149]. Use of statins in hypothyroidism with dyslipidaemia should be customised. In patients not known to have underlying CAD or risk for CAD, it is better to wait until the patient becomes euthyroid. A repeat lipid profile will help to proceed according to the existing guidelines [Citation107]. In presence of underlying CAD or acute coronary syndrome, statins should be started along with levothyroxine replacement [Citation150]. Use of statins or fibrates alone or in combination in patients with dyslipidaemia and uncontrolled hypothyroidism carries a higher risk of myopathy [Citation151]. Risk of myopathy may also be associated with the co-administration of statins with thyroxine [Citation152]. It is generally agreed that thyroid replacement therapy has beneficial effects on serum lipid profile and CVD risk in overt hypothyroid patients, but no clear consensus has been established regarding the treatment of sub-clinical hypothyroidism subjects. Some of the points put forth by researches in favour of providing treatment to patients with sub-clinical hypothyroidism are that treatment will prevent progression to overt hypothyroidism and will also reduce future risk for CVD [Citation153]. However, these findings are yet to be strengthened by definitive evidence. There is no clear consensus on whether thyroid replacement therapy with thyroxine has beneficial effects on serum lipid profile and CVD risk in subclinical hypothyroidism patients [Citation154]. Thyroid replacement in patients with subclinical hypothyroidism is recommended if serum TSH is >10 mIU/L, patients have high initial cholesterol levels or are elderly, are smokers or are positive for anti-thyroid peroxidase antibodies.
The treatment of hyperthyroidism is directed towards lowering the serum concentration of thyroid hormones and to re-establish a eumetabolic state. The management of lipid levels in patients with hyperthyroidism should be based on the existing guidelines [Citation107]. Sewerynek et al. suggested that treatment with methimazole in patients with hyperthyroidism can be protective against the oxidative stress induced by overproduction of thyroid hormones [Citation155].
7.6. LAI recommendations
All patients with dyslipidaemia should be screened for thyroid dysfunction.
Patients with dyslipidaemia and overt hypothyroidism should be treated initially with thyroxine.
In patients with underlying risk for CAD, start low dose thyroxine and titrate upwards slowly. Assess CVD risk and start lipid-lowering drugs as per LAI recommendation for primary prevention of CVD.
Patients without underlying risk for CAD: wait for the patient to become euthyroid.
Statins or fibrates alone or in combination in patients with dyslipidaemia and uncontrolled hypothyroidism carry a higher risk of myopathy.
Thyroid replacement in patients with sub-clinical hypothyroidism if serum TSH is >10 mIU/L, patients have high initial cholesterol levels or are elderly, are smokers or are positive for anti-thyroid peroxidase antibodies.
In patients with established vascular disease requiring thyroxine and lipid-lowering therapy, caution should be exercised with thyroxine dose to prevent onset of ischemic symptoms.
8. Management of ASCVD risk in inflammatory joint diseases
Inflammatory joint disorders (IJDs) include rheumatoid arthritis (RA), spondyloarthritis [including ankylosing spondylitis (AS)] and psoriaticarthritis (PsA) and many others. The first two are the main groups of joint disorders that have been evaluated and managed for CVD in clinical practice. These two conditions are associated with an excess of CVD morbidity and mortality. Systemic lupus erythematosus, progressive systemic sclerosis, etc. are other autoimmune conditions (not strictly “IJDs”) associated with excess CVD morbidity, but compared with IJDs, these conditions have been subjected to less scrutiny vis-a-vis their CVD potential. The following discussion therefore focuses on the CVD risk and its management in the IJDs only (namely RA, AS and PsA).
IJDs are being managed with increasingly effective medications like biologic disease modifying anti-rheumatic drugs (DMARDs) and targeted synthetic DMARDs (tsDMARDs) [Citation156, 157]. The excess morbidity and mortality of IJDs are no longer attributed to musculoskeletal factors, but are more due to increased CV events [Citation158, 159]. The main reasons for the excess CVD in IJDs include: (a) presence of systemic inflammation that also causes vascular inflammation [Citation160], (b) the deleterious effect of non-steroidal anti-inflammatory drugs (NSAIDs) and long-term steroids, used for managing IJDs [Citation161] and (c) a reduced ability of patients with IJD to exercise [Citation162] and lifestyle-related factors (smoking in particular). Indeed, the risk of CVD in RA patients rivals the risk of CVD in diabetic patients [Citation158]. Also, AS and PsA have now been studied for their CVD potential [Citation159, 163] and their CVD assessment and treatment may soon follow a path similar to the one followed for RA.
8.1. Evidence from clinical trials for the beneficial role of statin use in RA
One landmark study published in 2004 (the “TARA” trial) evaluated the disease-modifying effect of a statin in the treatment of RA. The authors postulated that statins might reduce inflammation, its associated CV (cardiovascular) implications and could therefore be potentially disease-modifying (akin to methotrexate (MTX) and prednisolone) in reducing arthritis. Patients (n = 116) with RA were randomised in a double-blind placebo-controlled trial to receive 40 mg of atorvastatin or placebo on the background of standard disease-modifying therapy. The findings at the end of 6 months revealed a statistically significant but clinically modest reduction in arthritis scores in the atorvastatin groups. However, these scores were not enough to qualify as “disease modification” by current Biologic and DMARD standards in RA treatment [Citation164].
In another trial, 30 patients with early RA received either MTX plus prednisolone or MTX plus prednisolone plus a statin (atorvastatin 40 mg/day). The parameters of inflammation that included tissue necrosis factor (TNF)-alpha levels, lipid profiles, serum malondialdehyde (MDA) and brachial artery flow-mediated dilatation (FMD) were evaluated before and 6 months after treatment. The results favoured the atorvastatin group for an overall improvement in the endothelial function and CV risk benefit (clinical parameters were not compared) [Citation165].
In another trial which studied the effect of intensive lipid-lowering strategies in patients with RA and a previous MI (“IDEAL trial”) showed that RA patients with a previous MI had comparable reduction in lipid levels and rates of CV events when compared with patients without RA [Citation166].
8.2. General recommendations
Recommendations for CVD risk assessment and its management in RA and AS or PsA have been published (“EULAR recommendations-2009”) [Citation167]. These have been updated with modifications based on new data from clinical and epidemiological studies [Citation168]. Briefly, the updated recommendations are: 1) TC/HDL-Cratio is preferred over individual lipid components when calculating CVD risk in RA patients and the values should be assessed when the patient is in complete remission or has stable activity, 2) The use of an RA-adapted risk prediction model was recommended over the use of an un-adapted general population model. Moreover, the general population risk CVD risk algorithm should have a multiplication factor of 1.5 for all RA patients (and not just with long-standing seropositive disease as was recommended by the 2009 EULAR recommendation), 3) CVD risk assessment should apply for RA as well as other IJDs like AS and PsA and assessments should be repeated every 5 years by the treating rheumatologist and 4) Disease control for all IJDs (RA, AS, PsA) is mandatory and should be the first step for reducing CVD risk.
Most rheumatologists will target complete remission or low disease activity, both for RA and SpA/PsA. A complete remission would logically reduce the disease-related vascular inflammation, and would therefore be the single most important factor for reducing CVD events in IJDs. Correction of hypertension and lipid abnormalities is only the next step for reducing CVD in IJDs, as discussed in the following paragraphs. Treatment with medications including NSAIDs, steroids, biologics DMARDs (bDMARDs) and tsDMARDs is followed based on guidelines published by the American College of Rheumatology [Citation169].
The recommendations for the management of RA and AS published from India [Citation169, 170] can also be referred to, but their relevance to current management is limited given their date of publication (2010 and 2012). Simpler algorithms have been published for the management of early RA [Citation171] and established RA [Citation172] and are inclusive of bDMARDs and conventional DMARDs. These may instead be used for management decisions especially for Indian patients. The rheumatologist/physician/musculoskeletal expert should evaluate and manage CVD risk in all patients of IJDs, with input from cardiologists/neurologists/physicians [Citation167, 168].
The treating physician should be wary of the low levels of LDL-C, HDL-C and total cholesterol in untreated and active disease with RA [Citation173], which when used for CVD risk calculation in these patients will underestimate the risk. This paradox of low lipid levels with a higher propensity to CVD is partly because the HDL is dysfunctional and its protective role is compromised in active inflammation [Citation174, 175]. Similar abnormalities (low LDL-C and low HDL-C) are also found in sepsis/infections, trauma, etc. and this phenomenon may be due to cytokine-mediated activation of the reticulo-endothelial system which mops up the lipid particles [Citation173]. Successful anti-inflammatory treatment increases both the LDL-C and HDL-C [Citation176], but the level of the HDL-C is raised more than the LDL-C and therefore there is a net anti-atherogenic shift in total cholesterol/HDL-C ratio. Also, the use of conventional synthetic DMARDs (csDMARDs) like MTX and hydroxychloroquine in RA patients has resulted in a consistent overall improvement in the lipid profile, insulin resistance and cIMT [Citation177]. The remission induction and successful treatment of RA with csDMARDs and bDMARDs have registered benefits for CVD morbidity and mortality in large populations of RA patients (real-world registry data from various countries, BIOBADASER database, biologic registries from UK, US, etc.) [Citation178, 179].
The Prospective Randomised Evaluation Of Celecoxib Integrated Safety VS Ibuprofen Or Naproxen (PRECISION) trial’s results bring new NSAID data to the forefront, in which it was found that celecoxib was non-inferior to naproxen as regards safety of CVD events in a large cohort of osteoarthritis and RA patients combined [Citation180]. Another important finding from the study includes: for all-cause mortality in patients with RA patients alone, treated with naproxen, (hitherto considered the safest NSAID as far as CVD risk is concerned) the risks for CVD and all-cause mortality were higher when compared with celecoxib [selective cyclooxygenase-2 (COX-2) inhibitor, hitherto considered higher CVD risk as compared with conventional NSAIDs]. Therefore, it may be pertinent to consider selective COX-2 inhibitor celecoxib as the NSAID of choice for treating at least the elderly RA patients with comorbid gastrointestinal risk. On the other hand, for most osteoarthritis patients regardless of age, either celecoxib or naproxen may be used depending on patient/physician preference. At the time of the baseline visit and at each follow-up visit of IJD patients, the rheumatologist should assess adherence to diet and exercise. Cessation of smoking should be discussed with the patient if applicable.
A study showed that various biomarkers were affected in an Indian population with RA. CRP, TNF-α, serum nitrite, DAS-28 and depleted EPC population predicted endothelial dysfunction. Age, IL-6, HDL, LDL and depleted EPC population predicted accelerated atherosclerosis. Also, flow-mediated dilation and cIMT were also impaired in RA [Citation181].
8.3. LAI recommendations
CVD risk assessment for IJD patients should be carried out [including total cholesterol and HDL-C (fasting or non-fasting)] during the stable disease state once every 5 years [Citation168].
The use of statins and CVD risk management should follow the same principles as discussed in the Part1 of this consensus document [Citation1]. As discussed, the Joint British Societies 3rd version (JBS3) risk calculator should be the preferred calculator as other conventional calculators when used for CVD risk assessment in active RA and IJD patients underestimate CVD outcomes over time [Citation182]. This is because the low lipid levels underlie the true state of vascular inflammation. Statins should not be used solely for disease modification since trials like the TARA trial have shown a clinically insignificant arthritis-reducing result in RA. Statins have to be used in AS/PsA patients with a lower threshold for lipid abnormalities (statins lowered CVD mortality by 32% in a population of AS/PsA patients from UK – The Health Improvement Network-database UCL) [Citation183]. For hypertension management, the Indian guidelines or the European Society Guidelines should be followed and minor adjustments may be made depending on the comorbid conditions of the patient. Use of steroids should be at the lowest dose possible for the shortest duration. At each follow-up visit, the rheumatologist should assess the need for continued steroid use.
All NSAIDs (selective and non-selective COX-2 inhibitors) have a CVD risk with chronic use. Naproxen sodium is considered safer than other NSAIDs as regards their potential for CVD toxicity, but celecoxib has been found to be non-inferior to naproxen in the PRECISION trial as regards CVD risk [Citation180]. Celecoxib may therefore be considered equal to naproxen for most musculoskeletal conditions, and is the NSAID of choice in elderly musculoskeletal disease patients who have high gastrointestinal risk and CVD risk. There is insufficient evidence for use of ezetimibe in these patients.
9. Dyslipidaemia in the elderly
The World Health Organization (WHO) defines elderly as an individual aged ≥ 65 years, although racial variations may exist. The absolute risk of CAD and death due to CAD rises in both men and women with increasing age. In males, after the onset of puberty, cholesterol levels keep increasing until 50 years of age and plateau until 70 years followed by a slight fall in the levels. In females between 20 and 25 years, the cholesterol levels are higher than in men, and thereafter, it rises at a slower rate than in males. Cholesterol levels are almost equal between males and females aged 55–60 years. LDL-C mainly contributes to the increase in total cholesterol with age [Citation184]. This is probably due to the decreased ability of hepatic LDL receptors to metabolise LDL with age [Citation185]. The HDL-C values do not change much with age in both males and females. CV risk is predicted by serum cholesterol levels. High LDL-C and low HDL-C have been shown to be risk factors in the elderly for CAD both in the Framingham Heart Study and the Systolic Hypertension in the Elderly Program (SHEP) studies [Citation186, 187]. The Framingham Heart Study showed that men and women had 1.5 and 2.3 relative risk of developing symptomatic CAD when the serum cholesterol was above 200 mg/dL [Citation188]. Low HDL-C (<35 mg/dL) is also a risk factor contributing to increased deaths due to CAD in the elderly above 70 years of age, with a relative mortality of 4.9 in males and 2.0 in females [Citation188].
Although the relative increase in mortality that is seen with elevated cholesterol levels declines with age, the absolute risk increases with increasing age because of other comorbidities [Citation187]. The higher absolute risk in older adults suggests that the benefit from cholesterol-lowering therapy should be greater than in younger individuals [Citation189, 190]. However, various randomised trials with statins have excluded older patients because of concerns regarding safety, life expectancy, comorbidities in the elderly and cost–benefit analysis. Further, statins are also withheld in elderly patients because of the belief that LDL-C-lowering therapy requires many years before the course of atherosclerosis can be altered. However, many studies have observed clinical benefits as early as 6 months to 2 years [Citation191, 192]. Similarly, another concern is that side effects are more common in the elderly. However, many trials have shown that there is no significant increase in side effects compared with placebo in the elderly taking statins [Citation193, 194].
9.1. Benefits of lipid lowering in older adults
Subgroup analysis of various secondary prevention trials of LDL-C lowering show that elderly patients derive similar benefit from lipid-lowering therapy as younger ones [Citation8, 195, 196]. The CARDS study and the HPS showed similar reductions in serum lipids in elderly and younger individuals, with significant reduction in all-cause mortality, mortality from CAD, major coronary events and the number of revascularisation procedures in elderly patients [Citation8, 197, 198]. The LIPID trial which included a large number of patients between the ages of 65 and 75 years (n = 3514) who had a prior MI or unstable angina with a baseline serum cholesterol of 155 to 271 mg/dL showed greater absolute benefit of pravastatin in elderly patients [Citation195]. This was due to increased baseline risk of CV events in the elderly. In the Treating to New Targets (TNT) study, atorvastatin 80 mg was compared with 10 mg in 10,003 patients (of these, 3809 were ≥ 65 years) with stable CAD. High dose atorvastatin therapy significantly reduced major CV events compared with low dose without any increase in creatine kinase (CK) levels suggesting a role for aggressive statin therapy in the elderly [Citation199]. In a study of 7220 patients with angiographically proven CAD followed up for a mean of 3.3 years, statin therapy reduced mortality across all age groups including the very elderly (≥80 years) [Citation200]. However, in this study, the elderly were significantly less likely to be prescribed statins. Similarly, the Prospective Study of Pravastatin in the Elderly at Risk (PROSPER) trial [Citation201] and the Study Assessing Goals in the Elderly (SAGE) trial [Citation202] showed mortality reduction with intensive statin therapy in patients aged ≥ 65 years with a history of or risk factors for vascular disease. Importantly, in the PROSPER study, pravastatin had no significant effect on cognitive function.
Primary prevention trials of statins, including the Air Force/Texas Coronary Atherosclerosis Prevention study (AFCAPS/TexCAPS) and Anglo-Scandinavian Cardiac Outcomes Trial-Lipid-Lowering Arm (ASCOT-LLA), found similar results on clinical endpoints in younger and older individuals [Citation203–Citation205]. The JUPITER trial in which rosuvastatin was administered to patients with low-to-average LDL-C levels with elevated CRP levels, patients 70 years and older (32% of total cohort) derived greater absolute risk reduction than younger patients [Citation205].
The secondary causes of dyslipidaemia, e.g. hypothyroidism, diabetes mellitus, CKD, thiazide diuretics, should be ruled out and managed. The prevalence of subclinical and overt hypothyroidism increases with age and hence needs to be routinely ruled out [Citation206]. Similarly, CKD is common in the elderly and kidney function should be evaluated in an elderly patient with dyslipidaemia [Citation207]. Lifestyle measures including smoking, exercise and dietary saturated fat restriction are as important in the elderly as in the younger population. However, aggressive dietary restrictions may be inappropriate in the elderly as they are at high risk of malnutrition as dementia and physical disabilities may limit their access to adequate nutrition. Elderly patients are frequently treated with multiple medications and so carry the risk of drug interactions (e.g. administration of macrolide antibiotics can raise statin levels and increased risk of muscle toxicity). The adherence to statins declines substantially with time in elderly patients [Citation208]. This occurs even when the cost is not an issue.
9.2. Summary
The event rates in elderly subjects are proportionately higher than for younger subjects in both primary prevention and secondary prevention studies emphasising increased absolute risk. Hence, instituting statin therapy is important for appropriate subjects. Both chronological and physiologic age should be taken into consideration before deciding to treat high or high-normal serum cholesterol in elderly patients [Citation209]. Fibric acid derivative should be used with caution in those with renal impairment. The treatment should be individualised – a patient with a projected limited life span due to comorbidities may not be a candidate for statins, whereas an otherwise healthy older adult individual should not be denied drug therapy simply on the basis of age alone [Citation190].
Many trials support the use of lipid-lowering therapy for secondary prevention in older patients with established CAD who do not have life-limiting comorbid disease [Citation8, 197, 199]. The data from the Cardiovascular Health Study suggest significant benefit from primary prevention in patients aged ≥65 years [Citation210]. However, despite proven benefit, statins are largely underutilised in elderly patients even in those with increased baseline risk [Citation211].
9.3. LAI recommendations
CVD is the most common cause of death in the elderly. As in the young, elderly patients with dyslipidaemia have an increased risk for CVD.
Secondary causes of dyslipidaemia such as hypothyroidism, diabetes, CKD and drug effects should be considered in elderly patients.
The RRR of lipid-lowering therapy in elderly patients is similar to that in younger patients; however, the absolute benefit is higher than in younger patients.
Reductions in events with statin therapy are apparent within a few weeks to months even in older patients.
In general, side effects of lipid-lowering therapy are similar in the old and young. However, in very elderly frail patients, lower doses may be appropriate.
Secondary prevention: Statin therapy should be instituted in all appropriate elderly patients as above with treatment goals similar to younger patients. Dose of statins may need to be individualised in the elderly based on frailty and other comorbidities.
Primary prevention: Elderly subjects with a reasonable life expectancy should be treated similar to younger patients as per risk stratification algorithm proposed in the previously published LAI expert consensus statement [Citation1].
10. Dyslipidaemia in women
Heart disease remains the leading killer of women. After MI, women are more likely than men to develop HF or die within 5 years of the event [Citation212].
Management of dyslipidaemia in women requires consideration of gender-specific differences in CV risk, evidence from the available trials and the potential risks and benefits of lipid-lowering therapies.
Despite the importance of ASCVD as a health concern for women, they have historically been under-represented in RCTs of lipid-lowering therapies. In the statin trials that have included women, there have been no gender-specific differences in the observed lipid responses to statin therapy, with similar reductions in total cholesterol, LDL-C and TG, and increases in HDL-C among men and women [Citation213].
10.1. Physiology of lipoproteins in women
Women experience a number of hormonal changes throughout their lifetime, beginning from puberty, through pregnancy and up to menopause. LDL-C levels tend to increase in both men and women after the age of 20 years, but do so more rapidly in men [Citation184]. There is a rise in the number of hormones including human chorionic gonadotropin hormone, beta-estradiol, insulin and progesterone during pregnancy which is associated with increases in total cholesterol, TG and LDL-C [Citation214, 215]. Additionally, pregnancy is also associated with an increase in total HDL-C and Apo A1 concentration, with maximum levels at week 25 of pregnancy [Citation214, 215].
After menopause, women have increased levels of LDL-C, total cholesterol and Apo B compared with premenopausal women [Citation216]. There is a transition in LDL particles to more atherogenic smaller and more dense particles [Citation217]. Total HDL-C and HDL2 also decrease in postmenopausal women [Citation218]. Lipoprotein (a) levels increase after menopause and decrease in those on hormone replacement therapy (HRT) [Citation219, 220].
10.2. Evidence for the role of lipid pharmacologic therapy in women
Studies have demonstrated gender differences in the burden of CV outcomes for patients with dyslipidaemia. Farahaniet al. investigated the evolving pattern of sex/gender disparity in participants of RCTs on statins between 1990 and 2010. This study observed a significant progress in the inclusion of women in RCTs involving statins [Citation221].
10.2.1. Statins in women
The JUPITER primary prevention trial included 6801 women and 11,001 men. The post-treatment changes in lipid parameters and hsCRP levels were similar in men and women as was the primary endpoint which was a composite of MI, stroke, hospitalisation for unstable angina, arterial revascularisation or ASCVD death [Citation222].
The Management of Elevated Cholesterol in the Primary Prevention Group of Adult Japanese (MEGA) study which included highest percentage of women in any statin trial to date (7832 subjects with 5356 women – 68% and 2476 men – 22%) studied the usefulness of pravastatin in primary prevention of CV events in adult Japanese women [Citation223]. The incidence of CV events was 2–3 times lower in women compared with men. There was a non-significant 25% risk reduction in CAD in women on pravastatin compared with a non-significant 35% CAD risk reduction in men.
The Cholesterol Treatment Trialists’ (CTT) collaborators conducted a meta-analysis of 27 primary and secondary prevention RCTs with 174,249 participants [46,675 (27%) women] to examine gender differences in the efficacy and safety of statin therapy [Citation224]. The meta-analysis showed similar reductions in events in men and women.
In a meta-analysis that included 40,275 women from 18 primary and secondary prevention RCTs with gender-specific outcomes, a statistically significant decrease in the primary endpoint was observed among women in both primary and secondary prevention [Citation225]. Another meta-analysis of 37 prospective, multinational studies demonstrated that the relative risk (RR) for fatal CHD associated with diabetes is nearly 50% higher in women (RR 3.5; 95% CI 2.7–4.5) than it is in men (RR 2.06; 95% CI 1.81–2.34) after adjustment for major coronary risk factors, although the absolute ASCVD risk may remain lower in women with diabetes compared with men with diabetes [Citation226].
Based on the above information, statins are the first-line cholesterol-lowering drug therapy for women as per the risk stratification algorithm of LAI [Citation1].
10.2.2. Non-statin therapy in women
10.2.2.1. Niacin
The Heart Protection Study 2-Treatment of HDL to Reduce the Incidence of Vascular Events (HPS2-THRIVE) [Citation227] was a large trial conducted in 25,673 patients with established vascular disease (4444 women, 17.3%). Participants were randomly assigned to receive 2 g of extended release niacin and 40 mg of laropiprant or a matching placebo daily. Women randomised to niacin and laropiprant had an excess of events compared with those randomised to placebo with a trend towards harm compared with benefit in men, but this gender difference was not statistically significant.
In the Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides: Impact on Global Health Outcomes (AIM-HIGH) [Citation228] study, 3414 patients (14.8% women) with established ASCVD were randomised to extended release niacin 1500 to 2000 mg/day or matching placebo in addition to simvastatin and ezetimibe in all the patients. Following a 36-month follow-up, the trial was terminated due to lack of clinical benefit from the addition of extended release niacin to statin therapy. There was no heterogeneity by gender.
Nicotinic acid preparations are available for use in some countries including India. Care should be taken when prescribing niacin in view of the side effects.
10.2.2.2. Fibrates
Two large RCTs of fenofibrate have included women as well as men: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) and Action to Control Cardiovascular Risk in Diabetes (ACCORD) studies [Citation229, 230]. The FIELD study included 9795 (37.3% women) type II diabetic patients aged 50–75 years of age and not taking statin therapy at study entry. Approximately 22% of patients had pre-existing ASCVD. Patients were randomly assigned to micronised fenofibrate 200 mg/day or matching placebo and followed-up for 5 years. Although the study was not adequately powered for gender-specific analysis, women had a statistically significant 18.9% relative reduction in total CV events (9.5% for placebo vs. 7.7% for fenofibrate, p = 0.04). There were no significant gender-specific differences in adverse effects of treatment with fenofibrate [Citation229].
The ACCORD trial included both primary and secondary prevention patients (women 1694(31%), men 3824) with type 2 diabetes [Citation230]. Participants were randomised to simvastatin plus fenofibrate 160 mg or placebo and followed for 4.7 years for the primary outcome, and 5.0 years for all-cause death. In the subgroup analysis, men seemed to benefit from fenofibrate therapy, whereas there was a trend towards harm among women. The ACCORD study results are in contrast to the FIELD study results where no significant interaction was seen between treatment and sex on outcomes.
10.2.2.3. Ezetimibe
A gender subset analysis (women, n = 1065; men, n = 796) on data pooled across four randomised, double-blind trials showed that statin/ezetimibe combination decreased LDL-C equally in men and women [Citation231]. The Improved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT) included 18,144 patients (24% women) with acute coronary syndrome < 10 days duration who were randomised to simvastatin 40 mg + ezetimibe 10 mg daily or simvastatin monotherapy [Citation232]. Incremental lowering of LDL-C levels and improved CV outcomes were observed in the combination group. No statistically significant gender difference was observed in CV events.
10.2.2.4. Omega-3 fatty acids/fish oils
The Japan EPA Lipid Intervention Study (JELIS) evaluated the effects of statin plus highly purified eicosapentaenoic acid (EPA) ethyl ester 1800 mg daily (600 mg three times a day after meals) or usual care (no EPA) in 18,645 (68% women) Japanese primary and secondary prevention patients [Citation233]. There was no heterogeneity by gender for the relative reduction in the occurrence of coronary events, although women had a lower absolute risk of events (2.7% control vs. 2.0% EPA group) compared with men (4.2% control vs. 3.5% EPA group).
The Alpha Omega trial randomised post-MI patients who were on the state-of-art treatment to margarine supplemented with a combination of EPA and docosahexaenoic acid (DHA), a margarine supplemented with alphalinolenic acid (ALA), a margarine supplemented with EPA-DHA and ALA or placebo margarine [Citation234]. Overall, there was no benefit; however, there was non-significant 27% reduction in major CV events with ALA among women (HR 0.73, p = 0.07).
Not much gender-specific data are available presently for PCSK9 inhibitors, microsomal triglyceride transfer protein inhibitors and Apo B inhibitors. In the recently published Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk (FOURIER) study, which included 27,564 patients (25% women), the beneficial effects of evolocumab were similar in men and women [Citation235].
10.2.3. Non-pharmacological approaches
Therapeutic lifestyle changes are required for the optimal management of dyslipidaemia in women, including dietary changes, increases in physical activity and exercise and smoking cessation, all of which have been shown to reduce overall CVD risk factors and to contribute to a decline in CAD. Among 84,129 women in the Nurses’ Health Study, 82% of CAD events were attributed to lack of adherence to lifestyle guidelines [Citation236].
Tobacco use is a major preventable cause of CAD in women. Moderate (e.g. 1–2 drinks/day) alcohol consumption increases HDL-C in women and may be cardio-protective, but because of other concerns (increased risk of breast cancer, liver disease, alcoholism, etc.), it is difficult to routinely recommend regular alcohol consumption to non-drinkers.
In addition to dietary restrictions in saturated fat, trans fatty acids and cholesterol, some studies have reported that increased intake of soy or soy protein, especially as a replacement for animal protein, resulted in small, but at times, statistically significant, lipid improvements in women [Citation237].
10.3. Gender differences in adverse effects
Meta-analyses have shown no evidence for an increase in non-CV mortality, specifically no increase in cancer, associated with statin therapy in women [Citation224]. In JUPITER, women (n = 6801) were significantly older, more likely to have hypertension and the metabolic syndrome and had a higher body–mass index and lower eGFR compared with men (n = 11,001). The rates of muscle-related adverse events were similar among women and men regardless of assignment to statin or placebo [Citation222].
Nevertheless, gender differences in body fat, muscle mass, CoQ10 levels, pain perception and reporting of adverse reactions and underlying genetic factors (polymorphisms associated with cytochrome P450 isoenzymes, drug transporters or myocyte metabolism) may play a role in the increased frequencies of adverse drug effects reported by women [Citation238].
10.4. Special situations
10.4.1. Pregnancy
A steady rise of lipoprotein levels occurs throughout pregnancy; however, in uncomplicated pregnancy, neither total cholesterol nor TG exceeds 250 mg/dL [Citation239]. TG levels exceeding 250 mg/dL are associated with pregnancy complications, including pregnancy-induced hypertension, preeclampsia, gestational diabetes and more for gestational age babies [Citation240]. Secondary causes of hypertriglyceridaemia should be ruled out as in non-pregnant women. The Amsterdam Born Children and Their Development (ABCD) study showed that atherogenic lipid profiles during the first trimester confer an increased risk of adverse pregnancy outcomes including maternal morbidity, mortality and preterm delivery [Citation240].
The American Association of Physicians/American College of Obstetricians and Gynecologists Guidelines for Perinatal Care recommend lipid assessment annually for all age groups [Citation241]. The best time to screen for dyslipidaemia is prior to pregnancy and monitor during the pregnancy if values are elevated. Follow-up should be performed routinely after the pregnancy is over, usually by the 6-week postpartum visit.
10.4.1.1. Treatment of dyslipidaemia during pregnancy
Appropriate diet, weight management and exercise are essential for management during pregnancy. For women on lipid-lowering medications prior to pregnancy, all except bile acid sequestrants are currently recommended to be stopped in preparation for pregnancy [Citation242]. The Food and Drug Administration (FDA) has classified statins as category X ().
Table 3. Lipid-lowering agents and their safety during pregnancy.
Controlling glycaemia is central to managing increased TG in women with gestational diabetes or diabetes present prior to pregnancy. Very high TG (≥500 mg/dL is associated with a higher risk for pancreatitis) may be treated with diet/lifestyle management plus omega-3-fatty acids and/or with fenofibrate early in the second trimester based on clinical judgement [Citation243].
It is recommended that lipids should be monitored every trimester, or within 6 weeks of an intervention. Hypercholesterolaemia during pregnancy, especially in women with familial hypercholesterolaemia (FH), may be treated with bile acid sequestrants as they are not absorbed systemically. Bile acid sequestrants like colesevelam may increase TG levels. However, diet and exercise continue to be the mainstay of treatment during pregnancy. Women with FH may also be treated with LDL apheresis.
10.4.2. Breast feeding
Appropriate diet and exercise should be continued following pregnancy during breast feeding. Patients with FH can receive bile acid sequestrants, and drug therapy for severe hypertriglyceridaemia may be continued (as above). In the Nurse’s Health Study, there was a 23% lower incidence of MI in women in their 60s if they had breast fed [Citation236].
10.4.3. Polycystic ovary syndrome (PCOS)
PCOS affects 4–22% of reproductive age Indian women [Citation244, 245]. Women with PCOS are at increased risk for metabolic syndrome, diabetes mellitus and complications of pregnancy [Citation246], as well as endometrial cancer [Citation247]. Insulin resistance is associated with dyslipidaemia in women with PCOS, independent of obesity and the lipid profile is significantly different (high TG, total cholesterol and lower HDL-C) compared with insulin-sensitive women [Citation248].
As per the National Lipid Association (NLA) Expert Panel, patients with PCOS irrespective of their age should undergo initial lipid and diabetes screening, typically with fasting glucose or glycated haemoglobin and should have screening (at least every 2 years) if initial values are normal [Citation249]. Dyslipidaemia should be treated as per the LAI recommendation[Citation1].
10.4.4. Menopause
Both LDL-C and Apo B increase in the few years prior to menopausal symptoms, peak and then plateau. HDL-C tends to decrease after menopause. The absolute risk for ASCVD increases substantially during the menopause transition for women [Citation250].
In postmenopausal women, use of HRT with unopposed oestrogen is associated with significantly higher HDL-C and lower LDL-C compared with premenopausal women [Citation251]. Combination HRT appears to negate the increases in HDL-C seen with oestrogen monotherapy. In the Postmenopausal Oestrogen and Progesterone Interventions trial, investigators noted that combination HRT, which included progesterone, was associated with a significantly smaller increase in HDL-C and TG as compared with oestrogen-only HRT [Citation252].
While epidemiologic and observational studies suggested a CV benefit of HRT, data from randomised controlled trials failed to confirm these benefits. In the Heart and Oestrogen/Progestin Replacement Study, 4 years follow-up of 2763 postmenopausal women, use of HRT was associated with an 11% reduction in LDL-C and a 10% increase in HDL-C, but this beneficial lipid change was not associated with significant improvement in CAD [Citation253]. The Women’s Health Initiative study [Citation254] was stopped early due to a significant increase in the risk of stroke in the oestrogen-only-treated women and the oestrogen and progesterone therapy terminated due to increase in breast cancer cases.
Based upon these randomised controlled trials, treatment guidelines were subsequently changed with removal of the recommendation for use of HRT in women to prevent CAD events [Citation255].
10.4.5. Contraception
Oral contraceptive pill (OCP) can have estrogenic, progestational, androgenic, anti-estrogenic and anti-androgenic effects. The major risk associated with their use is thrombotic, including venous and arterial events. Women with comorbidities, older age and, most importantly, current smokers are at greater risk for CV events.
The oestrogen contained in OCP will frequently increase TG and HDL-C, and lower LDL-C [Citation256]. Androgenic progestins (such as norgestrel and levonorgestrel) can raise LDL-C and lower HDL-C. The other progestational agents are usually lipid-neutral. The more oestrogen an OCP contains, the greater TG raising effect it will generally have; these agents may cause severe hypertriglyceridaemia in women with high baseline TG [Citation257].
Transdermal or vaginal contraceptives (oestrogen plus progestin) do not have a lower thrombotic risk compared with OCP, but transdermal contraceptives are less prone to producing clinically important elevations in TG concentration [Citation258]. Progestin-only containing oral contraceptives are available and may be used if estrogenic preparations are contraindicated. They are, however, associated with greater risk for breakthrough bleeding and are not as effective for contraceptive purposes.
10.5. LAI recommendations
Based on all the evidence from primary and secondary RCTs, meta-analysis and the various available guidelines, the recommendations in Indian women can be summarised as:
10.5.1. General recommendations
Women with collagen vascular disorders, PCOS, preeclampsia, pregnancy-induced hypertension and gestational diabetes are at higher ASCVD risk.
In general, the dyslipidaemia management is similar in both men and women. However, in elderly frail women, treatment should be started with low dose statin in view of higher incidence of muscle-related adverse effects except in the very high risk group.
Statins remain the first-line cholesterol-lowering drug therapy for primary and secondary prevention in women. Statin dosage should be increased to the maximally tolerated dose before adding a non-statin drug if goal levels of LDL-C and non-HDL-C are not achieved.
Non-statin drugs may be considered as a primary drug for women with contraindications for, or intolerance to, statin therapy.
10.5.2. Recommendations in pregnancy
Lipid-lowering drugs must be stopped if pregnancy is planned, during pregnancy and during breast feeding.
Diet and exercise continue to be the mainstay of treatment during pregnancy. Statins are contraindicated. Only the bile acid sequestrants are considered safe for use during pregnancy.
Very high TG (≥500 mg/dL) may be treated during pregnancy and breast feeding with diet/lifestyle management plus omega-3 fatty acids [Citation259]; fenofibrate or gemfibrozil may be considered [Citation260]. During pregnancy there is a high risk of developing pancreatitis. Although apheresis may be useful in pregnant women, it is not available in India
10.5.3. Recommendations for PCOS
All patients with PCOS should undergo initial lipid and diabetes screening and regular follow-up is recommended.
Therapeutic management of dyslipidaemia in PCOS should focus on diet, exercise and lipid-lowering medication (as above).
10.5.4. Recommendations for menopause
Hormone therapy should not be used for prevention or treatment of ASCVD.
Hormone therapy is an option for treatment of significant menopausal symptoms during menopause transition for women at minimal risk for ASCVD.
10.5.5. Recommendations for contraception
Contraceptive pills derange lipid profiles and should be used carefully especially by women who use tobacco/tobacco products as it increases risk of thrombosis. LAI recommends annual lipid assessment in women using oral contraceptive pills for long duration.
11. Familial hypercholesterolaemia (FH) – an Indian perspective
FH is the most common monogenic disorder of lipoprotein metabolism, mostly with an autosomal dominant inheritance [Citation261]. This condition, also called as Fredrickson type 2a hyperlipidaemia, is characterised by elevated total cholesterol and LDL-C concentration. An estimated 14–34 million people are likely to have this condition worldwide but <1% of those individuals have been diagnosed with only half of them receiving optimal treatment [Citation262]. The affected individuals have a high risk for developing premature ASCVD and through appropriate intervention to reduce LDL-C concentration, significant reduction in mortality can be achieved [Citation263].
CVD is the leading cause of death attributing to 28% of the total mortality in the Indian population with an average age of onset below 55 years [Citation264]. However, the contribution of FH to premature CVD in Indians is unknown, mainly due to the lack of awareness of this condition among both health care providers and the general population. A recent survey in Tamil Nadu suggested a substantial deficit in awareness and knowledge of FH among medical practitioners [Citation265].
11.1. Pathophysiology of FH
FH is caused by mutations in the genes of the LDL receptor (LDLR) or Apo B orPCSK9 that interfere with clearance of LDL-C by the liver [Citation266]. An autosomal recessive rare form affecting LDL receptor adaptor protein 1 gene has also been described as causing FH [Citation267]. The most common cause among Indians is a mutation in the LDLR gene, accounting for up to 85–90% of all cases of FH [Citation268]. Over 1000 mutations have been described; however, the Pro685Leu mutation in exon 14 of LDLR has been shown to be particularly common accounting for 50% of all FH mutations in Indian migrants from South Africa [Citation269]. The prevalence of FH is high in Indians living in South Africa and it is possible that it is due to the “Founder effect”. However, in a study of 16 homozygous subjects, no mutation was seen in exons 3, 9 and 14 of the LDLR gene, which were considered to be the hot spots in studies done on Asian Indians in South Africa [Citation270]. FH occurs clinically as heterozygous FH (HeFH) or homozygous FH (HoFH). HeFH occurs when the affected individual inherits one mutant gene and is characterised by LDL-C concentration that is 3–4-fold higher compared with the general population. Such patients typically develop lipid stigmata that include corneal arcus and tendon xanthomata and develop premature CVD in their 4th decade, although women may develop these later. In contrast, those with HoFH inherit the defective genes from both the parents, have an LDL-C concentration that exceeds 4–8-fold that of the general population and they develop cutaneous stigmata and atherosclerotic CVD in their teens [Citation271].
11.2. Diagnosis of FH
Different diagnostic criteria have been proposed by different authorities/investigators. The Simon Broome Criteria, commonly used in the United Kingdom, take into consideration total cholesterol and LDL-C concentrations, presence of tendon xanthomata, presence of family history of premature vascular disease and cholesterol concentration in the family members [Citation272] (). According to these criteria, the index patient is categorised into “definite FH” or “possible FH” depending on certain predefined parameters-
Diagnose definite FH if the following are met-
| |||||||||||||||||||||||
| • | Diagnose possible FH if the following are present-
| ||||||||||||||||||||||
Figure 1. Peripheral stigmata included in familial hypercholesterolemia.
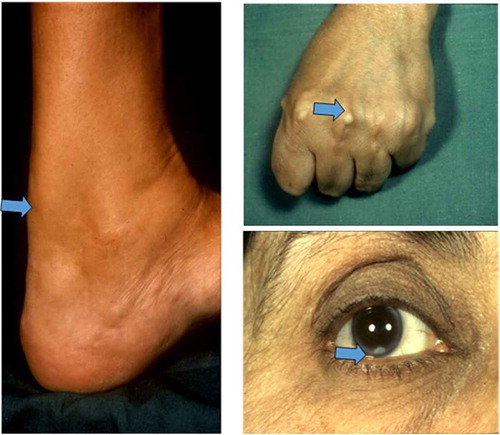
The Dutch criteria use a point system for LDL-C concentration, presence of xanthomata and presence of CVD and a total score of over 8 is considered as definite FH and 6–8 as probable FH [Citation273]. The US Make Early Diagnosis to Prevent Early Deaths (MEDPED) criteria uses age- and relative-specific LDL-C concentration for diagnosing FH [Citation274].
The LAI expert consensus recommends universal screening of all individuals prior to the age of 20 years or at the time of college entry [Citation1] or earlier if there is a family history of HeFH/HoFH or premature CAD. If all efforts are directed to implement this concept of screening, it may be possible to detect more cases of FH patients in India. This requires coordinated efforts from both government agencies and health care providers.
LAI recommends the Simon Broome criteria for diagnosis of FH because of ease of remembering the criteria in busy outpatient clinics. The Dutch criteria may be used but are more difficult in a busy outpatient setting. The Simon Broome diagnostic criteria require history taking for premature vascular disease in family, physical examination for lipid stigmata and measurement of the lipid profile. However, the other available criteria may be used as per physician preference.
11.2.1. Cascade screening
FH is an autosomal dominant condition with a 50% chance of inheritance of the condition in the siblings and therefore cascade testing (doing lipid profiles of relatives of an index case) is the most efficient way to identify new cases of FH [Citation275]. Cascade testing also facilitates early institution of preventive therapy in these high-risk individuals. The diagnosis of the index patient is carried out either clinically through history, physical examination and a lipid profile or by molecular diagnosis. Subsequently, the cascade testing of the family can be carried out similarly using lipid profiles followed by molecular testing in those meeting the criteria for FH. The National Institute for Health and Care Excellence (NICE) guideline for FH recommends a “3 generation” family history to guide and help with cascade screening [Citation276]. Health care providers should be trained to construct family trees with relevant information such as premature vascular disease in family members and cholesterol levels to allow assessment of the need for intervention among the family members ().
Figure 2. Constructing family tree in for a patient with familial hypercholesterolaemia.
Figure 3. Lipid stigmata in hypertriglyceridaemia.
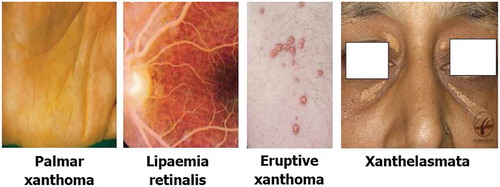
LAI recommends training health professionals in basic genetics as well as training in construction of the family tree. An online guidance will be available for that on the LAI website (www.lipid.net.in). Cascade testing requires time and in India, where most of the medical care is provided through a fee-paying structure, unless the government issues policies to assist case-finding, health professionals are likely to face barriers on financial grounds alone from individuals and family. Health professionals may also face difficulties in testing the young because their parents may not give consent to confirm a diagnosis for fear of not being able to marry their children due to the perceived stigma of a genetic diagnosis. LAI will work towards establishing awareness programmes within communities and provide access to information on the importance of testing family members. LAI would recommend testing children at age 2 if there is a family history of FH. Due to pubertal changes in cholesterol levels, the recommendation from different expert panels is to test children before age of 10 or after puberty [Citation277, 278].
11.2.2. Molecular diagnosis
A diagnosis of FH can be confirmed by genetic testing. LAI recommends that molecular testing should be available to confirm the diagnosis of FH. However, it should be understood by health care providers that failure to detect a mutation does not exclude a diagnosis of FH and intervention with lipid-lowering therapy is required if the diagnosis is made on clinical grounds [Citation1]. One should remember to “Treat the Phenotype and Counsel the Genotype”. Cost might become a deterrent in adoption of molecular testing given the fee-paying structure that is a norm in India.
11.2.3. Role of Lp(a)
Lp(a) is an independent risk predictor of ASCVD and in persons with FH [Citation279] and those with Lp(a) levels > 50 mg/dL have a higher risk of ASCVD. Accordingly, measurement of Lp(a) should be routinely performed in all individuals with premature ASCVD and particularly those with FH. These measurements do not need to be repeated as levels are mostly genetically determined. However, if therapies that can lower Lp(a) become available in future, monitoring may be required.
11.3. Management of FH (HoFH and HeFH)
Lifestyle issues such as smoking cessation should be addressed and other ASCVD risk factors should be meticulously looked for and treated.
Recommended diet should include no more than 30% of total calories from fat, <7–10% of total calories from saturated fat and dietary cholesterol restriction to 75–100 mg/1000 kcal, not to exceed 300 mg/day [Citation1]. For children >2 years of age, skimmed (fat free) unflavoured milk and limited sugar sweetened beverages should be encouraged [Citation280]. Food rich in saturated fat, i.e. butter, ghee, Indian sweets, cheese including paneer, ice cream, cakes, deserts and coconut oil, should be avoided and replaced with unsaturated fat such as nuts and vegetable oils. India is undergoing significant changes in the food industry with increased availability of processed food high in trans-fat and saturated fat. Though there is an upper allowable limit of 1% of trans fats in India, the LAI expert consensus recommends 0% trans fats in patients of HoFH. Food items such as burgers, pastries, pizzas and patties should be strictly avoided to reduce fat consumption. The meals should be based around high fibre-containing carbohydrates and proteins and should include plenty of fruits and vegetables.
A concern in paediatric patients consuming a low-fat and low-cholesterol diet is regarding the compromised growth, nutrition and psychosocial issues. Retarded growth rates were demonstrated in an eight-year study of growth patterns in 13 children with hypercholesterolaemia receiving <30% of total calories from fat. However, most of the recent studies suggest that a moderate dietary fat restriction does not affect growth [Citation281–Citation283]. Overall, paediatric subjects may need to be closely monitored for potential nutritional deficiencies because of a more limited food selection. Adolescents and adults with FH are encouraged to be physically active, avoid smoking and alcohol intake and strive to achieve the ideal body weight for their age.
There is no need to risk stratify this population as statin therapy is mandatory [Citation271, 272, 284]. The cumulative exposure to elevated LDL-Clevels from birth results in a high vascular risk. Statins at high doses is the mainstay of therapy with or without ezetimibe [Citation271, 285]. If intolerant to maximal doses of statin, low dose statin with bile acid resins is an option in other countries. The LAI expert consensus recommends pharmacological intervention with high doses of atorvastatin or rosuvastatin for adults with a diagnosis of HoFH [Citation1].
In children with FH with LDL-C of >160 mg/dL, treatment with statin should be started at the age of 8 years. Pravastatin or atorvastatin may be used with or without ezetimibe in children with a modest additional reduction of 15–20% [Citation286–Citation288]. There are controversies about earlier intervention with statins in children. The safety of statins in children as young as 8 years has been established [Citation289, 290]. At the same time, non-invasive evaluation using cIMT showed significant difference in children with FH compared with children without FH [Citation289].
Newer drugs are mipomersen [Citation291] and lomitapide [Citation292](not approved for HeFH) and PCSK9 inhibitors [Citation293]; the latter lower LDL-C levels by up to 60% in patients already on statins [Citation294, 295]. Both mipomersen and lomitapide have the potential for use in HoFH [Citation296–Citation298]. Thus, these drugs that lower the LDL-C concentration when added to existing therapy in adults give the opportunity to reach the LDL-C target that is optimal for this group [Citation293, 295].
Apheresis, a standard therapy until now used routinely in HoFH in many developed countries [Citation299–Citation301], is not available in India.
11.3.1. Drug therapy for LDL-C lowering can be summarised as
For Children
| |||||
| • | For Adults
| ||||
Liver transplantation corrects the defect in the LDLR-mediated metabolism leading to marked improvement in LDL-C levels. However, it has several inherent problems such as the risk of surgical complications, donor availability and exposure to lifelong immunosuppressive therapy [Citation271].
11.3.2. Annual review
All subjects diagnosed with FH should have a structured annual review when the family tree should be updated, lifestyle issues addressed and compliance with medication confirmed. Evidence of ASCVD should also be looked for.
11.3.3. Management of pregnant women with FH
At present, statins are contraindicated in pregnancy. Therefore, women in the reproductive age group should be advised to avoid pregnancy while taking statins and appropriate contraceptive cover should be provided. When planning for pregnancy, the health care professional should advise the patient to discontinue statins, and the same should be restarted after completing breast feeding [Citation302, 303]. If a woman with FH has significant CAD, she should be advised against pregnancy as appropriate in view of the risk of CV complications. Young women who need contraception should be advised regarding lipid-friendly contraceptives.
11.4. LAI recommendations
Lipid profile estimation of children to be done at 2 years of age in those with family history of FH and premature ASCVD.
Universal screening of lipids to be carried out at age 20 years or at the time of college admission.
LAI recommends the Simon Broome criteria for the diagnosis of FH.
In an established case of FH, LAI recommends estimation of Lp(a) levels.
Genetic testing and cascade screening should be performed wherever feasible.
Look for other ASCVD risk factors and manage them appropriately.
Strict dietary recommendations and lifestyle modifications as advised.
Drug therapy to be started at age 8 years or earlier in individualised cases.
LDL-C targets to be achieved:<70 mg/dL for HoFH and <100 mg/dL for HeFH in children and in adults <50 mg/dL in HoFH and 70 mg/dL in HeFH or at least 50% reduction in LDL-C from the baseline.
12. Inherited hypertriglyceridaemia
Hypertriglyceridaemia is defined as elevated fasting TG concentrations above the 95th percentile for age and gender in a given population [Citation304]. In all cases of raised TG levels, secondary causes such as obesity, diabetes, renal and liver diseases, as well as concomitant medication that may raise TG must be ruled out [Citation305–Citation308]. If the raised TG levels cannot be explained by a secondary cause, a diagnosis of primary hypertriglyceridaemia should be considered [Citation309]. Less than 5% of all hypertriglyceridaemia are primary and <1% are due to a monogenic cause [Citation310]. Loss of function mutations in apolipoprotein C3 (Apo C3) results in low levels of TG. A recent genetic study involving >75000 participants showed that heterozygosity for loss-of-function mutations in Apo C3 was associated with 44% lower levels of non-fasting TG levels with 41% reduction in the risk for CVD and 36% reduction in the risk for CAD [Citation311]. Apart from increased ASCVD risk with raised TG, TG levels > 1000 mg/dL are associated with an increased risk for acute pancreatitis [Citation312].
Dietary TG is absorbed and assembled as chylomicrons in the gut, which enter the circulation. These particles undergo a lipolytic process, whereby the free fatty acid and TG are removed through lipoprotein lipase (LPL) enzyme activity. After the initial phase of lipolysis, the chylomicron remnant that remains in the circulation is cleared by a receptor-mediated mechanism with the help of Apo E, which acts as a ligand [Citation313].
The majority of inherited lipoprotein disorders that fall under the Fredrickson classification are characterised by elevated TG concentrations except for FH (type IIahyperlipidaemia) [Citation314].
12.1. Classification of inherited forms of lipoprotein disorders
Inherited conditions with raised TG concentration include [Citation314]:
Chylomicronaemia syndrome or type 1 hyperlipidaemia
Familial combined hyperlipidaemiaor type 2b hyperlipidaemia
Dysbetalipoproteinaemia or type 3 hyperlipidaemia
Familial hypertriglyceridaemia or type 4 hyperlipidaemia
Type 5 hyperlipidaemia
These conditions are described in greater detail below.
All these conditions exacerbate the degree of hypertriglyceridaemia of pregnancy, especially the type 1 and type 5, with the added risk of complications resulting in maternal and fetal mortality due to acute pancreatitis [Citation315].
12.2. Diagnosis of hypertriglyceridaemia
Hypertriglyceridaemia results from increased production or reduced clearance of TG-rich particles in the circulation. It can either be a result of excessive circulating free fatty acids reaching the liver and increasing the synthesis or de novo synthesis of VLDL in the liver [Citation313, 316]. Insulin resistance contributes to both increased free fatty acid concentration and lipogenesis in the liver through the increased production of VLDL [Citation313, 316]. The relationship between VLDL production and insulin resistance can vary across different ethnicities and gender [Citation317].
LAI recommends assessing TG concentrations in a fasting state [Citation1]. A non-fasting lipid profile may be performed as an initial step in all individuals above 20 years of age or at the time of college admission. If the TG level is high in anon-fasting sample, then fasting TG estimation is needed, with a repeat measurement performed every 5 years [Citation189, 318]. Thresholds for diagnosis of hypertriglyceridaemia are the same as those mentioned in the LAI part 1 expert consensus statement, National Cholesterol Education Program or Endocrine Society Clinical Practice Guideline for the management of hypertriglyceridaemia [Citation189, 318] ( ).
Table 4. Classification of fasting serum triglyceride concentration (National Cholesterol Education Programme Adult Treatment Panel III classification) [Citation189].
12.2.1. Physical examination in a patient with severe hypertriglyceridaemia
All patients with elevated TG levels should undergo a full clinical examination and assessment to evaluate their CV risk, and those with very high TG levels should also be assessed for acute pancreatitis. Careful search for any underlying secondary cause of hypertriglyceridaemia should be made (). Signs indicating a secondary cause, such as pedal oedema as part of nephrotic syndrome, should not be missed. Physical assessment should include evaluation of any lipid stigmata. Any individual presenting with lipid stigmata as described below should have a full fasting lipid profile, and if necessary, further biochemical investigations should be carried out to evaluate the possibility of an inherited form of hypertriglyceridaemia
Table 5. Secondary causes for hypertriglyceridaemia.
12.2.1.1. Lipid stigmata in hypertriglyceridaemia (Figure 3) [Citation316]
Eruptive xanthomata: These erupt as small yellow papules found most commonly on the buttocks, shoulders, back, arms and legs. They do not appear on the face. They can be painful, tender and are often itchy. These lesions resolve when TG concentrations fall, as a result of treatment or aggressive lifestyle changes. They are often seen in type 1 hyperlipidaemia. Type 4 and 5 hyperlipidaemia may also present eruptive xanthomata. Infants and children with type 1 hyperlipidaemia or chylomicronaemia syndrome can manifest these lesions.
Palmar xanthoma: Found on palmar creases; they are pathognomonic of Type 3 hyperlipidaemia.
Tubo-eruptive xanthomata: Appear as clusters of small papules on elbows, knees and buttocks. They can grow to the size of grapes. They are pathognomonic of type 3 hyperlipidaemia.
Xanthelasma: Distributed as soft, velvety, yellow and flat topped papules and plaques in the upper and lower eyelids. These can also occur in people with normal lipids. Often these lesions are seen in those with hypothyroidism or liver disease, with or without increased lipid levels.
Lipaemiaretinalis: This is a milky appearance of the retinal vein when viewed through a fundoscope and is commonly seen in patients with severe hypertriglyceridaemia and can be associated with visual problems.
12.2.2. General investigations
12.2.2.1. Following investigations are required to exclude secondary causes [Citation319]
Urine dipstick test to rule out proteinuria, thereby excluding nephrotic syndrome and some other forms of renal disease
Thyroid function tests to rule out hypothyroidism
Liver tests (including gamma GT) to rule out liver disease, excessive alcohol intake and primary biliary cirrhosis. Further liver tests such as anti-mitochondrial antibody concentration and other autoantibody screens may be required
Glucose and glycated haemoglobin to rule out diabetes
Protein electrophoresis may be required as the presence of monoclonal proteins can raise TG.
12.2.3. Special Investigations
The various investigations to rule out different types of primary hyperlipidaemias are discussed below.
12.2.3.1. Type 1 hyperlipidaemia or chylomicronaemia syndrome
This is an autosomal recessive condition characterised by excessive circulating chylomicrons due to defective or absent LPL activity [Citation320, 321]. Usually it is seen in children or in the adolescent age group. The patients present with recurrent acute pancreatitis and very high TG levels (values reaching to about >1000 mg/dL), mostly contributed by chylomicrons. They develop extensive eruptive xanthomata and carry a high risk for recurrent acute pancreatitis. They may also present with hepatosplenomegaly that resolves with reduction in TG levels [Citation320–Citation322]. As the patient ages, the phenotype changes and subjects may present as type V hyperlipidaemia.
The prevalence is one in a million, but higher prevalence is possible in communities in India where history of consanguinity is not uncommon.
Genetic testing is available for the diagnosis of the condition. Additional proteins involved in lipolysis are Apo CII, Apo A5, Lipase Maturation factor 1 (LMF1) and glycosylphosphatidyl inositol anchoring high-density lipoprotein binding protein 1 (GPIHBP1). Mutations involving these proteins can also cause type 1 hyperlipidaemia [Citation323].
12.2.3.1.1. Investigations
The simplest test involves placing the tube in which the blood sample was centrifuged in a refrigerator to demonstrate the creamy layer with a clear serum layer separating it from the red cells. High TG levels interfere with measurement of LDL-C and other analytes like sodium concentration depending on the method used [Citation324].
A full lipid profile: often calculated LDL-C is not available as the TG level is high and the Friedewald formula for calculating LDL-C is inaccurate.
Lipoprotein Lipase: Mass or activity measurement of LPL is rarely available in routine laboratories. This measurement requires administration of 60 to 100 IU of heparin/kg to mobilise the LPL bound to the endothelium. These assays are only available in specialist centres [Citation325].
Genetic testing.
The United Kingdom Genetic Testing Network recommends that patients with type 1 hyperlipidaemia be tested to confirm the molecular defect, if they satisfy the following criteria (https://ukgtn.nhs.uk/)-
TG > 2000 mg/dL (>20 mmol/L)
History of acute pancreatitis
Secondary causes excluded
Known family mutation
Genetic testing is available in many international centres for diagnosis of this condition. LPL gene sequencing is essential for confirming a diagnosis; however, as mentioned above, genetic defects in other proteins involved in the lipolysis such as Apo C2 and A5, LMF1 and GPIHBP1 can also cause type 1 hyperlipidaemia. Therefore, current recommendation is for full gene sequencing of LPL and the genes for other four co-factors involved in lipolysis [Citation323].
Genetic testing is not widely available in India, and should be set up (perhaps in a reference centre) to provide a diagnostic service for these patients.
12.2.3.2. Type 2b hyperlipidaemia or familial combined hyperlipidaemia
Prevalence ranges from 1 in 50 to 1 in 200. Many genes are implicated. Often this condition is confused with FH and familial hypertriglyceridaemia. Age of presentation is middle age and beyond. This condition is associated with insulin resistance. There is evidence supporting its association with increased risk for ASCVD [Citation326]. Genetic testing is not available. A nomogram including Apo B levels, total cholesterol and TG concentration for diagnosis of familial combined hyperlipidaemia has been suggested but not routinely used as it needs to be more widely evaluated [Citation327].
12.2.3.3. Type 3 hyperlipidaemia or dysbetalipoproteinaemia
This condition is more common than that the above-mentioned lipoprotein disorders. The defect is in the Apo E, a ligand that facilitates removal of chylomicron remnant particles to the liver through a receptor-mediated mechanism [Citation316]. It is an autosomal recessive condition, often the phenotypic expression requiring the presence of a second hit mechanism such as obesity, hypothyroidism, diabetes and excessive alcohol consumption in addition to the genotype [Citation328]. A rare autosomal dominant form also exists [Citation329]. Raised chylomicron remnants’ level in this condition is associated with an increased risk for all forms of ASCVD, including MI, stroke, peripheral arterial disease as well as renal artery disease [Citation330]. Palmar xanthomas are pathognomonic of this condition. Soft tissue xanthomata may also be seen. Cholesterol and TG increases are more often in the equimolar range. The gene for Apo E exists in three isoforms – 2, 3 and 4. Each individual inherits two genes in any combination, one from each of the parents. Apo 3, 3 is considered as normal with all others being the mutant form with the 2, 2 isoform causing the dysbetalipoproteinaemia syndrome [Citation316].
12.2.3.3.1. Investigations
A full lipid profile is often the first line of investigation. Inability to report LDL-C, and sometimes interference to measurement of other analytes, which is well known with high TG is also seen with this type of hyperlipidaemia but not as common as in the type 1 hyperlipidaemia.
There are no simple tests for diagnosis of dysbetalipoproteinaemia. It can only be done by measurement of remnant particle number or characterisation of Apo E. Ultracentrifugation is essential, followed by electrophoresis to demonstrate beta-VLDL or chylomicron remnants that accumulate in this condition. A ratio of ApoB to total cholesterol below 0.15 has also been shown to differentiate this condition from other mixed hyperlipidaemias [Citation327, 331].
For remnant particle measurement, no routine assays are available but are under evaluation and available for research purposes [Citation332].
Apo E phenotyping using isoelectric focusing can be set up and/or genotyping can be made available in specialist laboratories.
12.2.3.4. Type 4 hyperlipidaemia
This disorder, often called familial hypertriglyceridaemia, is associated with mutations in the Apo A5 or lipase 1 gene and is of polygenic origin [Citation328]. The patients often present with low HDL-C, obesity or diabetes. Association with elevated ASCVD risk has been established [Citation333]. This has a pseudo-autosomal dominant inheritance. No genetic testing is available. With additional metabolic risk factors such as obesity and diabetes, these subjects can deteriorate to type 5, but the TG concentration rarely rises to levels that increase the risk for acute pancreatitis.
12.2.3.4.1. Investigations
Investigations are limited, except for a full lipid profile. The VLDL fraction is increased along with raised TG and a low HDL-C. The concentration of TG usually ranges from 300 to 900 mg/dL. Total cholesterol and non-HDL-Care are high due the increased VLDL component that also explains the association with increased risk for vascular disease [Citation333].
12.2.3.5. Type 5 hyperlipidaemia
This condition is characterised by raised VLDL fraction and chylomicrons. There is increased risk for acute pancreatitis due to high concentration of chylomicrons [Citation313].
12.3. Management
Patient with hypertriglyceridaemia should be referred to a specialist centre in the following situations:
Primary hypertriglyceridaemia
Treatment failure in primary care (a minority of patients with very high levels of TG may require multiple drugs)
Pregnancy: high TG during treatment prior to pregnancy and hypertriglyceridaemia identified during pregnancy
Pharmacological intervention leading to increase in:
TG levels, e.g. anti-retroviral therapy (ART), where important interactions between anti-retroviral drugs and lipid-lowering drugs are possible.
Raised CK, induced by drugs such as lipid-lowering drugs in any muscle disease, as these patients may require combination therapy with statin and fibrate which further increases the potential for muscle side effects.
Liver function test abnormalities – increase in transaminase activity (>3 times ULN).
12.3.1. Non-pharmacological therapy of hypertriglyceridaemia
The following are non-pharmacological treatment options for management of hypertriglyceridaemia [Citation189, 318]:
Weight loss for patients who are obese (diet and exercise)
Strict glycaemic control if diabetes is present
Treatment of hypothyroidism
Avoidance of excessive alcohol consumption
Avoidance of TG raising medications
Low fat diet – <30% of calories as fat, and in severe hypertriglyceridaemia reduce to <15%
Refrain from simple sugars and high carbohydrate diet
A diet moderate in total fats, low in saturated fats, low in simple sugars, with high fibre content and high omega-3 fatty acids is beneficial in hypertriglyceridaemia. A dietary pattern that emphasises intake of vegetables, fruits and whole grains, low-fat dairy products, poultry, fish, legumes, non-tropical vegetable oils and nuts with limited intake of sweets, sugar-sweetened beverages and red meat should be encouraged [Citation334, 335].
Including fresh vegetables, complex carbohydrates, fatty fish for non-vegetarians and fenugreek seeds, mustard seeds, flax seeds, soybean oil, mustard oil, etc. for vegetarians (as sources of omega-3 fatty acids) and nuts in the diet can be a step towards a good dietary pattern in these patients [Citation334, 335].
In patients with type 1 hyperchylomicronaemia syndrome, a very low fat diet < 15 g/day and caloric supplementation with oils rich in medium chain TG should be used, as they bypass intestinal absorption and directly enter portal circulation [Citation1].
The harmful effects of partially hydrogenated vegetable oil containing high proportion of trans-fats are well-known. There is an increased consumption of such oils by the Indian population. Although, there is a legal requirement for the manufacturers to reduce the amount of trans-fat content, it is not strictly followed [Citation336]. Many commonly used bakery products and Indian snacks that are commercially fried are rich in trans-fat [Citation335, 336]. Patients should be advised to specifically avoid these products. Use of saturated fat such as coconut oil and palm oil should also be discouraged [Citation337].
12.3.2. Pharmacotherapy
The basic principles of pharmacotherapy are described below [Citation338]:
In subjects with TG concentrations between 150 and 199 mg/dL, non-pharmacological therapy (as described above) may be sufficient. ASCVD risk assessment is essential and statin use indicated if ASCVD risk is high.
TG concentrations between 199 and 500 mg/dL with a high non-HDL-C concentration will require lifestyle changes along with pharmacotherapy. LDL-C reduction should be the primary aim. Statin is the drug of choice; appropriate doses of atorvastatin or rosuvastatin should be used. The effect of statins on TG levels depends on the LDL-C-lowering effect and initial TG level.
When TG concentrations is > 500 mg/dL, start with a fibric acid derivative such as fenofibrate and add a statin if non-HDL-C is high as defined in the LAI consensus Part 1 statement [Citation1].
Additional TG reduction can be obtained by adding an omega-3 acid ester concentrate.
Nicotinic acid use is debatable.
Controlling TG levels in patients with type 1 hyperlipidaemia can be challenging. New therapies are being investigated in type 1 hyperlipidaemia, including lomitapide, which is a microsomal TG transfer protein inhibitor, and also gene therapy [Citation338–Citation340].
Fibric acid derivatives [Citation338, 341] are the drug of choice in type 3 hyperlipidaemia. When combination therapy with a statin and a fibric acid derivative is prescribed, the risk for muscle-related side effects such as myalgia, myositis and rhabdomyolysis is increased. Thus, appropriate monitoring with CK and renal function tests should be performed after the initial 8 weeks, and if necessary, with every dose escalation [Citation338, 341, 342].
Treatment of severely high TG is more challenging than severe hypercholesterolaemia. Differentiating subjects who might develop ASCVD and/or acute pancreatitis is difficult. Very strict dietary practice may be essential in managing subjects with very high TG. Secondary factors can exacerbate a primary hyperlipidaemia. It is essential that physicians managing these patients are advised to obtain or seek specialist knowledge and skill essential for managing patients with high TG [Citation189, 318].
In patients of acute pancreatitis and severe hypertriglyceridaemia (TG > 1000 mg/dL), plasma apheresis is a treatment option [Citation343].
12.4. LAI recommendations
All subjects should be screened for hypertriglyceridaemia with a fasting lipid profile. A non-fasting lipid profile may be performed as an initial step, but fasting TG estimation will be needed if TG is found to be high in the non-fasting sample
Exclude and treat secondary causes
Aggressive therapeutic lifestyle changes should be implemented
Subjects with TG > 200 mg/dL and <500 mg/dL – treat with a statin
Subjects with TG ≥ 500 mg/dL-treat with fibrates to prevent acute pancreatitis and later add statin to achieve non-HDL-C goal
Preferred drugs: Fibrates and high dose omega fatty acids; the role of niacin is controversial.
13. Dyslipidaemia in heart transplantation
13.1. General considerations
Dyslipidaemia is common in patients after heart transplantation, with a prevalence of 60–80% [Citation344]. Dyslipidaemia can occur as a pre-existing condition or can be a side effect of immunosuppressive therapy, leading to an increased risk for CV morbidity and mortality. Typically, dyslipidaemia worsens in post-transplantation period as immunosuppressive therapy with cyclosporine, sirolimus and corticosteroids and to a lesser extent tacrolimus and everolimus adversely affects the lipid levels [Citation345–Citation348]. Immunosuppression also increases the incidence of hypertension, diabetes and can cause nephrotoxicity thus increasing the overall CV risk (). A significant association has been shown between increased total cholesterol and CV events after heart transplantation and dyslipidaemia represents one of the major modifiable risk factors [Citation349]. Interestingly, for the same level of dyslipidaemia, there is a significant increase in intimal thickness, greater angiogenesis and higher accumulation of T-lymphocytes within the allograft vasculature compared with native vessels. This suggests an additional role of immune mechanisms in the pathogenesis of allograft vasculopathy beyond that of high cholesterol [Citation350].
Table 6. Immunosuppressive potency and side effects of various immunosuppressive drugs [Citation345–348, 351].
Cardiac allograft vasculopathy (CAV) is an accelerated form of obliterative CAD that occurs in heart transplant recipients. It is one of the leading causes of mortality among long-term cardiac transplant recipients. CAV affects approximately 8, 30 and 50% of patients who survive 1, 5 and 10 years post-transplant [Citation352]. Histologically, CAV is characterised by concentric intimal thickening, smooth muscle cell proliferation, inflammatory cell infiltration, along with lipid deposition resulting in diffuse narrowing along entire length of the vessel [Citation353]. Although the pathogenesis of CAV involves both immunological and non-immunological mechanisms, dyslipidaemia is an important factor in the development of CAV [Citation354].
As CAV involves both epicardial and intramural arteries, procedures to reopen epicardial coronary arteries like angioplasty and bypass graft surgery have only limited efficacy. CAV is irreversible and is associated with poor prognosis, therefore the key is prevention. Even though various pre-transplant and post-transplant strategies can be used to prevent the development of CAV, dyslipidaemia management remains one of the most important and essential approaches in heart transplant recipients [Citation355, 356].
13.2. Role of immunosuppressive drugs
Calcineurin inhibitor-based therapy (cyclosporine and tacrolimus) remains the mainstay in immunosuppressive protocols used after heart transplantation because of its proven efficacy. Maintenance therapy should include a calcineurin inhibitor in all paediatric heart transplant recipients as per International Society for Heart and Lung Transplantation guidelines [Citation357]. Mycophenolate mofetil, everolimus or sirolimus as tolerated should be included in contemporary immunosuppressive regimens as these drugs reduce onset and progression of CAV [Citation357].
Cyclosporine inhibits the enzyme 26-hydroxylase and also inhibits binding of LDL to LDLR resulting in an increase in circulating LDL-C of up to 31% (). Lovastatin, simvastatin and atorvastatin are mainly metabolised by the CYP3A4 pathway. Fluvastatin metabolism predominantly occurs via the CYP2C9 enzyme with minimal need for CYP3A4 activity. Since cyclosporine inhibits CYP3A4 activity, markedly increased levels of statins metabolised by CYP3A4 may occur with concomitant administration of cyclosporine [Citation54, 358]. Thus, cyclosporine not only increases LDL-C levels but it also inhibits the metabolism of statins. The higher statin levels increase the risk of myopathy and rhabdomyolysis. Rhabdomyolysis has been reported with all statins except pravastatin and fluvastatin. Cyclosporine increases plasma concentrations of statins by manifold; atorvastatin: 6-fold, pravastatin: 5-fold, simvastatin: 3-fold and lovastatin: 20-fold [Citation54].
Table 7. Lipid abnormalities with immunosuppressive drugs [Citation345–Citation348].
Despite both cyclosporine and tacrolimus inhibiting CYP3A4, only the cyclosporine–statin interaction may be clinically relevant. Although data are limited on tacrolimus, its interaction with statin may not raise plasma levels of statins to the same degree as cyclosporine [Citation359, 360].
The mTOR (mammalian target of rapamycin) inhibitors sirolimus and everolimus are potent immunosuppressive agents, allowing the dose of nephrotoxic calcineurin inhibitors cyclosporine and tacrolimus to be decreased in solid organ transplant recipients. Dyslipidaemia is very common with mTOR inhibitors, with an estimated prevalence of up to 75% [Citation345]. The mTOR inhibitors reduce the catabolism of circulating lipoproteins by inhibiting the activity of lipases, resulting in dyslipidaemia. Of the mTOR inhibitors, everolimus has less deleterious effects on lipids than sirolimus [Citation346]. Everolimus and sirolimus with cyclosporine increase serum cholesterol by about 16% and 20–46%, respectively, and serum TG by 4% and 21–57%, respectively, in heart transplant recipients. The incidence of new onset diabetes (NOD) is 5–32% and 20–27% with everolimus and sirolimus, respectively, adding to the CV risk [Citation347]. Corticosteroids induce peripheral insulin resistance and an ensuing inability to activate lipid storage, which stimulates a reflex increase in total cholesterol [Citation348, 361]. The diabetogenic potential of these drugs needs to be considered in the presence of statins which can also cause NOD [Citation362].
13.3. Statins in heart transplant recipients
The beneficial effects of statins after heart transplantation correspond to cholesterol control, anti-inflammatory and immunomodulatory effects. Statins inhibit the production of proinflammatory cytokines and reduce the expression of several immunoregulatory molecules, including major histocompatibility complex class II (MHC-II) molecules. Statins effectively suppress the induction of MHC-II expression by interferon-gamma and inhibit MHC class II-dependent activation of T-lymphocytes [Citation363, 364].
Statins cause a significant reduction in incidence of severe acute rejection and CAV and increase long-term survival [Citation365, 366]. Unfortunately, statins, which rarely cause myopathy in non-transplant general population, may do so in transplant patients. In fact, in early experience, 40–80 mg lovastatin in heart transplant patients resulted in 4 out of 5 developing rhabdomyolysis [Citation367]. Low doses of statins are however relatively well tolerated [Citation367] but it is at the cost of decreased cholesterol-lowering efficacy.
In a landmark study, which paved the way for routine statin therapy in post-transplant patients, 97 heart transplants were randomly assigned to pravastatin 40 mg/day (47 patients) or no pravastatin (50 patients) in addition to their immunosuppressive treatment with cyclosporine, prednisolone and azathioprine [Citation365]. The pravastatin group had lower mean cholesterol levels than the control group (193 ± 36 vs. 248 ± 49 mg/dL, p < 0.001), decreased incidence of coronary vasculopathy (3 vs. 10 patients, p = 0.049) and improved survival (94 vs. 78%, p = 0.025) at 12 months follow-up. Pravastatin did not change the overall incidence of cardiac rejection, but it decreased the rate of rejection accompanied by haemodynamic compromise, resulting in better survival. During long-term follow-up, 81% of the control patients were eventually placed on statin therapy. In the intention-to-treat analysis, pravastatin group had increased 10-year survival (68 vs. 48%, p = 0.026). The 10-year freedom from angiographic CAV and/or death in pravastatin group was significantly greater compared with the control group (43 vs. 20%, p = 0.009) [Citation366].
Similar salutary benefits of statins have also been demonstrated with simvastatin [Citation368] and rosuvastatin [Citation369]. Seventy-two heart transplant recipients receiving standard triple immuno suppression (cyclosporin, azathioprine and prednisolone) were randomly assigned to simvastatin (n = 35) or a control group (n = 37). Over 4 years follow-up, the simvastatin group had significantly lower LDL-C than the control group (115 ± 14 vs. 156 ± 17 mg/dL, p = 0.002), a significantly better long-term survival (88.6 vs. 70.3%, p = 0.05) and a lower incidence of CAV (16.6 vs. 42.3%, p = 0.045). Intracoronary ultrasound performed after 4 years in a subgroup of 27 patients showed less intimal thickening in patients with LDL-C levels of < 110 mg/dL [Citation368].
The long-term safety of rosuvastatin 10 mg was evaluated in 30 heart transplant patients who failed to achieve cholesterol goals (LDL-C > 100 mg/dL) despite statin therapy. All patients received oral prednisone and a combination of ≥2 of the following three drug groups: (i) cyclosporine or tacrolimus, (ii) everolimus or sirolimus and (iii) mycophenolate formulation. At baseline, median total cholesterol was 236 mg/dL (range: 214–264) and median LDL-C was 142 mg/dL (range: 125–162). The average absolute reduction of total cholesterol was −48.7 mg/dL and LDL-C was −46.6 mg/dL, with 57% patients achieving a serum LDL-C < 100 mg/dL. Nearly 90% patients continued on long-term therapy with rosuvastatin over a median of 3.6 years. Rosuvastatin was discontinued in only two (6.7%) patients due to muscle toxicity and in one (3.3%) patient due to liver toxicity [Citation369].
Two head-to-head comparisons between pravastatin and equipotent dosages of simvastatin in heart transplant recipients did not find any statistically significant differences in rejection or survival outcomes [Citation370, 371]. Despite known drug interactions with calcineurin inhibitors, the use of statins is highly recommended in current guidelines [Citation357]. Hydrophilic statins (e.g. pravastatin, rosuvastatin) undergo limited metabolism by the cytochrome P3A4 isoenzyme system and are less likely to cause toxicity seen with lipophilic statins (e.g. simvastatin, atorvastatin, lovastatin). The recommended daily doses of statins are: pravastatin (20–40 mg), simvastatin (5–20 mg), atorvastatin (10–20 mg), lovastatin 20 mg androsuvastatin (5–20 mg) [Citation54, 357].
The 2013 ACC/AHA guidelines on cholesterol management recommend high intensity statin therapy in patients with established ASCVD, thus lowering LDL-C by approximately ≥50% [Citation107]. The role of such an approach in solid organ transplantation is unclear.
13.4. LAI recommendations
Baseline lipid levels should be obtained for all patients after heart transplantation.
The strict control of modifiable risk factors including hypertension, diabetes, dyslipidaemia, smoking and obesity should be reinforced.
In adults, the use of statins beginning 1 to 2 weeks after heart transplantation is recommended regardless of cholesterol levels [Citation357].
Pravastatin shows the least interaction with cyclosporine to produce myopathy, making it the drug of choice. Rosuvastatin up to 10 mg/day may be given as a second choice. Addition of a statin to a cyclosporine–sirolimus regimen produces multiple beneficial effects like reduced cholesterol levels, decreased acute rejection episodes, decreased incidence of CAV and improved survival.
In heart transplant recipients, a strategy of lowest achievable LDL-C levels with maximally tolerated dose of statin by slowly up-titrating the statin dose is justified.
Owing to pharmacologic interactions with cyclosporine and risk for toxicity, initial statin doses should be lower. In paediatric patients with evidence of hyperlipidaemia, CAV or after re-transplantation and increased risk of rejection use of statins is recommended [Citation357]. All patients should be actively monitored for development of myopathy with regular monitoring of CK.
In the absence of RCTs and very limited data, use of non-statin lipid-lowering drugs is not recommended for routine use [Citation372]. Their use may be warranted in certain scenarios like severe hypertriglyceridaemia.
Although statins are the drug of choice, ezetimibe can be used as a second-line drug, predominantly to decrease LDL-C levels, but needs monitoring of calcineurin inhibitors. Concomitant use of cyclosporine with ezetimibe may increase the levels of both the drugs [Citation373].
Fibrates may be used, after discussing risks and benefit with each patient, to prevent acute pancreatitis in situations with predominant hypertriglyceridaemia, as can occur with mTOR inhibitors and especially if TG levels are >500 mg/dL [Citation351]. As fenofibrate is predominantly metabolised by the kidneys, dose adjustment is necessary in patients with moderate renal dysfunction [Citation374]. Further, fenofibrate-induced nephrotoxicity is more common in renal transplant recipients, signifying the need for serum creatinine monitoring [Citation374]. Combination of fibrates with statins should be avoided for fear of myotoxicity. The role of niacin is controversial.
Cholestyramine is contraindicated due to interference with absorption of calcineurin inhibitors.
14. Dyslipidaemia in other solid organ transplantation
Dyslipidaemia is common after solid organ transplantation with a prevalence of 30–70%. CVD is the most important cause of death in solid organ transplant recipients who have a functioning graft [Citation375]. The incidence of CVD in renal transplant recipients is 3–5 times higher and in chronic haemodialysis patients 10–20 times higher than that of general population [Citation376]. Although several factors (e.g. hypertension and left ventricular hypertrophy) contribute to CVD morbidity and mortality, dyslipidaemia plays an important role. The factors contributing to dyslipidaemia include abnormal renal function and impaired glucose metabolism, in addition to those mentioned above for heart transplantation [Citation377].
In the ALERT study, 2102 cyclosporine-treated stable renal transplant recipients were randomised to fluvastatin 40–80 mg daily or placebo. Although on initial analysis the primary composite cardiac endpoint was not significantly reduced, the extended follow-up for up to8 years was associated with a 35% reduction in the composite endpoint of cardiac death or non-fatal MI [Citation38, 49]. Total mortality and graft loss did not differ between the two groups. Importantly, early introduction of statin therapy was associated with increased survival benefit [Citation38, 49]. Interestingly, the CV events in renal transplant recipients may be not only because of conventional CVD risk factors but also due to a high prevalence of renal dysfunction, hypertension and left ventricular hypertrophy. The management of dyslipidaemia in solid transplant recipients has three major components – lifestyle modification, lipid-lowering drugs and fine tuning the immunosuppressive regimen. The KDIGO 2013 lipid management guidelines [Citation378] recommend statins in all adult renal transplant recipients above the age 30 years.
The 2012 Practice Guideline of the American Association for the Study of Liver Diseases and the American Society of Transplantation recommends an annual fasting lipid profile for healthy liver transplant recipients and recommend cut-off of LDL-C levels > 100 mg/dL irrespective of presence of hypertriglyceridaemia as the target for initiation of statins [Citation379].
14.1. LAI recommendations
In the absence of available robust data for heart transplantation, measurement of fasting serum total cholesterol, LDL-C and TG is recommended after a solid organ transplant and at 6 months post-transplant, thereafter annually or if there is a change in immunosuppressive medication.
Statin therapy should be initiated in all adult renal and in liver transplant recipients if LDL-C levels are >100 mg/dL. For second-line drug use and for management of hypertriglyceridaemia, the same recommendations as given for heart transplantation should be followed.
15. Dyslipidaemia in human immunodeficiency virus infection
15.1. Introduction
There is an increased mortality and morbidity associated with human immunodeficiency virus (HIV)-related conditions [Citation380] and ART has changed the course of HIV to a chronic disease. With this change, the treatment-induced risk for comorbidities has increased, in particular CVD. Patients with HIV infection have been shown to have an increased risk for CVD compared with the general population [Citation381, 382]. Furthermore, dyslipidaemia in HIV infection is well recognised and its treatment is associated with abnormalities in lipid metabolism. The hall mark findings are high TGs and low HDL-C [Citation383]. Increased TNF and IL-6 are the predominant cytokines involved in pathogenesis of dyslipidaemia; HIV in addition has an independent effect on cardiovascular risk [Citation384]. ART may also increase the incidence of metabolic risk factors like insulin resistance, lipoatrophy, dyslipidaemia and abnormalities of fat distribution in HIV patients [Citation385]. It is important to recognise that as patients live longer, they became vulnerable to age-related complications, prominently CVD.
Although there are many different lipoprote in particles, the cornerstone of lipid therapy has traditionally focused on LDL-C. Years of research have demonstrated that LDL-C is the primary atherogenic particle which is directly associated with the development of CVD and therefore the target of lipid-lowering therapy [Citation304, 385]. There are currently no long-term epidemiologic studies or randomised clinical trials demonstrating the efficacy of lipid medication or benefits of lowering LDL-C on cardiovascular event outcomes in patients with HIV.
Dyslipidaemia is being increasingly recognised in patients on Protease Inhibitor (PI) based regimens [Citation386]. The majority (70–80%) have high TGs and LDL-C and decreased HDL-C and accumulation of ApoE and ApoC III. There is also central obesity, lipoatrophy and insulin resistance [Citation387]. This is due to stimulation of hepatic synthesis of TGs and inhibition of lipogenesis, adipocyte differentiation and decreased hepatocyte clearance of chylomicrons and VLDL [Citation388, 389]. Nucleoside Reverse Transcriptase Inhibitors (NRTIs) also cause high TGs and lipoatrophy although less seen with tenofovir [Citation390]. Abacavir and didanosine (NRTIs) were found to be independent risk factors for MI in the data collection on adverse events of Anti-HIV Drugs (DAD) study. Nevirapine has been shown to increase the HDL-C concentration and efavirnez produces increase in total cholesterol and TGs [Citation391] Dyslipidaemia, insulin resistance and diabetes has been shown with use of ART medication in HIV patients [Citation392].
Unfortunately, there are scant data on the effect of HIV infection on CHD prevalence and outcomes in resource-poor countries like India. Because of the connection of lipid abnormalities with CVD, the understanding of the pathophysiology of the disorders in lipid metabolism and the management of lipid disorders in HIV infected individuals is important.
15.2. Epidemiology
Dyslipidaemia is highly prevalent among patients with HIV infection with and without antiretroviral therapy and can contribute to the increased CVD risk [Citation393, 394]. CVD is an increasing health problem affecting HIV-infected patients [Citation395, 396]. Antiretroviral therapy has remarkably improved the survival of HIV-infected patients [Citation397, 398]; however, the mortality rates in HIV patients are still higher than in the general population also due to non-HIV-related causes including CVD [Citation399, 400]. Patients with HIV have increased traditional cardiovascular risk factors including dyslipidaemia, smoking and glucose abnormalities [Citation396]. Studies have shown that children receiving antiretroviral therapy had higher total cholesterol and TGs, and increased cIMT; thus, cardiovascular risk may be higher among HIV patients even at a young age [Citation393, 401, 402]. In a multicenter cross-sectional study of elderly individuals with HIV, dyslipidaemia was found in 54%, CVD in 23% and lipodystrophy in 58% [Citation403]. Hejazi et al. found that dyslipidaemia is common in HIV subjects receiving antiretroviral medication: it reached 82.3% among 1583 patients in their Malaysian study [Citation404].
15.3. Dyslipidaemia in HIV a challenge to manage
Cardiovascular morbidities are observed in HIV/AIDS especially in older and obese patients. Chu et al. found prevalence rates of 26, 48 and 13% for hypertension, dyslipidaemia and diabetes, respectively. Hence, the treatment of the comorbidities may improve outcome due to the modifiable nature of cardiovascular risk [Citation405]. Increased prevalence of dyslipidaemia is also seen in infected HIV children [Citation396] and they are at increased risk of premature CAD.
Before the availability of highly active antiretroviral therapy, Grunfeldet al. found that patients with AIDS had elevated plasma TG and free fatty acid levels, whereas HIV-infected patients without AIDS had decreased total cholesterol and HDL-C.
In another multicentre study, total cholesterol, HDL-C and LDL-C levels declined following HIV seroconversion [Citation406]. In these patients, total cholesterol and LDL-C levels rose but HDL-C remained decreased after initiation of highly active antiretroviral therapy.
In the Swiss HIV Cohort Study, HIV protease inhibitor (PI) use was found to be associated with an increase in plasma total cholesterol and TGs [Citation407]. Discontinuation of antiretroviral therapy in the The Strategies for Management of Antiretroviral Therapy (SMART) Study resulted in a decline in total cholesterol and LDL-C, but HDL-C declined as well, leading to an unfavourable increase in the total cholesterol/HDL-C ratio [Citation408]. Recently, Duprezetal [Citation409] showed that cardiovascular risk in HIV patients was associated with lower levels of small and large HDL-particle concentration independently of other cardiovascular risk factors. Patients with HIV-associated fat redistribution are even more likely to have dyslipidaemia, possibly related to excess visceral adipose tissue accumulation [Citation397].
15.4. Pathogenesis of HIV-associated CAD
This is primarily due to interaction of inflammation, endothelial dysfunction and coagulation disorders leading to atherosclerosis. The typical plaque is non-calcified in HIV-infected individuals. However, they may have calcium oxalate crystal while on ART therapy [Citation406]. cIMT and coronary CT angiography studies show increased prevalence of subclinical atherosclerosis in HIV-positive patients [Citation410–Citation412]. More vulnerable plaques in patients with low CD4 counts have also been observed [Citation382]. C-reactive protein, IL-6, TNF levels are high in HIV-positive patients [Citation413, 414]. Patients with complete virological suppression may still continue to have inflammation and increased immune activation. This predisposes risk of CAD [Citation415]. Similarly endothelial function improvement after suppression of HIV viraemia proves that it is one of the significant factors for atherosclerosis [Citation416]. Coagulopathy reflected as increased D-dimer, fibrinogen, factor VII, etc. was shown to be associated with risk for CVD deaths in the SMART study [Citation410]. Tissue factor activation is also seen. Platelet activation is another observation made in HIV patients [Citation417].
15.5. Evaluation and treatment of dyslipidaemia in HIV patients
Guidelines and recommendations for the evaluation and management of dyslipidaemia in HIV-infected adults put forth by the Infectious Disease Society of America (IDSA) and the Adult AIDS Clinical Trials Group (ACTG) in 2003 [Citation418] support the use of the updated recommendations from the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults [Adult Treatment Panel III (ATP III)] [Citation419]. Of note, since the IDSA/ACTG Guidelines were written, a more recent update was published for the NCEP ATP III Guidelines in 2004 [Citation420] that should be taken into consideration when relevant. As recommended in the NCEP ATP III and IDSA/ACTG Guidelines [Citation418], lifestyle counselling on diet and exercise should generally be instituted first. Other modifiable cardiovascular risk factors such as smoking, hypertension and diabetes mellitus should also be addressed. When medications become necessary, then identification of the lipid abnormality needs to be made and the choice of therapy should target the specific abnormality. Silverberg et al. [Citation421] found that lipid-lowering therapies are generally well tolerated in HIV-infected patients but hypertriglyceridaemia can be more difficult to treat in HIV-infected patients compared with non-HIV-infected patients.
15.6. Risk scores
Traditional scores either underestimate or overestimate cardiovascular risk in HIV patients irrespective of being on ART or not. Hence, there is a need to identify specific cardiovascular risk equations [Citation422]. ART use in the past, including duration of use of protease inhibitors and non-nucleoside reverse transcriptase inhibitors, was not associated with myocardial infraction [Citation423]. A risk calculator including both traditional risk and exposure to individual ART therapy has been developed [Citation424]; the Veterans Aging Cohort study (VACS) is one such score. A model using traditional risks and HIV specific risks like CD4 + Count, abacavir use exposure to Protease Inhibitors (PIs) and NRTI has better prediction. But validation is not yet confirmed [Citation425]. Non-invasive risk stratification using coronary computed tomography angiography (CCTA) and cIMT can help in identifying HIV patients at risk [Citation392, 426, 427]. The ASCVD risk stratification algorithm in Indians () given by LAI can be used in HIV patients to take into account conventional risk factors in addition to ART.
Figure 4. ASCVD risk stratification in Indians.
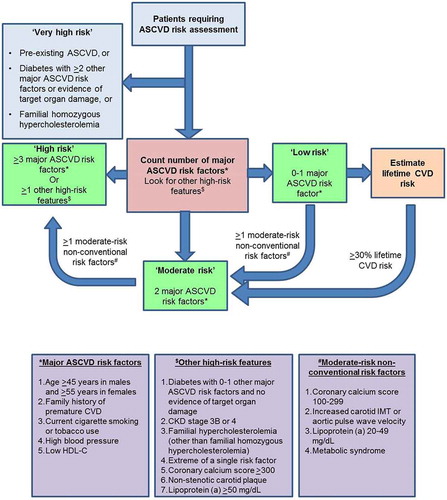
15.7. Lipid targets for patients living with HIV
The NCEP ATP III recommends achieving LDL-C targets as a “first priority” unless TG are >500 mg/dL at baseline when the first priority is to lower TG levels to prevent acute pancreatitis. The European AIDS Society has set up a target of <155 mg/dl for total cholesterol and <80 mg/dLfor LDL-C [Citation383]. However, the IDSA and others emphasise the need for more stringent targets as in non-HIV patients [Citation384]. However, HIV status is not considered in the NCEPATPIII guidelines [Citation428] lists the lipid targets recommended by LAI to be used in HIV patients.
Table 8. Lipids targets in HIV patients as recommended by LAI.
15.8. Management of hyperlipidaemia in HIV patients
Most of the patients of HIV with dyslipidaemia require treatment. Lifestyle modifications like weight reduction with caloric restriction should be an important component for management. HMG-Co-A reductase inhibitors, known as statins, are the most widely used and researched medications for the treatment of dyslipidaemia. They are primarily used to lower LDL-C and have a modest effect on raising HDL-C and lowering TGs. In addition, they also lead to improvement in endothelial function, decreased platelet aggregation and reduced inflammation [Citation429]. As statins differ in their pharmacokinetic properties, drug interaction profiles, risk of myopathy, many issues regarding the use in patients infected with HIV are still unclear including metabolism and efficacy, not only for LDL-C lowering but also regarding their effect on CVD events and mortality ().
Table 9. Effect of different statins in HIV patients with dyslidaemia [Citation431].
In a recently published meta-analysis [Citation430], the authors concluded that in HIV-positive patients on combination antiretroviral therapy (cART), statins prescribed for primary prevention showed a good safety profile and were effective for improving dyslipidaemia. Rosuvastatin 10 mg showed greater reductions of TC, LDL-C and TGs and significantly increased HDL-C; pravastatin 40 mg mildly decreased TC, LDL-C and triglycerides, but was unable to significantly increase HDL-C. Atorvastatin showed controversial safety results and its prescription should be carefully monitored. Simvastatin, despite proving to be safe and effective, should not be prescribed concomitantly with PIs due to pharmacokinetic interactions. Fluvastatin appeared to be safe and efficacious, though the results were based on a single study. Combination antiretroviral therapy does not appear to affect the safety of dose-adjusted statins.
15.9. Other lipid-lowering medications
HIV and ART therapy both contribute to dyslipidaemia. The interaction between HIV treatment and statins and HIV-induced conditions like NAFLD, hypothyroidism, early menopause, diabetes and CKD poses a unique challenge. An ageing population further contributes to the challenge. Statin-unresponsive dyslipidaemia can be treated with ezetimibe; mixed hyperlipidaemia predominantly with TG increase can be treated with fenofibrate, niacin and fish oil.
Ezetimibe reduces cholesterol absorption at the intestinal brush border. Unlike statins, ezetimibe does not interact with cytochrome CYP3A and does not cause drug–drug interactions with ART. Ezetimibe can be used safely in patients with HIV-related dyslipidaemia [Citation432, 433].
Niacin has a variety of effects on lipid metabolism because it inhibits the hepatic production of VLDL and consequently LDL. It can also raise HDL-C by reducing lipid transfer from HDL to VLDL [Citation434] as well as lower TG by 15–20% when used in higher doses (2000 mg/day) [Citation435]. Use of niacin is limited by side effects, mainly flushing but also itching and headache. Because niacin is not a potent LDL-lowering medication and its use for lowering TG requires high doses, it is not an ideal medication for patients with HIV. This drug has been withdrawn from use in some countries.
Fenofibrate lacks significant interactions with ART and is the most commonly prescribed fibrate for HIV-infected patients with elevated TG. In a study of 635 HIV-infected patients on PI-based ART with TG > 300 mg/dL, patients were treated with bezafibrate, gemfibrozil or fenofibrate. All fibrates showed a similar and significant efficacy [Citation436]. Fenofibrate is eliminated by the kidneys and should be dose-adjusted in patients with reduced renal function or on other nephrotoxic drugs [Citation374].
Fish oils that are long-chain omega-3 polyunsaturated fatty acids (PUFAs) which are present in cold water fish can lower TG [Citation437] and reduce CVD events in the general population [Citation438].
15.10. Conclusions
Prevention of CVD in HIV patients is an increasing priority. The challenge of more CAD in HIV patients as longevity increases on account of ART is considerable. Our understanding of the complex interaction of traditional risk factors and HIV-specific factors has improved. Diagnosis and treatment of dyslipidaemia remains an integral part of this prevention effort. Current guidelines including the NCEP ATP III [Citation428] as well as the IDSA/ACTG Guidelines [Citation439] for the evaluation and management of lipid disorders in HIV patients can be used to help guide therapy. Lifestyle counselling on diet and exercise should be emphasised in every patient with dyslipidaemia. Special consideration to potential drug–drug interactions needs to be taken when lipid-lowering therapy is initiated in patients with HIV.
15.11. LAI recommendations
Lifestyle modifications with special emphasis on smoking cessation, weight reduction and calorie restriction are important.
The recommendations for the evaluation and treatment of dyslipidaemia as suggested by LAI regarding target goals for lipids should be strictly followed to help guide therapy. At least, evidence available in the Indian population suggests the same.
Different types of statins are available to lower plasma lipids to guideline levels in patients with HIV, but they differ in their pharmacokinetic properties and drug interaction profiles. Simvastatin and lovastatin are contraindicated in patients taking PIs. The other statins atorvastatin and rosuvastatin (apart from simvastatin and lovastatin) have better therapeutic effect in lowering LDL-C in HIV dyslipidaemia. The addition of ezetimibe is another option [Citation432, 433]. Fenofibrate [Citation440] and fish oil [Citation441, 442] can be used in statin-unresponsive HIV dyslipidaemia.
16. Concluding statement
The ASCVD risk stratification should be completed as per the LAI recommendation part 1 and the treatment goals and statin intervention thresholds should consider the additional risk for all special populations. In inherited hyperlipidaemia, the recommended levels of <50 mg/dl should be reached if ASCVD risk is very high and <70 mg/dl if risk is high. Other targets and threshold for familial forms have been discussed in the relevant sections. This comprehensive recommendation developed by the expert committee has tried to base all recommendations on the available data from Indian population, while also critically reviewing and incorporating the available evidence worldwide. In this context, if Indian data were lacking, inferences have been drawn from appropriate international studies and guidelines to formulate these recommendations.
Conflicts of interest of the writing committee
S. S. Iyengar, research funding, including grants from: Sanofi. Consultancy or speaker fees over the past 12 months: Boehringer Ingelheim, CIPLA and MANKIND Pharma.
R. Puri, Consultancy or speaker fees over the past 12 months: Boehringer Ingelheim, Zydus, Novartis and Bayer.
S. N. Narasingan, companies/organisations providing travel expenses, registration fees or accommodation at professional meetings: Sun Pharmaceutical industries and Dr. Reddy’s Laboratories Ltd. Consultancy or speaker fees over the past 12 months: Sun Pharmaceutical Industries, Glaxo SmithKline Pharmaceuticals Ltd., Cadila Healthcare Ltd., Dr. Reddy’s Laboratories Ltd., Unichem Laboratories Ltd., Boehringer Ingelheim, Glenmark and Abbott Healthcare Ltd.
D. R. Nair has received grants from Pfizer (Pfizer Foundation award 2008), Solvay, Merck Sharp & Dohme and Astra Zeneca for this service. DRN has advisory board membership for Merck Sharp & Dohme, Sanofi and Amgen. DRN is a speaker for Merck Sharp & Dohme, Sanofi and Amgen.
V. Mehta, no conflicts of interest.
J. C. Mohan, companies/organisations providing travel expenses, registration fees or accommodation at professional meetings: “APEX” society for CME. Consultancy or speaker fees over the past 12 months: Boehringer Ingelheim, Janssen Pharma, AstraZeneca, MSD, CIPLA, USV, LUPIN, INTAS Pharma, Glenmark and ENCURE.
S. K. Wangnoo, no conflicts of interest.
J. J. Dalal, Consultancy or speaker fees over the past 12 months: AstraZeneca, Sun Pharmaceutical Industries, Boehringer Ingelheim, Sedia, ERIS, CIPLA, MANKIND, Novartis, Glenmark, JB Chemicals, India Bulls, Pfizer and Bayer.
V. Jha, research funding, including grants from: Glaxo SmithKline Pharmaceuticals Ltd. and Baxter Healthcare. Companies/organisations providing travel expenses, registration fees or accommodation at professional meetings: Baxter Healthcare. Consultancy or speaker fees over the past 12 months: NephroPlus
S. Puri, no conflicts of interest.
A. Misra, research funding, including grants from: MEBALIFE, Almond Board of California and Abbott India. Companies/organisations providing travel expenses, registration fees or accommodation at professional meetings: Janssen Pharma, Boehringer Ingelheim, Dr. Reddy’s Laboratories Ltd., Novartis, AstraZeneca, US Vitamin LUPIN, INTAS Pharma. Consultancy or speaker fees over the past 12 months: as above.
M. K. Daga, Consultancy or speaker fees over the past 12 months: ASAHI KASEI Pharma, USA.
M. Varma, no conflicts of interest.
S. Jasuja, Consultancy or speaker fees over the past 12 months: Dr. Reddy’s Laboratories Ltd and Novartis.
S. Upadhyaya, no conflicts of interest.
R. R. Kasliwal, no conflicts of interest.
M. Bansal, no conflicts of interest.
R. Mehrotra, Consultancy or speaker fees over the past 12 months: INTAS Pharmaceuticals Ltd., Cadila Healthcare Ltd., USV Pvt. Ltd. and Abbott Healthcare Ltd.
A. Jain, no conflicts of interest.
K. K. Talwar, no conflicts of interest.
R. Rajput, no conflicts of interest.
A. Pradhan, no conflicts of interest.
S. Seth, no conflicts of interest.
D. Kapoor, Consultancy or speaker fees over the past 12 months: Novo Nordisk, Eli Lilly, AstraZeneca, Novartis and MSD.
R. P. Melinkeri, no conflicts of interest.
S. Ramakrishnan, no conflicts of interest.
N. N. Khanna, Consultancy or speaker fees over the past 12 months: Merril.
R. Khadgawat, no conflicts of interest.
S. Puri, no conflicts of interest.
A. Shaikh, Consultancy or speaker fees over the past 12 months: Abbott Healthcare Ltd, AstraZeneca, Novartis, MSD and Sanofi.
N. Kovalipati, Consultancy or speaker fees over the past 12 months: Sanofi-Aventis, AstraZeneca, Novartis, MSD and Sun Pharmaceutical Industries.
N. Bordoloi, Companies/organisations providing travel expenses, registration fees or accommodation at professional meetings: Public Health Foundation of India, Lipid Association of India, Sun Pharmaceutical Industries, CSI-WB Chapter and West Bengal Academy of Echocardiography. Consultancy or speaker fees over the past 12 months: USB Pvt. Ltd., Boehringer Ingelheim and Abbott Healthcare Ltd.
A. H. Zargar, Consultancy or speaker fees over the past 12 months: Biocon, Dr. Reddy’s Laboratories Ltd., Boehringer Ingelheim, AstraZeneca and Johnson and Johnson.
R. K. Agarwal, Consultancy or speaker fees over the past 12 months: Cadila Healthcare Ltd. ENCURE Pharmaceuticals and Abbott Healthcare Ltd.
A. Rastogi, no conflicts of interest.
M. Chag, no conflicts of interest.
D. Prabhakar, Consultancy or speaker fees over the past 12 months: Boehringer Ingelheim.
S. K. Mathur, no conflicts of interest.
H. Rehan, no conflicts of interest.
P. K. Sahoo, Consultancy or speaker fees over the past 12 months: SUN Pharma, Boston Scientific, MANKIND Pharma and Torrent Pharma.
A. Dutta, no conflicts of interest.
A. Sharma, Consultancy or speaker fees over the past 12 months: ABBOTT, Novartis and LUPIN.
A. K. Pancholia, no conflicts of interest.
K. U. Natarajan, Consultancy or speaker fees over the past 12 months: Medtronic, Boston Scientific, St. Jude Medical and Abbott.
A. Mishra, no conflicts of interest.
K. Singh, no conflicts of interest.
References
- Iyengar SS, Puri R, Narasingan SN, et al. Lipid association of India expert consensus statement on management of dyslipidemia in Indians 2016: part 1. J Assoc Physicians India. 2016;64:S7–S52.
- Puri R, Narasingan S, Iyengar S. Lipid association of India expert consensus statement on management of dyslipidemia in Indians 2016: part 1 – executive summary. J Clin Preventive Cardiol. 2016;5:51–61.10.4103/2250-3528.186492
- Rauchhaus M, Clark AL, Doehner W, et al. The relationship between cholesterol and survival in patients with chronic heart failure. J Am Coll Cardiol. 2003;42:1933–1940.10.1016/j.jacc.2003.07.016
- Horwich TB, Hamilton MA, MacLellan WR, et al. Low serum total cholesterol is associated with marked increase in mortality in advanced heart failure. J Card Fail. 2002;8:216–224.10.1054/jcaf.2002.0804216
- Yoon CH, Youn TJ, Ahn S, et al. Low serum total cholesterol level is a surrogate marker, but not a risk factor, for poor outcome in patients hospitalized with acute heart failure: a report from the Korean Heart Failure Registry. J Card Fail. 2012;18:194–201.10.1016/j.cardfail.2011.12.006
- Anker SD, Ponikowski P, Varney S, et al. Wasting as independent risk factor for mortality in chronic heart failure. Lancet 1997;349:1050–1053.10.1016/S0140-6736(96)07015-8
- Jankowska EA, Biel B, Majda J, et al. Anabolic deficiency in men with chronic heart failure: prevalence and detrimental impact on survival. Circulation 2006;114:1829–1837.10.1161/CIRCULATIONAHA.106.649426
- Group HPSC. MRC/BHF heart protection study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002;360:7–22.
- Levy WC. Observational studies of statins in systolic heart failure. Heart Fail Clin. 2008;4:201–208.10.1016/j.hfc.2008.01.006
- Kjekshus J, Apetrei E, Barrios V, et al. Rosuvastatin in older patients with systolic heart failure. N Engl J Med. 2007;357:2248–2261.10.1056/NEJMoa0706201
- Tavazzi L, Maggioni AP, Marchioli R, et al. Effect of rosuvastatin in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet 2008;372:1231–1239.
- Feinstein MJ, Jhund P, Kang J, et al. Do statins reduce the risk of myocardial infarction in patients with heart failure? A pooled individual-level reanalysis of CORONA and GISSI-HF. Eur J Heart Fail. 2015;17:434–441.10.1002/ejhf.2015.17.issue-4
- Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure [practice guideline]. J Am Coll Cardiol. 2013;62:e147–e239.10.1016/j.jacc.2013.05.019
- Writing C, Lloyd-Jones DM, Morris PB, et al. 2016 ACC expert consensus decision pathway on the role of non-statin therapies for LDL-cholesterol lowering in the management of atherosclerotic cardiovascular disease risk: a report of the American College of Cardiology Task Force on clinical expert consensus documents. J Am Coll Cardiol. 2016;68:92–125.
- Takagi H, Umemoto T. Atorvastatin, not rosuvastatin, improves cardiac function in heart failure: A meta-analysis of randomized trials. Int J Cardiol. 2012;155:296–299.10.1016/j.ijcard.2011.11.079
- Banach M, Serban C, Ursoniu S, et al. Statin therapy and plasma coenzyme Q10 concentrations – a systematic review and meta-analysis of placebo-controlled trials. Pharmacol Res. 2015;99:329–336.10.1016/j.phrs.2015.07.008
- McMurray JJ, Dunselman P, Wedel H, et al. Coenzyme Q10, rosuvastatin, and clinical outcomes in heart failure. J Am Coll Cardiol. 2010;56:1196–1204.10.1016/j.jacc.2010.02.075
- Marcoff L, Thompson PD. The role of coenzyme Q10 in statin-associated myopathy. J Am Coll Cardiol. 2007;49:2231–2237.10.1016/j.jacc.2007.02.049
- Banach M, Serban C, Sahebkar A, et al. Effects of coenzyme Q10 on statin-induced myopathy. Mayo Clin Proc. 2015;90:24–34.10.1016/j.mayocp.2014.08.021
- Ridker PM. LDL cholesterol: controversies and future therapeutic directions. Lancet 2014;384:607–617.10.1016/S0140-6736(14)61009-6
- Prospective Studies C, Lewington S, Whitlock G, et al. Blood cholesterol and vascular mortality by age, sex, and blood pressure: a meta-analysis of individual data from 61 prospective studies with 55,000 vascular deaths. Lancet 2007;370:1829–1839.
- Cleland JG, McMurray JJ, Kjekshus J, et al. Plasma concentration of amino-terminal pro-brain natriuretic peptide in chronic heart failure: prediction of cardiovascular events and interaction with the effects of rosuvastatin. J Am Coll Cardiol. 2009;54:1850–1859.10.1016/j.jacc.2009.06.041
- Sharma UC, Pokharel S, van Brakel TJ, et al. Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation 2004;110:3121–3128.10.1161/01.CIR.0000147181.65298.4D
- Ueland T, Aukrust P, Broch K, et al. Galectin-3 in heart failure: high levels are associated with all-cause mortality. Int J Cardiol. 2011;150:361–364.10.1016/j.ijcard.2011.05.081
- Gullestad L, Ueland T, Kjekshus J, et al. Galectin-3 predicts response to statin therapy in the Controlled Rosuvastatin Multinational Trial in Heart Failure (CORONA). Eur Heart J. 2012;33:2290–2296.10.1093/eurheartj/ehs077
- Al-Gobari M, Le HH, Fall M, et al. No benefits of statins for sudden cardiac death prevention in patients with heart failure and reduced ejection fraction: a meta-analysis of randomized controlled trials. PLOS ONE. 2017;12:e0171168.10.1371/journal.pone.0171168
- National Kidney F. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis. 2002;39:S1–266.
- Go AS, Chertow GM, Fan D, et al. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004;351:1296–1305.10.1056/NEJMoa041031
- Sarnak MJ, Levey AS, Schoolwerth AC, et al. Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation 2003;108:2154–2169.10.1161/01.CIR.0000095676.90936.80
- Sakhuja V, Sud K. End-stage renal disease in India and Pakistan: burden of disease and management issues. Kidney Int Suppl. 2003;S115–S118.
- Vaziri ND. Causes of dysregulation of lipid metabolism in chronic renal failure. Semin Dial. 2009;22:644–651.10.1111/sdi.2009.22.issue-6
- Kilpatrick RD, McAllister CJ, Kovesdy CP, et al. Association between serum lipids and survival in hemodialysis patients and impact of race. J Am Soc Nephrol. 2007;18:293–303.10.1681/ASN.2006070795
- Chu M, Wang AY, Chan IH, et al. Serum small-dense LDL abnormalities in chronic renal disease patients. Br J Biomed Sci. 2012;69:99–102.
- Mikhailidis DP, Elisaf M, Rizzo M, et al. “European panel on low density lipoprotein (LDL) subclasses”: a statement on the pathophysiology, atherogenicity and clinical significance of LDL subclasses. Curr Vasc Pharmacol. 2011;9:533–571.10.2174/157016111796642661
- Wanner C, Krane V, März W, et al. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N Engl J Med. 2005;353:238–248.10.1056/NEJMoa043545
- Fellström BC, Jardine AG, Schmieder RE, et al. Rosuvastatin and cardiovascular events in patients undergoing hemodialysis. N Engl J Med. 2009;360:1395–1407.10.1056/NEJMoa0810177
- Baigent C, Landray MJ, Reith C, et al. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (study of heart and renal protection): a randomised placebo-controlled trial. Lancet 2011;377:2181–2192.10.1016/S0140-6736(11)60739-3
- Holdaas H, Fellström B, Jardine AG, et al. Effect of fluvastatin on cardiac outcomes in renal transplant recipients: a multicentre, randomised, placebo-controlled trial. Lancet 2003;361:2024–2031.10.1016/S0140-6736(03)13638-0
- Wanner C, Tonelli M. KDIGO clinical practice guideline for lipid management in CKD: summary of recommendation statements and clinical approach to the patient. Kidney Int. 2014;85:1303–1309.10.1038/ki.2014.31
- Joint British Societies’ consensus recommendations for the prevention of cardiovascular disease (JBS3). Heart 2014;100(Suppl 2):ii1–ii67.
- Anderson TJ, Grégoire J, Pearson GJ, et al. 2016 Canadian Cardiovascular Society guidelines for the management of dyslipidemia for the prevention of cardiovascular disease in the adult. Can J Cardiol. 2016;32:1263–1282.10.1016/j.cjca.2016.07.510
- Enas EA, Yusuf S, Mehta JL. Prevalence of coronary artery disease in Asian Indians. Am J Cardiol. 1992;70:945–949.10.1016/0002-9149(92)90744-J
- Joshi P, Islam S, Pais P, et al. Risk factors for early myocardial infarction in south asians compared with individuals in other countries. JAMA 2007;297:286–294.10.1001/jama.297.3.286
- Massy ZA, de Zeeuw D. LDL cholesterol in CKD–to treat or not to treat? Kidney Int. 2013;84:451–456.10.1038/ki.2013.181
- Elisaf M, Filippas-Ntekouan S. Pathophysiological mechanisms of dyslipidemia in patients with nephrotic syndrome: a fresh look. Hellenic J Atherosclerosis. 2016;7.
- Su X, Zhang L, Lv J, et al. Effect of statins on kidney disease outcomes: a systematic review and meta-analysis. Am J Kidney Dis. 2016;67:881–892.10.1053/j.ajkd.2016.01.016
- Vaziri ND, Anzalone DA, Catini J. Statins in chronic kidney disease: when and when not to use them. J Fam Pract. 2016;65.
- Epstein M, Vaziri ND. Statins in the management of dyslipidemia associated with chronic kidney disease. Nat Rev Nephrol. 2012;8:214–223.10.1038/nrneph.2012.33
- Holdaas H, Fellström B, Cole E, et al. Long-term cardiac outcomes in renal transplant recipients receiving fluvastatin: the ALERT extension study. Am J Transplant. 2005;5:2929–2936.10.1111/j.1600-6143.2005.01105.x
- Launay-Vacher V, Izzedine H, Deray G. Statins’ dosage in patients with renal failure and cyclosporine drug-drug interactions in transplant recipient patients. Int J Cardiol. 2005;101:9–17.10.1016/j.ijcard.2004.04.005
- Tonelli M, Isles C, Curhan GC, et al. Effect of pravastatin on cardiovascular events in people with chronic kidney disease. Circulation 2004;110:1557–1563.10.1161/01.CIR.0000143892.84582.60
- Farbakhsh K, Kasiske BL. Dyslipidemias in patients who have chronic kidney disease. Med Clin North Am. 2005;89:689–699.10.1016/j.mcna.2004.11.002
- AstraZeneca. Crestor, Rosuvastatin calcium, prescribing information. AstraZeneca; 2006 [cited 2017 May 21]. Available from: www.astrazeneca_us.com/pi/crestor.pdf
- Ballantyne CM, Corsini A, Davidson MH, et al. Risk for myopathy with statin therapy in high-risk patients. Arch Intern Med. 2003;163:553–564.10.1001/archinte.163.5.553
- Hyogo H, Ikegami T, Tokushige K, et al. Efficacy of pitavastatin for the treatment of non-alcoholic steatohepatitis with dyslipidemia: an open-label, pilot study. Hepatol Res. 2011;41:1057–1065.10.1111/hep.2011.41.issue-11
- Hyogo H, Tazuma S, Arihiro K, et al. Efficacy of atorvastatin for the treatment of nonalcoholic steatohepatitis with dyslipidemia. Metabolism 2008;57:1711–1718.10.1016/j.metabol.2008.07.030
- de Keyser CE, Koehler EM, Schouten JN, et al. Statin therapy is associated with a reduced risk of non-alcoholic fatty liver in overweight individuals. Dig Liver Dis. 2014;46:720–725.10.1016/j.dld.2014.04.002
- Duseja A, Singh SP, Saraswat VA, et al. Non-alcoholic fatty liver disease and metabolic syndrome-position paper of the Indian National Association for the Study of the Liver, Endocrine Society of India, Indian College of Cardiology and Indian Society of Gastroenterology. J Clin Exp Hepatol. 2015;5:51–68.10.1016/j.jceh.2015.02.006
- Pfeffer MA, Keech A, Sacks FM, et al. Safety and tolerability of pravastatin in long-term clinical trials: prospective pravastatin pooling (PPP) project. Circulation 2002;105:2341–2346.10.1161/01.CIR.0000017634.00171.24
- Shepherd J, Cobbe SM, Ford I, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. N Engl J Med. 1995;333:1301–1308.10.1056/NEJM199511163332001
- Sacks FM, Pfeffer MA, Moye LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. N Engl J Med. 1996;335:1001–1009.10.1056/NEJM199610033351401
- Group L-TIwPiIDLS. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med. 1998;339:1349–1357.
- Denus S, Spinler SA, Miller K, et al. Statins and liver toxicity: a meta-analysis. Pharmacotherapy 2004;24:584–591.10.1592/phco.24.6.584.34738
- Alsheikh-Ali AA, Maddukuri PV, Han H, et al. Effect of the magnitude of lipid lowering on risk of elevated liver enzymes, rhabdomyolysis, and cancer. J Am Coll Cardiol. 2007;50:409–418.10.1016/j.jacc.2007.02.073
- Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34:274–285.10.1111/apt.2011.34.issue-3
- Calderon RM, Cubeddu LX, Goldberg RB, et al. Statins in the treatment of dyslipidemia in the presence of elevated liver aminotransferase levels: a therapeutic dilemma. Mayo Clin Proc. 2010;85:349–356.10.4065/mcp.2009.0365
- Athyros VG, Tziomalos K, Gossios TD, et al. Safety and efficacy of long-term statin treatment for cardiovascular events in patients with coronary heart disease and abnormal liver tests in the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) study: a post hoc analysis. Lancet 2010;376:1916–1922.10.1016/S0140-6736(10)61272-X
- Egan M, Prasad S. PURLs: statins for patients with nonalcoholic fatty liver? J Fam Pract. 2011;60:536–538.
- Browning JD. Statins and hepatic steatosis: perspectives from the Dallas Heart Study. Hepatology 2006;44:466–471.10.1002/(ISSN)1527-3350
- Chalasani N, Aljadhey H, Kesterson J, et al. Patients with elevated liver enzymes are not at higher risk for statin hepatotoxicity. Gastroenterology 2004;126:1287–1292.10.1053/j.gastro.2004.02.015
- Lewis JH, Mortensen ME, Zweig S, et al. Efficacy and safety of high-dose pravastatin in hypercholesterolemic patients with well-compensated chronic liver disease: results of a prospective, randomized, double-blind, placebo-controlled, multicenter trial. Hepatology 2007;46:1453–1463.10.1002/hep.21848
- Antonopoulos S, Mikros S, Mylonopoulou M, et al. Rosuvastatin as a novel treatment of non-alcoholic fatty liver disease in hyperlipidemic patients. Atherosclerosis 2006;184:233–234.10.1016/j.atherosclerosis.2005.08.021
- Gomez-Dominguez E, Gisbert JP, Moreno-Monteagudo JA, et al. A pilot study of atorvastatin treatment in dyslipemid, non-alcoholic fatty liver patients. Aliment Pharmacol Ther. 2006;23:1643–1647.10.1111/apt.2006.23.issue-11
- Han KH, Rha SW, Kang HJ, et al. Evaluation of short-term safety and efficacy of HMG-CoA reductase inhibitors in hypercholesterolemic patients with elevated serum alanine transaminase concentrations: PITCH study (PITavastatin versus atorvastatin to evaluate the effect on patients with hypercholesterolemia and mild to moderate hepatic damage). J Clin Lipidol. 2012;6:340–351.10.1016/j.jacl.2012.01.009
- Del Ben M, Polimeni L, Carnevale R, et al. NOX2-generated oxidative stress is associated with severity of ultrasound liver steatosis in patients with non-alcoholic fatty liver disease. BMC Gastroenterol. 2014;14:1221.10.1186/1471-230X-14-81
- Athyros VG, Ganotakis E, Kolovou GD, et al. Assessing the treatment effect in metabolic syndrome without perceptible diabetes (ATTEMPT): a prospective-randomized study in middle aged men and women. Curr Vasc Pharmacol. 2011;9:647–657.10.2174/157016111797484080
- Athyros VG, Katsiki N, Karagiannis A, et al. Are statins ‘IDEAL’ for non-alcoholic fatty liver disease? Curr Med Res Opin. 2014;30:229–231.10.1185/03007995.2013.855192
- Athyros VG, Giouleme O, Ganotakis ES, et al. Safety and impact on cardiovascular events of long-term multifactorial treatment in patients with metabolic syndrome and abnormal liver function tests: a post hoc analysis of the randomised ATTEMPT study. Arch Med Sci. 2011;5:796–805.10.5114/aoms.2011.25554
- Eslami L, Merat S, Malekzadeh R, et al. Statins for non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Cochrane Database Syst Rev. 2013:CD008623.
- Dongiovanni P, Petta S, Mannisto V, et al. Statin use and non-alcoholic steatohepatitis in at risk individuals. J Hepatol. 2015;63:705–712.10.1016/j.jhep.2015.05.006
- Nelson A, Torres DM, Morgan AE, et al. A pilot study using simvastatin in the treatment of nonalcoholic steatohepatitis. J Clin Gastroenterol. 2009;43:990–994.10.1097/MCG.0b013e31819c392e
- Pastori D, Polimeni L, Baratta F, et al. The efficacy and safety of statins for the treatment of non-alcoholic fatty liver disease. Dig Liver Dis. 2015;47:4–11.10.1016/j.dld.2014.07.170
- Tikkanen MJ, Fayyad R, Faergeman O, et al. Effect of intensive lipid lowering with atorvastatin on cardiovascular outcomes in coronary heart disease patients with mild-to-moderate baseline elevations in alanine aminotransferase levels. Int J Cardiol. 2013;168:3846–3852.10.1016/j.ijcard.2013.06.024
- Singh S, Kuftinec GN, Sarkar S. Non-alcoholic fatty liver disease in South Asians: a review of the literature. J Clin Translational Hepatol. 2017;5:76–81.
- Banerjee TK, Das SK. Fifty years of stroke researches in India. Ann Indian Acad Neurol. 2016;19:1–8.
- Lipska K, Sylaja PN, Sarma PS, et al. Risk factors for acute ischaemic stroke in young adults in South India. J Neurol Neurosurg Psychiatry. 2007;78:959–963.10.1136/jnnp.2006.106831
- Amarenco P, Bogousslavsky J, Callahan A 3rd, et al. High-dose atorvastatin after stroke or transient ischemic attack. N Engl J Med. 2006;355:549–559.
- Goldstein LB, Amarenco P, Szarek M, et al. Hemorrhagic stroke in the stroke prevention by aggressive reduction in cholesterol levels study. Neurology 2008;70:2364–2370.10.1212/01.wnl.0000296277.63350.77
- Amarenco P, Goldstein LB, Szarek M, et al. Effects of intense low-density lipoprotein cholesterol reduction in patients with stroke or transient ischemic attack: the Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) trial. Stroke 2007;38:3198–3204.10.1161/STROKEAHA.107.493106
- Amarenco P, Goldstein LB, Callahan A 3rd, et al. Baseline blood pressure, low- and high-density lipoproteins, and triglycerides and the risk of vascular events in the Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) trial. Atherosclerosis 2009;204:515–520.10.1016/j.atherosclerosis.2008.09.008
- Amarenco P, Goldstein LB, Messig M, et al. Relative and cumulative effects of lipid and blood pressure control in the Stroke Prevention by Aggressive Reduction in Cholesterol Levels trial. Stroke 2009;40:2486–2492.10.1161/STROKEAHA.108.546135
- Elkind MS, Sacco RL, MacArthur RB, et al. The Neuroprotection with Statin Therapy for Acute Recovery Trial (NeuSTART): an adaptive design phase I dose-escalation study of high-dose lovastatin in acute ischemic stroke. Int J Stroke. 2008;3:210–218.10.1111/j.1747-4949.2008.00200.x
- Hong KS, Lee JS. Statins in acute ischemic stroke: a systematic review. J Stroke. 2015;17:282–301.10.5853/jos.2015.17.3.282
- Jauch EC, Saver JL, Adams HP Jr, et al. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013;44:870–947.10.1161/STR.0b013e318284056a
- Blanco M, Nombela F, Castellanos M, et al. Statin treatment withdrawal in ischemic stroke: a controlled randomized study. Neurology 2007;69:904–910.10.1212/01.wnl.0000269789.09277.47
- Ni Chroinin D, Asplund K, Asberg S, et al. Statin therapy and outcome after ischemic stroke: systematic review and meta-analysis of observational studies and randomized trials. Stroke 2013;44:448–456.10.1161/STROKEAHA.112.668277
- Cappellari M, Bovi P, Toni D, et al. Cardioembolic stroke in the THRombolysis and STatins (THRaST) study. Int J Stroke. 2015;10:E22.10.1111/ijs.12355
- Orken DN, Kenangil G, Celik M, et al. Association of low cholesterol with primary intracerebral haemorrhage: a case control study. Acta Neurol Scand. 2009;119:151–154.10.1111/ane.2009.119.issue-3
- Valappil AV, Chaudhary NV, Praveenkumar R, et al. Low cholesterol as a risk factor for primary intracerebral hemorrhage: A case-control study. Ann Indian Acad Neurol. 2012;15:19–22.10.4103/0972-2327.93270
- Woodward M, Barzi F, Feigin V, et al. Associations between high-density lipoprotein cholesterol and both stroke and coronary heart disease in the Asia Pacific region. Eur Heart J. 2007;28:2653–2660.10.1093/eurheartj/ehm427
- McKinney JS, Kostis WJ. Statin therapy and the risk of intracerebral hemorrhage: a meta-analysis of 31 randomized controlled trials. Stroke 2012;43:2149–2156.10.1161/STROKEAHA.112.655894
- Dowlatshahi D, Demchuk AM, Fang J, et al. Association of statins and statin discontinuation with poor outcome and survival after intracerebral hemorrhage. Stroke 2012;43:1518–1523.10.1161/STROKEAHA.111.645978
- Colhoun HM, Betteridge DJ, Durrington PN, et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet 2004;364:685–696.10.1016/S0140-6736(04)16895-5
- Goldstein LB, Bushnell CD, Adams RJ, et al. Guidelines for the primary prevention of stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2011;42:517–584.10.1161/STR.0b013e3181fcb238
- De Caterina R, Scarano M, Marfisi R, et al. Cholesterol-lowering interventions and stroke. J Am Coll Cardiol. 2010;55:198–211.10.1016/j.jacc.2009.07.062
- Kernan WN, Ovbiagele B, Black HR, et al. Guidelines for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2014;45:2160–2236.10.1161/STR.0000000000000024
- Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults [practice guideline]. Circulation 2014;129:S1–S45.10.1161/01.cir.0000437738.63853.7a
- Scheitz JF, Nolte CH, Endres M. Should statins be paused or discontinued after thrombolysis or acute intracerebral hemorrhage? No!. Stroke 2013;44:1472–1476.10.1161/STROKEAHA.111.000001
- Van Matre ET, Sherman DS, Kiser TH. Management of intracerebral hemorrhage–use of statins. Vasc Health Risk Manag. 2016;12:153–161.
- Westover MB, Bianchi MT, Eckman MH, et al. Statin use following intracerebral hemorrhage: a decision analysis. Arch Neurol. 2011;68:573–579.
- Epstein AA, Lande H. Studies on blood lipoids. Arch Intern Med. 1922;30:563.10.1001/archinte.1922.00110110034005
- Turner KB. Studies on the prevention of cholesterol atherosclerosis in rabbits: I. The effects of whole thyroid and of potassium iodide. J Exp Med. 1933;58:115–125.10.1084/jem.58.1.115
- O’Brien T, Dinneen SF, O’Brien PC, et al. Hyperlipidemia in patients with primary and secondary hypothyroidism. Mayo Clin Proc. 1993;68:860–866.10.1016/S0025-6196(12)60694-6
- Stone NJ. Secondary causes of hyperlipidemia. Med Clin North Am. 1994;78:117–141.10.1016/S0025-7125(16)30179-1
- Kanaya AM, Harris F, Volpato S, et al. Association between thyroid dysfunction and total cholesterol level in an older biracial population. Arch Intern Med. 2002;162:773–779.10.1001/archinte.162.7.773
- Elder J, McLelland A, O’Reilly DS, et al. The relationship between serum cholesterol and serum thyrotropin, thyroxine and tri-iodothyronine concentrations in suspected hypothyroidism. Ann Clin Biochem. 1990;27(2):110–113.10.1177/000456329002700204
- Gluvic Z, Sudar E, Tica J, et al. Effects of levothyroxine replacement therapy on parameters of metabolic syndrome and atherosclerosis in hypothyroid patients: a prospective pilot study. Int J Endocrinol. 2015;2015:147070.
- Tanis BC, Westendorp RGJ, Smelt AHM. Effect of thyroid substitution on hypercholesterolaemia in patients with subclinical hypothyroidism: a reanalysis of intervention studies. Clin Endocrinol. 1996;44:643–649.10.1046/j.1365-2265.1996.739560.x
- Illingworth DR, McClung MR, Connor WE, et al. Familial hypercholesterolaemia and primary hypothyroidism: coexistence of both disorders in a young woman with severe hypercholesterolaemia. Clin Endocrinol. 1981;14:145–152.10.1111/j.1365-2265.1981.tb00609.x
- Peters JP, Man EB. The interrelations of serum lipids in patients with thyroid disease. J Clin Invest. 1943;22:715–720.10.1172/JCI101444
- Rizos CV, Elisaf MS, Liberopoulos EN. Effects of thyroid dysfunction on lipid profile. Open Cardiovasc Med J. 2011;5:76–84.10.2174/1874192401105010076
- Lee WY, Suh JY, Rhee EJ, et al. Plasma CRP, apolipoprotein A-1, apolipoprotein B and Lp(a) levels according to thyroid function status. Arch Med Res. 2004;35:540–545.10.1016/j.arcmed.2004.08.003
- Nikkilä EA, Kekki M. Plasma triglyceride metabolism in thyroid disease. J Clin Invest. 1972;51:2103–2114.10.1172/JCI107017
- Al-Tonsi AA, Abdel-Gayoum AA, Saad M. The secondary dyslipidemia and deranged serum phosphate concentration in thyroid disorders. Exp Mol Pathol. 2004;76:182–187.10.1016/j.yexmp.2003.10.006
- Hazzard WR, Bierman EL. Aggravation of broad-ß disease (type 3 hyperlipoproteinemia) by hypothyroidism. Arch Intern Med. 1972;130:822–828.10.1001/archinte.1972.03650060018004
- Althaus BU, Staub JJ, Lèche A, et al. LDL/HDL-changes in subclinical hypothyroidism: possible risk factors for coronary heart disease. Clin Endocrinol. 1988;28:157–163.10.1111/j.1365-2265.1988.tb03651.x
- Parle JV, Franklyn JA, Cross KW, et al. Circulating lipids and minor abnormalities of thyroid function. Clin Endocrinol. 1992;37:411–414.10.1111/j.1365-2265.1992.tb02351.x
- Zhao M, Liu L, Wang F, et al. A worthy finding: decrease in total cholesterol and low-density lipoprotein cholesterol in treated mild subclinical hypothyroidism. Thyroid 2016;26:1019–1029.10.1089/thy.2016.0010
- Bakker SJ, ter Maaten JC, Popp-Snijders C, et al. The relationship between thyrotropin and low density lipoprotein cholesterol is modified by insulin sensitivity in healthy euthyroid subjects. J Clin Endocrinol Metab. 2001;86:1206–1211.
- Aviram M, Luboshitzky R, Brook JG. Lipid and lipoprotein pattern in thyroid dysfunction and the effect of therapy. Clin Biochem. 1982;15:62–66.10.1016/S0009-9120(82)90529-X
- Kung AW, Pang RW, Lauder I, et al. Changes in serum lipoprotein(a) and lipids during treatment of hyperthyroidism. Clin Chem. 1995;41:226–231.
- Mullur R, Liu YY, Brent GA. Thyroid hormone regulation of metabolism. Physiol Rev. 2014;94:355–382.10.1152/physrev.00030.2013
- Bakker O, Hudig F, Meijssen S, et al. Effects of triiodothyronine and amiodarone on the promoter of the human LDL receptor gene. Biochem Biophys Res Commun. 1998;249:517–521.10.1006/bbrc.1998.9174
- Shin DJ, Osborne TF. Thyroid hormone regulation and cholesterol metabolism are connected through Sterol Regulatory Element-Binding Protein-2 (SREBP-2). J Biol Chem. 2003;278:34114–34118.10.1074/jbc.M305417200
- Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev. 2010;31:139–170.10.1210/er.2009-0007
- Brent GA. Mechanisms of thyroid hormone action. J Clin Invest. 2012;122:3035–3043.10.1172/JCI60047
- Trost SU, Swanson E, Gloss B, et al. The thyroid hormone receptor-β-selective agonist GC-1 differentially affects plasma lipids and cardiac activity. Endocrinology 2000;141:3057–3064.10.1210/endo.141.9.7681
- Lu C, Cheng SY. Thyroid hormone receptors regulate adipogenesis and carcinogenesis via crosstalk signaling with peroxisome proliferator-activated receptors. J Mol Endocrinol. 2010;44:143–154.10.1677/JME-09-0107
- Verhoeven AJ, Kamer P, Groen AK, et al. Effects of thyroid hormone on mitochondrial oxidative phosphorylation. Biochem J. 1985;226:183–192.10.1042/bj2260183
- Skarulis MC, Celi FS, Mueller E, et al. Thyroid hormone induced brown adipose tissue and amelioration of diabetes in a patient with extreme insulin resistance. J Clin Endocrinol Metab. 2010;95:256–262.10.1210/jc.2009-0543
- Mayer O Jr, Šimon J, Filipovský J, et al. Hypothyroidism in coronary heart disease and its relation to selected risk factors. Vasc Health Risk Manag. 2006;2:499–506.10.2147/vhrm.2006.2.issue-4
- Peixoto de Miranda ÉJF., Bittencourt MS, Pereira AC, et al. Subclinical hypothyroidism is associated with higher carotid intima-media thickness in cross-sectional analysis of the Brazilian Longitudinal Study of Adult Health (ELSA-Brasil). Nutr Metab Cardiovasc Dis. 2016;26:915–921.10.1016/j.numecd.2016.06.005
- Hueston WJ, Pearson WS. Subclinical hypothyroidism and the risk of hypercholesterolemia. Ann Fam Med. 2004;2:351–355.10.1370/afm.79
- Unnikrishnan A, Kalra S, John M, et al., editors. Prevalence of hypothyroidism dyslipidemia (abstract DA-025). Asian Oceanic Thyroid Association Congress; 2012; Bali, Indonesia.
- Enas EA, Chacko V, Pazhoor SG, et al. Dyslipidemia in South Asian patients. Curr Atheroscler Rep. 2007;9:367–374.10.1007/s11883-007-0047-y
- Sawant AM, Shetty D, Mankeshwar R, et al. Prevalence of dyslipidemia in young adult Indian population. J Assoc Physicians India. 2008;56:99–102.
- Marwaha RK, Tandon N, Garg MK, et al. Dyslipidemia in subclinical hypothyroidism in an Indian population. Clin Biochem. 2011;44:1214–1217.10.1016/j.clinbiochem.2011.07.003
- Verdugo C, Perrot L, Ponsin G, et al. Time-course of alterations of high density lipoproteins (HDL) during thyroxine administration to hypothyroid women. Eur J Clin Invest. 1987;17:313–316.10.1111/j.1365-2362.1987.tb02193.x
- Clinical issues and management guidelines for thyroid disorders and dyslipidemia. [cited 2017 Apr 6]. Available from: https://www.apiindia.org/pdf/pdf_index/dyslipidemia_and_thyroid_dysfunction_guidelines.pdf
- Gammage M, Franklyn J. Hypothyroidism, thyroxine treatment, and the heart. Heart 1997;77:189–190.10.1136/hrt.77.3.189
- Bar SL, Holmes DT, Frohlich J. Asymptomatic hypothyroidism and statin-induced myopathy. Can Fam Physician. 2007;53:428–431.
- Tokinaga K, Oeda T, Suzuki Y, et al. HMG-CoA reductase inhibitors (statins) might cause high elevations of creatine phosphokinase (CK) in patients with unnoticed hypothyroidism. Endocr J. 2006;53:401–405.10.1507/endocrj.K04-144
- Asranna A, Taneja RS, Kulshreshta B. Dyslipidemia in subclinical hypothyroidism and the effect of thyroxine on lipid profile. Indian J Endocrinol Metab. 2012;16:S347–S349.
- Bogner U, Arntz HR, Peters H, et al. Subclinical hypothyroidism and hyperlipoproteinaemia: indiscriminate L-thyroxine treatment not justified. Acta Endocrinol. 1993;128:202–206.
- Sewerynek J, Wiktorska J, Nowak D, et al. Methimazole protection against oxidative stress induced by hyperthyroidism in Graves disease. Endocr Regul. 2000;34:83–89.
- Hamilton L, Barkham N, Bhalla A, et al. BSR and BHPR guideline for the treatment of axial spondyloarthritis (including ankylosing spondylitis) with biologics. Rheumatology 2017;56:313–316.10.1093/rheumatology/kew223
- Nam JL, Winthrop KL, van Vollenhoven RF, et al. Current evidence for the management of rheumatoid arthritis with biological disease-modifying antirheumatic drugs: a systematic literature review informing the EULAR recommendations for the management of RA. Ann Rheum Dis. 2010;69:976–986.10.1136/ard.2009.126573
- van Halm VP, Peters MJ, Voskuyl AE, et al. Rheumatoid arthritis versus diabetes as a risk factor for cardiovascular disease: a cross-sectional study, the CARRE Investigation. Ann Rheum Dis. 2009;68:1395–1400.10.1136/ard.2008.094151
- Haroon NN, Paterson JM, Li P, et al. Patients with ankylosing spondylitis have increased cardiovascular and cerebrovascular mortality. Ann Intern Med. 2015;163:409–416.10.7326/M14-2470
- Sherer Y, Shoenfeld Y. Mechanisms of disease: atherosclerosis in autoimmune diseases. Nat Clin Pract Rheumatol. 2006;2:99–106.10.1038/ncprheum0092
- Boots JM, Christiaans MH, van Hooff JP. Effect of immunosuppressive agents on long-term survival of renal transplant recipients. Drugs 2004;64:2047–2073.10.2165/00003495-200464180-00004
- Turesson C, Jarenros A, Jacobsson L. Increased incidence of cardiovascular disease in patients with rheumatoid arthritis: results from a community based study. Ann Rheum Dis. 2004;63:952–955.10.1136/ard.2003.018101
- Papagoras C, Voulgari PV, Drosos AA. Atherosclerosis and cardiovascular disease in the spondyloarthritides, particularly ankylosing spondylitis and psoriatic arthritis. Clin Exp Rheumatol. 2013;31:612–620.
- McCarey DW, McInnes IB, Madhok R, et al. Trial of atorvastatin in rheumatoid arthritis (TARA): double-blind, randomised placebo-controlled trial. Lancet 2004;363:2015–2021.10.1016/S0140-6736(04)16449-0
- El-Barbary AM, Hussein MS, Rageh EM, et al. Effect of atorvastatin on inflammation and modification of vascular risk factors in rheumatoid arthritis. J Rheumatol. 2011;38:229–235.10.3899/jrheum.100582
- Semb AG, Holme I, Kvien TK, et al. Intensive lipid lowering in patients with rheumatoid arthritis and previous myocardial infarction: an explorative analysis from the incremental decrease in endpoints through aggressive lipid lowering (IDEAL) trial. Rheumatology 2011;50:324–329.10.1093/rheumatology/keq295
- Peters MJ, Symmons DP, McCarey D, et al. EULAR evidence-based recommendations for cardiovascular risk management in patients with rheumatoid arthritis and other forms of inflammatory arthritis. Ann Rheum Dis. 2010;69:325–331.10.1136/ard.2009.113696
- Agca R, Heslinga SC, Rollefstad S, et al. EULAR recommendations for cardiovascular disease risk management in patients with rheumatoid arthritis and other forms of inflammatory joint disorders: 2015/2016 update. Ann Rheum Dis. 2017;76:17–28.10.1136/annrheumdis-2016-209775
- Singh JA, Furst DE, Bharat A, et al. 2012 update of the 2008 American College of Rheumatology recommendations for the use of disease-modifying antirheumatic drugs and biologic agents in the treatment of rheumatoid arthritis. Arthritis Care Res. 2012;64:625–639.10.1002/acr.21641
- Malaviya AN, Shankar S, Arya V, et al. Indian Rheumatology Association consensus statement on the diagnosis and treatment of axial spondyloarthropathies. Indian J Rheumatol. 2010;5:16–34.10.1016/S0973-3698(10)60531-6
- Upadhyaya S. Treatment of early RA. Apollo Med. 2012;9:345–346.10.1016/j.apme.2012.09.005
- Upadhyaya S. Treatment of established RA. Apollo Med. 2012;9:347–348.10.1016/j.apme.2012.09.006
- Choy E, Sattar N. Interpreting lipid levels in the context of high-grade inflammatory states with a focus on rheumatoid arthritis: a challenge to conventional cardiovascular risk actions. Ann Rheum Dis. 2009;68:460–469.10.1136/ard.2008.101964
- Kim JY, Lee EY, Park JK, et al. Patients with rheumatoid arthritis show altered lipoprotein profiles with dysfunctional high-density lipoproteins that can exacerbate inflammatory and atherogenic process. PLOS ONE. 2016;11:e0164564.10.1371/journal.pone.0164564
- Bag-Ozbek A, Giles JT. Inflammation, adiposity, and atherogenic dyslipidemia in rheumatoid arthritis: is there a paradoxical relationship? Curr Allergy Asthma Rep. 2015;15:415.10.1007/s11882-014-0497-6
- Soubrier M, Jouanel P, Mathieu S, et al. Effects of anti-tumor necrosis factor therapy on lipid profile in patients with rheumatoid arthritis. Joint Bone Spine. 2008;75:22–24.10.1016/j.jbspin.2007.04.014
- Kisiel B, Kruszewski R, Juszkiewicz A, et al. Methotrexate, cyclosporine A, and biologics protect against atherosclerosis in rheumatoid arthritis. J Immunol Res. 2015;2015:759610.
- Zink A, Askling J, Dixon WG, et al. European biologicals registers: methodology, selected results and perspectives. Ann Rheum Dis. 2009;68:1240–1246.10.1136/ard.2008.091926
- Dixon WG, Watson KD, Lunt M, et al. Reduction in the incidence of myocardial infarction in patients with rheumatoid arthritis who respond to anti–tumor necrosis factor α therapy: Results from the British Society for Rheumatology Biologics Register. Arthritis Rheum. 2007;56:2905–2912.10.1002/(ISSN)1529-0131
- Nissen SE, Yeomans ND, Solomon DH, et al. Cardiovascular safety of celecoxib, naproxen, or ibuprofen for arthritis. N Engl J Med. 2016;375:2519–2529.10.1056/NEJMoa1611593
- Verma I, Syngle A, Krishan P. Predictors of endothelial dysfunction and atherosclerosis in rheumatoid arthritis in Indian population. Indian Heart J. 2017;69:200–206.10.1016/j.ihj.2016.10.013
- Hollan I, Dessein PH, Ronda N, et al. Prevention of cardiovascular disease in rheumatoid arthritis. Autoimmun Rev. 2015;14:952–969.10.1016/j.autrev.2015.06.004
- The Health Improvement Network (THIN) [cited 2017 Apr]. Available from: https://www.ucl.ac.uk/pcph/research-groups-themes/thin-pub/database
- Kreisberg RA, Kasim S. Cholesterol metabolism and aging. Am J Med. 1987;82:54–60.10.1016/0002-9343(87)90272-5
- Ericsson S, Eriksson M, Vitols S, et al. Influence of age on the metabolism of plasma low density lipoproteins in healthy males. J Clin Invest. 1991;87:591–596.10.1172/JCI115034
- Castelli WP, Wilson PW, Levy D, et al. Cardiovascular risk factors in the elderly. Am J Cardiol. 1989;63:12–19.10.1016/0002-9149(89)90110-0
- Frost PH, Davis BR, Burlando AJ, et al. Serum lipids and incidence of coronary heart disease: findings from the systolic hypertension in the elderly program (SHEP). Circulation 1996;94:2381–2388.10.1161/01.CIR.94.10.2381
- Corti MC, Guralnik JM, Salive ME, et al. HDL cholesterol predicts coronary heart disease mortality in older persons. JAMA 1995;274:539–544.10.1001/jama.1995.03530070037026
- National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation 2002;106:3143–3421.
- Grundy SM, Cleeman JI, Rifkind BM, et al. Cholesterol lowering in the elderly population. Arch Intern Med. 1999;159:1670–1678.10.1001/archinte.159.15.1670
- Ornish D, Brown SE, Billings JH, et al. Can lifestyle changes reverse coronary heart disease? Lancet 1990;336:129–133.10.1016/0140-6736(90)91656-U
- Randomised trial of cholesterol lowering in. 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994;344:1383–1389.
- Santinga JT, Rosman HS, Rubenfire M, et al. Efficacy and safety of pravastatin in the long-term treatment of elderly patients with hypercholesterolemia. Am J Med. 1994;96:509–515.10.1016/0002-9343(94)90090-6
- LaRosa JC, Applegate W, Crouse JR 3rd, et al. Cholesterol lowering in the elderly. Arch Intern Med. 1994;154:529–539.10.1001/archinte.1994.00420050081008
- Hunt D. Benefits of pravastatin on cardiovascular events and mortality in older patients with coronary heart disease are equal to or exceed those seen in younger patients: results from the LIPID trial. Ann Intern Med. 2001;134:931.10.7326/0003-4819-134-10-200105150-00007
- Miettinen TA, Pyorala K, Olsson AG, et al. Cholesterol-lowering therapy in women and elderly patients with myocardial infarction or angina pectoris: findings from the Scandinavian Simvastatin Survival Study (4S). Circulation 1997;96:4211–4218.10.1161/01.CIR.96.12.4211
- Lewis SJ, Moye LA, Sacks FM, et al. Effect of pravastatin on cardiovascular events in older patients with myocardial infarction and cholesterol levels in the average range: results of the cholesterol and recurrent events (CARE) trial. Ann Intern Med. 1998;129:681–689.10.7326/0003-4819-129-9-199811010-00002
- Neil HA, DeMicco DA, Luo D, et al. Analysis of efficacy and safety in patients aged 65-75 years at randomization: Collaborative Atorvastatin Diabetes Study (CARDS). Diabetes Care. 2006;29:2378–2384.10.2337/dc06-0872
- Wenger NK. Outcomes of using high- or low-dose atorvastatin in patients 65 years of age or older with stable coronary heart disease. Ann Intern Med. 2007;147:1.10.7326/0003-4819-147-1-200707030-00002
- Allen Maycock CA, Muhlestein JB, Horne BD, et al. Statin therapy is associated with reduced mortality across all age groups of individuals with significant coronary disease, including very elderly patients. J Am Coll Cardiol. 2002;40:1777–1785.10.1016/S0735-1097(02)02477-4
- Shepherd J, Blauw GJ, Murphy MB, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet 2002;360:1623–1630.10.1016/S0140-6736(02)11600-X
- Deedwania P, Stone PH, Bairey Merz CN, et al. Effects of intensive versus moderate lipid-lowering therapy on myocardial ischemia in older patients with coronary heart disease: results of the study assessing goals in the elderly (SAGE). Circulation 2007;115:700–707.10.1161/CIRCULATIONAHA.106.654756
- Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels. JAMA 1998;279:1615–1622.10.1001/jama.279.20.1615
- Sever PS, Dahlöf B, Poulter NR, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial-Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet 2003;361:1149–1158.10.1016/S0140-6736(03)12948-0
- Glynn RJ, Koenig W, Nordestgaard BG, et al. Rosuvastatin for primary prevention in older persons with elevated C-reactive protein and low to average low-density lipoprotein cholesterol levels: exploratory analysis of a randomized trial. Ann Intern Med. 2010;152:488–496, W174.
- Canaris GJ, Manowitz NR, Mayor G, et al. The Colorado thyroid disease prevalence study. Arch Intern Med. 2000;160:526–534.10.1001/archinte.160.4.526
- Stevens LA, Viswanathan G, Weiner DE. Chronic kidney disease and end-stage renal disease in the elderly population: current prevalence, future projections, and clinical significance. Adv Chronic Kidney Dis. 2010;17:293–301.10.1053/j.ackd.2010.03.010
- Jackevicius CA. Adherence with statin therapy in elderly patients with and without acute coronary syndromes. JAMA 2002;288:462.10.1001/jama.288.4.462
- Strandberg TE, Kolehmainen L, Vuorio A. Evaluation and treatment of older patients with hypercholesterolemia. JAMA 2014;312:1136–1144.10.1001/jama.2014.10924
- Lemaitre RN, Psaty BM, Heckbert SR, et al. Therapy with hydroxymethylglutaryl coenzyme a reductase inhibitors (statins) and associated risk of incident cardiovascular events in older adults. Arch Intern Med. 2002;162:1395–1400.10.1001/archinte.162.12.1395
- Ko DT, Mamdani M, Alter DA. Lipid-lowering therapy with statins in high-risk elderly patients. JAMA 2004;291:1864–1870.10.1001/jama.291.15.1864
- National Institutes of Health. National Heart, Lung, and Blood Institute. Incidence and preva-lence: 2006 chart book on cardiovascular and lung diseases. Bethesda (MD): National Heart, Lung, and Blood Institute; 2006.
- Lewis SJ, Sacks FM, Mitchell JS, et al. Effect of pravastatin on cardiovascular events in women after myocardial infarction: the cholesterol and recurrent events (CARE) trial. J Am Coll Cardiol. 1998;32:140–146.10.1016/S0735-1097(98)00202-2
- Desoye G, Schweditsch MO, Pfeiffer KP, et al. Correlation of hormones with lipid and lipoprotein levels during normal pregnancy and postpartum. J Clin Endocrinol Metab. 1987;64:704–712.10.1210/jcem-64-4-704
- Lippi G, Albiero A, Montagnana M, et al. Lipid and lipoprotein profile in physiological pregnancy. Clin Lab. 2007;53:173–177.
- Akahoshi M, Soda M, Nakashima E, et al. Effects of menopause on trends of serum cholesterol, blood pressure, and body mass index. Circulation 1996;94:61–66.10.1161/01.CIR.94.1.61
- Carr MC, Kim KH, Zambon A, et al. Changes in LDL density across the menopausal transition. J Investig Med. 2000;48:245–250.
- Matthews KA, Meilahn E, Kuller LH, et al. Menopause and risk factors for coronary heart disease. N Engl J Med. 1989;321:641–646.10.1056/NEJM198909073211004
- Meilahn EN KL, Matthews KA, Stein EA. Lp(a) concentrations among pre- and postmenopausal women over time: the healthy women study. Circulation 1991;84(Suppl II):536.
- Kim CJ, Jang HC, Cho DH, et al. Effects of hormone replacement therapy on lipoprotein(a) and lipids in postmenopausal women. Arterioscler Thromb Vasc Biol. 1994;14:275–281.10.1161/01.ATV.14.2.275
- Farahani P. Sex/gender disparities in randomized controlled trials of statins: the impact of awareness efforts. Clin Invest Med. 2014;37:163.10.25011/cim.v37i3.21383
- Mora S, Glynn RJ, Hsia J, et al. Statins for the primary prevention of cardiovascular events in women with elevated high-sensitivity c-reactive protein or dyslipidemia: results from the Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) and meta-analysis of women from primary prevention trials. Circulation 2010;121:1069–1077.10.1161/CIRCULATIONAHA.109.906479
- Mizuno K, Nakaya N, Ohashi Y, et al. Usefulness of pravastatin in primary prevention of cardiovascular events in women: analysis of the Management of Elevated Cholesterol in the Primary Prevention Group of Adult Japanese (MEGA Study). Circulation 2008;117:494–502.10.1161/CIRCULATIONAHA.106.671826
- Cholesterol Treatment Trialists C, Fulcher J, O’Connell R, et al. Efficacy and safety of LDL-lowering therapy among men and women: meta-analysis of individual data from 174,000 participants in 27 randomised trials. Lancet 2015;385:1397–1405.
- Kostis WJ, Cheng JQ, Dobrzynski JM, et al. Meta-analysis of statin effects in women versus men. J Am Coll Cardiol. 2012;59:572–582.10.1016/j.jacc.2011.09.067
- Huxley R, Barzi F, Woodward M. Excess risk of fatal coronary heart disease associated with diabetes in men and women: meta-analysis of 37 prospective cohort studies. BMJ 2006;332:73–78.10.1136/bmj.38678.389583.7C
- Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371:203–212.
- Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–2267.
- Keech A, Simes RJ, Barter P, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet 2005;366:1849–1861.
- Ginsberg HN, Elam MB, Lovato LC, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362:1563–1574.
- Bennett S, Sager P, Lipka L, et al. Consistency in efficacy and safety of ezetimibe coadministered with statins for treatment of hypercholesterolemia in women and men. J Womens Health. 2004;13:1101–1107.10.1089/jwh.2004.13.1101
- Cannon CP, Blazing MA, Giugliano RP, et al. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387–2397.10.1056/NEJMoa1410489
- Yokoyama M, Origasa H, Matsuzaki M, et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet 2007;369:1090–1098.10.1016/S0140-6736(07)60527-3
- Kromhout D, Giltay EJ, Geleijnse JM, et al. n–3 Fatty acids and cardiovascular events after myocardial infarction. N Engl J Med. 2010;363:2015–2026.10.1056/NEJMoa1003603
- Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713–1722.10.1056/NEJMoa1615664
- Stampfer MJ, Hu FB, Manson JE, et al. Primary prevention of coronary heart disease in women through diet and lifestyle. N Engl J Med. 2000;343:16–22.10.1056/NEJM200007063430103
- Crouse JR, Morgan T, Terry JG, et al. A randomized trial comparing the effect of casein with that of soy protein containing varying amounts of isoflavones on plasma concentrations of lipids and lipoproteins. Arch Intern Med. 1999;159:2070.10.1001/archinte.159.17.2070
- Bhardwaj S, Selvarajah S, Schneider EB. Muscular effects of statins in the elderly female: a review. Clin Interv Aging. 2013;8:47–59.
- Wiznitzer A, Mayer A, Novack V, et al. Association of lipid levels during gestation with preeclampsia and gestational diabetes mellitus: a population-based study. Am J Obstet Gynecol. 2009;201(482):e1–e8.
- Vrijkotte TG, Krukziener N, Hutten BA, et al. Maternal lipid profile during early pregnancy and pregnancy complications and outcomes: the ABCD study. J Clin Endocrinol Metab. 2012;97:3917–3925.10.1210/jc.2012-1295
- American Academy of Pediatrics, American College of Obstetricians and Gynecologists. Guidelines for perinatal care. 7th ed. Elk Grove Village (IL)/Washington (DC): American Academy of Pediatrics/American College of Obstetricians and Gynecologists; 2012.
- Kusters DM, Lahsinoui H, van de Post JA, et al. Statin use during pregnancy: a systematic review and meta-analysis. Expert Rev Cardiovasc Ther. 2012;10:363–378.10.1586/erc.11.196
- Goldberg AS, Hegele RA. Severe hypertriglyceridemia in pregnancy. J Clin Endocrinol Metab. 2012;97:2589–2596.10.1210/jc.2012-1250
- Gill H, Tiwari P, Dabadghao P. Prevalence of polycystic ovary syndrome in young women from North India: a community-based study. Indian J Endocrinol Metab. 2012;16:S389–S392.
- Joshi B, Mukherjee S, Patil A, et al. A cross-sectional study of polycystic ovarian syndrome among adolescent and young girls in Mumbai, India. Indian J Endocrinol Metab. 2014;18:317–324.10.4103/2230-8210.131162
- Kjerulff LE, Sanchez-Ramos L, Duffy D. Pregnancy outcomes in women with polycystic ovary syndrome: a metaanalysis. Am J Obstet Gynecol. 2011;204(558):e1–e6.
- Barry JA, Azizia MM, Hardiman PJ. Risk of endometrial, ovarian and breast cancer in women with polycystic ovary syndrome: a systematic review and meta-analysis. Hum Reprod Update. 2014;20:748–758.10.1093/humupd/dmu012
- Kalra A, Nair S, Rai L. Association of obesity and insulin resistance with dyslipidemia in Indian women with polycystic ovarian syndrome. Indian J Med Sci. 2006;60:447–453.
- Legro RS, Arslanian SA, Ehrmann DA, et al. Diagnosis and treatment of polycystic ovary syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2013;98:4565–4592.10.1210/jc.2013-2350
- Kolovou GD, Bilianou HG. Influence of aging and menopause on lipids and lipoproteins in women. Angiology 2008;59:54S–57S.10.1177/0003319708319645
- Cauley JA, LaPorte RE, Kuller LH, et al. Menopausal estrogen use, high density lipoprotein cholesterol subfractions and liver function. Atherosclerosis 1983;49:31–39.10.1016/0021-9150(83)90005-9
- Randomised comparison of oestrogen versus oestrogen plus progestogen hormone replacement therapy in women with hysterectomy. Medical Research Council’s General Practice Research Framework. BMJ 1996;312:473–478.
- Hulley S, Grady D, Bush T, et al. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. JAMA 1998;280:605–613.10.1001/jama.280.7.605
- Anderson GL, Limacher M, Assaf AR, et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA 2004;291:1701–1712.
- NHLBI Stops Trial of Estrogen Plus Progestin Due to Increased Breast Cancer Risk, Lack of Overall Benefit. NIH News Release, Tuesday, 2002 Jul 9: 1–4. [cited 2017 Apr 6]. https://www.nhlbi.nih.gov/news/press-releases/2002/nhlbi-stops-trial-of-estrogen-plus-progestin-due-to-increased-breast-cancer-risk-lack-of-overall-benefit
- Sitruk-Ware R, Nath A. Characteristics and metabolic effects of estrogen and progestins contained in oral contraceptive pills. Best Pract Res Clin Endocrinol Metab. 2013;27:13–24.10.1016/j.beem.2012.09.004
- Goldenberg NM, Wang P, Glueck CJ. An observational study of severe hypertriglyceridemia, hypertriglyceridemic acute pancreatitis, and failure of triglyceride-lowering therapy when estrogens are given to women with and without familial hypertriglyceridemia. Clin Chim Acta. 2003;332:11–19.10.1016/S0009-8981(03)00129-3
- Goodman MP. Are all estrogens created equal? A review of oral vs. transdermal therapy. J Womens Health. 2012;21:161–169.10.1089/jwh.2011.2839
- Takaishi K, Miyoshi J, Matsumura T, et al. Hypertriglyceridemic acute pancreatitis during pregnancy: prevention with diet therapy and ω-3 fatty acids in the following pregnancy. Nutrition 2009;25:1094–1097.10.1016/j.nut.2009.04.009
- Saadi HF, Kurlander DJ, Erkins JM, et al. Severe hypertriglyceridemia and acute pancreatitis during pregnancy: treatment with gemfibrozil. Endocr Pract. 1999;5:33–36.10.4158/EP.5.1.33
- Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J Clin Invest. 2003;111:1795–1803.10.1172/JCI200318925
- Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478–3490.10.1093/eurheartj/eht273
- Neil A, Cooper J, Betteridge J, et al. Reductions in all-cause, cancer, and coronary mortality in statin-treated patients with heterozygous familial hypercholesterolaemia: a prospective registry study. Eur Heart J. 2008;29:2625–2633.10.1093/eurheartj/ehn422
- Joshi SR, Anjana RM, Deepa M, et al. Prevalence of dyslipidemia in urban and rural India: the ICMR–INDIAB study. PLoS ONE. 2014;9:e96808.10.1371/journal.pone.0096808
- Rangarajan N, Balasubramanian S, Pang J, et al. Knowledge and awareness of familial hypercholesterolaemia among registered medical practitioners in Tamil Nadu: are they suboptimal? J Clin Diagn Res. 2016;10:OC52–OC56.
- Austin MA, Hutter CM, Zimmern RL, et al. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol. 2004;160:407–420.10.1093/aje/kwh236
- Arca M, Zuliani G, Wilund K, et al. Autosomal recessive hypercholesterolaemia in Sardinia, Italy, and mutations in ARH: a clinical and molecular genetic analysis. Lancet 2002;359:841–847.10.1016/S0140-6736(02)07955-2
- Kotze MJ, Loubser O, Thiart R, et al. CpG hotspot mutations at the LDL receptor locus are a frequent cause of familial hypercholesterolemia among South African Indians. Clin Genet. 2008;51:394–398.10.1111/j.1399-0004.1997.tb02497.x
- Ashavaid TF, Altaf AK, Nair KG. Molecular basis of familial hypercholesterolemia: an Indian experience. Indian J Clin Biochem. 2000;15:11–19.10.1007/BF02867540
- Setia N, Saxena R, Arora A, et al. Spectrum of mutations in homozygous familial hypercholesterolemia in India, with four novel mutations. Atherosclerosis 2016;255:31–36.10.1016/j.atherosclerosis.2016.10.028
- Cuchel M, Bruckert E, Ginsberg HN, et al. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J. 2014;35:2146–2157.10.1093/eurheartj/ehu274
- Goldberg AC, Hopkins PN, Toth PP, et al. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients. J Clin Lipidol. 2011;5:S1–S8.10.1016/j.jacl.2011.04.003
- Civeira F. Guidelines for the diagnosis and management of heterozygous familial hypercholesterolemia. Atherosclerosis 2004;173:55–68.10.1016/j.atherosclerosis.2003.11.010
- Watts GF, Gidding S, Wierzbicki AS, et al. Integrated guidance on the care of familial hypercholesterolaemia from the International FH Foundation. Int J Cardiol. 2014;171:309–325.10.1016/j.ijcard.2013.11.025
- Setia N, Sawhney JP, Saxena R, et al. Successful role of cascade screening in familial hypercholesterolemia in Indian population. Cardiology 2015;131:151.
- Guideline NC. Familial hypercholesterolaemia: Identification and management; 2008. [cited 2017 7 Apr]. Available from: https://www.nice.org.uk/guidance/cg71
- Pang J, Martin AC, Mori TA, et al. Prevalence of familial hypercholesterolemia in adolescents: potential value of universal screening? J Pediatr. 2016;170:315–316.10.1016/j.jpeds.2015.11.019
- Eissa MA, Mihalopoulos NL, Holubkov R, et al. Changes in fasting lipids during puberty. J Pediatr. 2016;170:199–205.10.1016/j.jpeds.2015.11.018
- Alonso R, Andres E, Mata N, et al. Lipoprotein(a) levels in familial hypercholesterolemia. J Am Coll Cardiol. 2014;63:1982–1989.10.1016/j.jacc.2014.01.063
- Shamir R, Fisher EA. Dietary therapy for children with hypercholesterolemia. Am Fam Physician. 2000;61:675–682, 685–686.
- Kaplan RM, Toshima MT. Does a reduced fat diet cause retardation in child growth? Prev Med. 1992;21:33–52.10.1016/0091-7435(92)90004-2
- Lavigne JV, Brown KM, Gidding S, et al. A cholesterol-lowering diet does not produce adverse psychological effects in children: three-year results from the Dietary Intervention Study in Children. Health Psychol. 1999;18:604–613.10.1037/0278-6133.18.6.604
- Hansen D, Michaelsen KF, Skovby F. Growth during treatment of familial hypercholesterolemia. Acta Paediatr. 1992;81:1023–1025.10.1111/apa.1992.81.issue-12
- Marais AD, Blom DJ, Firth JC. Statins in homozygous familial hypercholesterolemia. Curr Atheroscler Rep. 2002;4:19–25.10.1007/s11883-002-0058-7
- Gagne C, Gaudet D, Bruckert E, et al. Efficacy and safety of ezetimibe coadministered with atorvastatin or simvastatin in patients with homozygous familial hypercholesterolemia. Circulation 2002;105:2469–2475.10.1161/01.CIR.0000018744.58460.62
- Vuorio A, Kuoppala J, Kovanen PT, et al. Cochrane database of systematic reviews. Cochrane Database of Systematic Reviews. Wiley-Blackwell; 2010.10.1002/14651858.CD006401.pub2
- Stein EA, Illingworth DR, Kwiterovich PO Jr, et al. Efficacy and safety of lovastatin in adolescent males with heterozygous familial hypercholesterolemia. JAMA 1999;281:137–144.10.1001/jama.281.2.137
- Kusters DM, Caceres M, Coll M, et al. Efficacy and safety of ezetimibe monotherapy in children with heterozygous familial or nonfamilial hypercholesterolemia. J Pediatr. 2015;166:1377–1384 e1–3.
- Kusters DM, Wiegman A, Kastelein JJ, et al. Carotid intima-media thickness in children with familial hypercholesterolemia. Circ Res. 2014;114:307–310.10.1161/CIRCRESAHA.114.301430
- Duplaga BA. Treatment of childhood hypercholesterolemia with HMG-CoA reductase inhibitors. Ann Pharmacother. 1999;33:1224–1227.10.1345/aph.19078
- Bell DA, Hooper AJ, Watts GF, et al. Mipomersen and other therapies for the treatment of severe familial hypercholesterolemia. Vasc Health Risk Manag. 2012;8:651–659.
- Cuchel M, Meagher EA, du Toit Theron H, et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet 2013;381:40–46.10.1016/S0140-6736(12)61731-0
- Raal FJ, Honarpour N, Blom DJ, et al. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double-blind, placebo-controlled trial. Lancet 2015;385:341–350.10.1016/S0140-6736(14)61374-X
- Raal FJ, Stein EA, Dufour R, et al. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. Lancet 2015;385:331–340.10.1016/S0140-6736(14)61399-4
- Kastelein JJ, Ginsberg HN, Langslet G, et al. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur Heart J. 2015;36:2996–3003.
- Raal FJ, Santos RD, Blom DJ, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet 2010;375:998–1006.10.1016/S0140-6736(10)60284-X
- Stein EA, Dufour R, Gagne C, et al. Apolipoprotein B synthesis Inhibition with mipomersen in heterozygous familial hypercholesterolemia: results of a randomized, double-blind, placebo-controlled trial to assess efficacy and safety as add-on therapy in patients with coronary artery disease. Circulation 2012;126:2283–2292.10.1161/CIRCULATIONAHA.112.104125
- Rader DJ, Kastelein JJ. Lomitapide and mipomersen: two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation 2014;129:1022–1032.10.1161/CIRCULATIONAHA.113.001292
- Lee WP, Datta BN, Ong BB, et al. Defining the role of lipoprotein apheresis in the management of familial hypercholesterolemia. Am J Cardiovasc Drugs. 2011;11:363–370.10.2165/11594970-000000000-00000
- Thompson GR. Lipoprotein apheresis. Curr Opin Lipidol. 2010;21:487–491.10.1097/MOL.0b013e32833e13fd
- Thompson GR, Catapano A, Saheb S, et al. Severe hypercholesterolaemia: therapeutic goals and eligibility criteria for LDL apheresis in Europe. Curr Opin Lipidol. 2010;21:492–498.10.1097/MOL.0b013e3283402f53
- Costantine MM, Cleary K, Hebert MF, et al. Safety and pharmacokinetics of pravastatin used for the prevention of preeclampsia in high-risk pregnant women: a pilot randomized controlled trial. Am J Obstet Gynecol. 2016;214:720 e1–720 e17.
- Taguchi N, Rubin ET, Hosokawa A, et al. Prenatal exposure to HMG-CoA reductase inhibitors: effects on fetal and neonatal outcomes. Reprod Toxicol. 2008;26:175–177.10.1016/j.reprotox.2008.06.009
- Miller M, Stone NJ, Ballantyne C, et al. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation 2011;123:2292–2333.10.1161/CIR.0b013e3182160726
- Chait A, Brunzell JD. Severe hypertriglyceridemia: role of familial and acquired disorders. Metabolism 1983;32:209–214.10.1016/0026-0495(83)90184-1
- Casey DE. Dyslipidemia and atypical antipsychotic drugs. J Clin Psychiatry. 2004;65(Suppl 18):27–35.
- Kronenberg F. Dyslipidemia and nephrotic syndrome: recent advances. J Ren Nutr. 2005;15:195–203.10.1053/j.jrn.2004.10.003
- Kaysen GA. Lipid and lipoprotein metabolism in chronic kidney disease. J Ren Nutr. 2009;19:73–77.10.1053/j.jrn.2008.10.011
- Johansen CT, Hegele RA. Genetic bases of hypertriglyceridemic phenotypes. Curr Opin Lipidol. 2011;22:247–253.10.1097/MOL.0b013e3283471972
- Hegele RA, Ginsberg HN, Chapman MJ, et al. The polygenic nature of hypertriglyceridaemia: implications for definition, diagnosis, and management. Lancet Diabetes Endocrinol. 2014;2:655–666.10.1016/S2213-8587(13)70191-8
- Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG, et al. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med. 2014;371:32–41.10.1056/NEJMoa1308027
- Murad MH, Hazem A, Coto-Yglesias F, et al. The association of hypertriglyceridemia with cardiovascular events and pancreatitis: a systematic review and meta-analysis. BMC Endocr Disord. 2012;12:572.10.1186/1472-6823-12-2
- Yuan G, Al-Shali KZ, Hegele RA. Hypertriglyceridemia: its etiology, effects and treatment. CMAJ 2007;176:1113–1120.10.1503/cmaj.060963
- Fredrickson DS, Lees RS. Editorial: a system for phenotyping hyperlipoproteinemia. Circulation 1965;31:321–327.10.1161/01.CIR.31.3.321
- Jeon HR, Kim SY, Cho YJ, et al. Hypertriglyceridemia-induced acute pancreatitis in pregnancy causing maternal death. Obstet Gynecol Sci. 2016;59:148–151.10.5468/ogs.2016.59.2.148
- Morganroth J, Levy RI, Fredrickson DS. The biochemical, clinical, and genetic features of type III hyperlipoproteinemia. Ann Intern Med. 1975;82:158–174.10.7326/0003-4819-82-2-158
- Heiss G, Tamir I, Davis CE, et al. Lipoprotein-cholesterol distributions in selected North American populations: the lipid research clinics program prevalence study. Circulation 1980;61:302–315.10.1161/01.CIR.61.2.302
- Berglund L, Brunzell JD, Goldberg AC, et al. Evaluation and treatment of hypertriglyceridemia: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2012;97:2969–2989.10.1210/jc.2011-3213
- Ferns G, Keti V, Griffin B. Investigation and management of hypertriglyceridaemia. J Clin Pathol. 2008;61:1174–1183.10.1136/jcp.2008.055756
- Stroes E, Moulin P, Parhofer KG, et al. Diagnostic algorithm for familial chylomicronemia syndrome. Atheroscler Suppl. 2017;23:1–7.10.1016/j.atherosclerosissup.2016.10.002
- Brahm AJ, Hegele RA. Chylomicronaemia–current diagnosis and future therapies. Nat Rev Endocrinol. 2015;11:352–362.10.1038/nrendo.2015.26
- Gotoda T, Shirai K, Ohta T, et al. Diagnosis and management of type I and type V hyperlipoproteinemia. J Atheroscler Thromb. 2012;19:1–12.10.5551/jat.10702
- Surendran RP, Visser ME, Heemelaar S, et al. Mutations in LPL, APOC2, APOA5, GPIHBP1 and LMF1 in patients with severe hypertriglyceridaemia. J Intern Med. 2012;272:185–196.10.1111/j.1365-2796.2012.02516.x
- Cox RA, García-Palmieri MR. Cholesterol, triglycerides, and associated lipoproteins. In: Walker HK, Hall WD, Hurst JW, editors. Clinical methods: the history, physical, and la-boratory examinations. 3rd ed. Boston (MA): Butterworths; 1990. Chapter 31. Available from: https://www.ncbi.nlm.nih.gov/books/NBK351/
- Kobayashi J, Nohara A, Kawashiri MA, et al. Serum lipoprotein lipase mass: clinical significance of its measurement. Clin Chim Acta. 2007;378:7–12.10.1016/j.cca.2006.12.003
- Do R, Willer CJ, Schmidt EM, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45:1345–1352.10.1038/ng.2795
- van Himbergen TM, Otokozawa S, Matthan NR, et al. Familial combined hyperlipidemia is associated with alterations in the cholesterol synthesis pathway. Arterioscler Thromb Vasc Biol. 2010;30:113–120.10.1161/ATVBAHA.109.196550
- Mahley RW, Huang Y, Rall SC Jr. Pathogenesis of type III hyperlipoproteinemia (dysbetalipoproteinemia). Questions, quandaries, and paradoxes. J Lipid Res. 1999;40:1933–1949.
- Smit M, de Knijff P, van der Kooij-Meijs E, et al. Genetic heterogeneity in familial dysbetalipoproteinemia. The E2(lys146 – gln) variant results in a dominant mode of inheritance. J Lipid Res. 1990;31:45–53.
- Ginsberg HN. New perspectives on atherogenesis: role of abnormal triglyceride-rich lipoprotein metabolism. Circulation 2002;106:2137–2142.10.1161/01.CIR.0000035280.64322.31
- Sniderman A, Tremblay A, Bergeron J, et al. Diagnosis of type III hyperlipoproteinemia from plasma total cholesterol, triglyceride, and apolipoprotein B. J Clin Lipidol. 2007;1:256–263.10.1016/j.jacl.2007.07.006
- Hanada H, Mugii S, Okubo M, et al. Establishment of chemiluminescence enzyme immunoassay for apolipoprotein B-48 and its clinical applications for evaluation of impaired chylomicron remnant metabolism. Clin Chim Acta. 2012;413:160–165.10.1016/j.cca.2011.09.013
- Brahm A, Hegele RA. Hypertriglyceridemia. Nutrients. 2013;5:981–1001.10.3390/nu5030981
- Willett WC. The Mediterranean diet: science and practice. Public Health Nutr. 2006;9:105–110.
- Misra A, Sharma R, Gulati S, et al. Consensus dietary guidelines for healthy living and prevention of obesity, the metabolic syndrome, diabetes, and related disorders in Asian Indians. Diabetes Technol Ther. 2011;13:683–694.10.1089/dia.2010.0198
- Gulati S, Misra A, Sharma M. Dietary fats and oils in India. Curr Diabetes Rev. 2016. PMID: 27501784.
- National Nutrition Monitoring Bureau: diet and nutritional status of rural population, prevalence of hypertension and diabetes among adults and infant and young child feeding practices; report of third repeat survey. Hyderabad: National Institute of Nutrition; Indian Council of Medical Research; 2012. (Technical report no. 26).
- Watts GF, Ooi EM, Chan DC. Demystifying the management of hypertriglyceridaemia. Nat Rev Cardiol. 2013;10:648–661.10.1038/nrcardio.2013.140
- Goulooze SC, Cohen AF, Rissmann R. Lomitapide. Br J Clin Pharmacol. 2015;80:179–181.10.1111/bcp.v80.2
- Gaudet D, Méthot J, Kastelein J. Gene therapy for lipoprotein lipase deficiency. Curr Opin Lipidol. 2012;23:310–320.10.1097/MOL.0b013e3283555a7e
- Tenenbaum A, Klempfner R, Fisman EZ. Hypertriglyceridemia: a too long unfairly neglected major cardiovascular risk factor. Cardiovasc Diabetol. 2014;13:159.10.1186/s12933-014-0159-y
- Feher MD, Foxton J, Banks D, et al. Long-term safety of statin-fibrate combination treatment in the management of hypercholesterolaemia in patients with coronary artery disease. Heart 1995;74:14–17.10.1136/hrt.74.1.14
- Nasa P, Alexander G, Kulkarni A, et al. Early plasmapheresis in patients with severe hypertriglyceridemia induced acute pancreatitis. Indian J Crit Care Med. 2015;19:487–489.10.4103/0972-5229.162472
- Miller LW, Schlant RC, Kobashigawa J, et al. Task force 5: complications. J Am Coll Cardiol. 1993;22:41–54.10.1016/0735-1097(93)90814-H
- Pallet N, Legendre C. Adverse events associated with mTOR inhibitors. Expert Opin Drug Saf. 2013;12:177–186.10.1517/14740338.2013.752814
- Tenderich G, Fuchs U, Zittermann A, et al. Comparison of sirolimus and everolimus in their effects on blood lipid profiles and haematological parameters in heart transplant recipients. Clin Transplant. 2007;21:536–543.10.1111/ctr.2007.21.issue-4
- Kaplan B, Qazi Y, Wellen JR. Strategies for the management of adverse events associated with mTOR inhibitors. Transplant Rev. 2014;28:126–133.10.1016/j.trre.2014.03.002
- Ettinger WH, Klinefelter HF, Kwiterovitch PO. Effect of short-term, low-dose corticosteroids on plasma lipoprotein lipids. Atherosclerosis 1987;63:167–172.10.1016/0021-9150(87)90117-1
- Arora S, Aukrust P, Andreassen A, et al. The prognostic importance of modifiable risk factors after heart transplantation. Am Heart J. 2009;158:431–436.10.1016/j.ahj.2009.05.036
- Raisanen-Sokolowski A, Tilly-Kiesi M, Ustinov J, et al. Hyperlipidemia accelerates allograft arteriosclerosis (chronic rejection) in the rat. Arterioscler Thromb. 1994;14:2032–2042.10.1161/01.ATV.14.12.2032
- Lentine KL, Naik AS, Schnitzler M, et al. Variation in comedication use according to kidney transplant immunosuppressive regimens: application of integrated registry and pharmacy claims data. Transplant Proc. 2016;48:55–58.10.1016/j.transproceed.2015.12.024
- Lund LH, Edwards LB, Kucheryavaya AY, et al. The registry of the international society for heart and lung transplantation: thirty-first official adult heart transplant report – 2014; focus theme: retransplantation. J Heart Lung Transplant. 2014;33:996–1008.10.1016/j.healun.2014.08.003
- Chih S, Chong AY, Mielniczuk LM, et al. Allograft vasculopathy. J Am Coll Cardiol. 2016;68:80–91.10.1016/j.jacc.2016.04.033
- Sipahi I, Starling RC. Cardiac allograft vasculopathy: an update. Heart Fail Clin. 2007;3:87–95.10.1016/j.hfc.2007.02.007
- Labarrere CA, Jaeger BR. Biomarkers of heart transplant rejection: the good, the bad, and the ugly!. Transl Res. 2012;159:238–251.10.1016/j.trsl.2012.01.018
- Perrault LP, Mahlberg F, Breugnot C, et al. Hypercholesterolemia increases coronary endothelial dysfunction, lipid content, and accelerated atherosclerosis after heart transplantation. Arterioscler Thromb Vasc Biol. 2000;20:728–736.10.1161/01.ATV.20.3.728
- Costanzo MR, Dipchand A, Starling R, et al. The International society of heart and lung transplantation guidelines for the care of heart transplant recipients. J Heart Lung Transplant. 2010;29:914–956.10.1016/j.healun.2010.05.034
- Ballantyne CM, Podet EJ, Patsch WP, et al. Effects of cyclosporine therapy on plasma lipoprotein levels. JAMA 1989;262:53–56.10.1001/jama.1989.03430010065032
- Lemahieu WP, Hermann M, Asberg A, et al. Combined therapy with atorvastatin and calcineurin inhibitors: no interactions with tacrolimus. Am J Transplant. 2005;5:2236–2243.10.1111/ajt.2005.5.issue-9
- Sieg A, Weeks P, Krustchinsky L, et al. Statin therapy in cardiac allograft vasculopathy progression in heart transplant patients: does potency matter? Transplant Rev. 2016;30:178–186.10.1016/j.trre.2016.01.001
- Lee KF, Pierce JD, Hess ML, et al. Cardiac transplantation with corticosteroid-free immunosuppression: long-term result. Ann Thorac Surg. 1991;52:211–218; discussion 218. 10.1016/0003-4975(91)91338-V
- Bloom RD, Crutchlow MF. Transplant-associated hyperglycemia. Transplant Rev. 2008;22:39–51.10.1016/j.trre.2007.06.001
- Kuipers HF, Biesta PJ, Groothuis TA, et al. Statins affect cell-surface expression of major histocompatibility complex class II molecules by disrupting cholesterol-containing microdomains. Hum Immunol. 2005;66:653–665.10.1016/j.humimm.2005.04.004
- Mach F. Statins as immunomodulatory agents. Circulation 2004;109:II15–II17.
- Kobashigawa JA, Katznelson S, Laks H, et al. Effect of pravastatin on outcomes after cardiac transplantation. N Engl J Med. 1995;333:621–627.10.1056/NEJM199509073331003
- Kobashigawa JA, Moriguchi JD, Laks H, et al. Ten-year follow-up of a randomized trial of pravastatin in heart transplant patients. J Heart Lung Transplant. 2005;24:1736–1740.10.1016/j.healun.2005.02.009
- Ballantyne CM, Radovancevic B, Farmer JA, et al. Hyperlipidemia after heart transplantation: report of a 6-year experience, with treatment recommendations. J Am Coll Cardiol. 1992;19:1315–1321.10.1016/0735-1097(92)90340-S
- Wenke K, Meiser B, Thiery J, et al. Simvastatin reduces graft vessel disease and mortality after heart transplantation: a four-year randomized trial. Circulation 1997;96:1398–1402.10.1161/01.CIR.96.5.1398
- Barge-Caballero G, Barge-Caballero E, Marzoa-Rivas R, et al. Clinical evaluation of rosuvastatin in heart transplant patients with hypercholesterolemia and therapeutic failure of other statin regimens: short-term and long-term efficacy and safety results. Transpl Int. 2015;28:1034–1041.10.1111/tri.2015.28.issue-9
- Mehra MR, Uber PA, Vivekananthan K, et al. Comparative beneficial effects of simvastatin and pravastatin on cardiac allograft rejection and survival. J Am Coll Cardiol. 2002;40:1609–1614.10.1016/S0735-1097(02)02340-9
- Keogh A, Macdonald P, Kaan A, et al. Efficacy and safety of pravastatin vs simvastatin after cardiac transplantation. J Heart Lung Transplant. 2000;19:529–537.10.1016/S1053-2498(00)00077-2
- Shah MK, Critchley WR, Yonan N, et al. Second line options for hyperlipidemia management after cardiac transplantation. Cardiovasc Ther. 2013;31:138–146.10.1111/cdr.2013.31.issue-3
- Kosoglou T, Statkevich P, Johnson-Levonas AO, et al. Ezetimibe. Clin Pharmacokinet. 2005;44:467–494.10.2165/00003088-200544050-00002
- Attridge RL, Frei CR, Ryan L, et al. Fenofibrate-associated nephrotoxicity: a review of current evidence. Am J Health Syst Pharm. 2013;70:1219–1225.10.2146/ajhp120131
- Gillis KA, Patel RK, Jardine AG. Cardiovascular complications after transplantation: treatment options in solid organ recipients. Transplant Rev. 2014;28:47–55.10.1016/j.trre.2013.12.001
- Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis. 1998;32:S112–S119.10.1053/ajkd.1998.v32.pm9820470
- Neale J, Smith AC. Cardiovascular risk factors following renal transplant. World J Transplant. 2015;5:183–195.10.5500/wjt.v5.i4.183
- Chapter 1: assessment of lipid status in adults with CKD. Kidney Int Suppl. 2013;3:268–270.
- Lucey MR, Terrault N, Ojo L, et al. Long-term management of the successful adult liver transplant: 2012 practice guideline by the American Association for the Study of Liver Diseases and the American Society of Transplantation. Liver Transpl. 2013;19:3–26.10.1002/lt.23566
- Bittar R, Giral P, Aslangul E, et al. Determinants of low-density lipoprotein particle diameter during antiretroviral therapy including protease inhibitors in HIV-1-infected patients. Antivir Ther. 2012;17:855–860.10.3851/IMP2065
- Lundgren JD, Battegay M, Behrens G, et al. European AIDS Clinical Society (EACS) guidelines on the prevention and management of metabolic diseases in HIV. HIV Med. 2008;9:72–81.10.1111/hiv.2008.9.issue-2
- Aberg JA, Gallant JE, Ghanem KG, et al. Primary care guidelines for the management of persons infected with HIV: 2013 update by the HIV Medicine Association of the Infectious Diseases Society of America. Clin Infect Dis. 2014;58:e1–e34.10.1093/cid/cit665
- Grinspoon S, Carr A. Cardiovascular risk and body-fat abnormalities in HIV-infected adults. N Engl J Med. 2005;352:48–62.10.1056/NEJMra041811
- Lo J, Grinspoon S. Cardiovascular disease in HIV-infected patients: does HIV infection in and of itself increase cardiovascular risk? Curr Opin HIV AIDS. 2008;3:207–213.10.1097/COH.0b013e3282fb7ba6
- Dubé MP, Parker RA, Tebas P, et al. Glucose metabolism, lipid, and body fat changes in antiretroviral-naive subjects randomized to nelfinavir or efavirenz plus dual nucleosides. AIDS. 2005;19:1807–1818.10.1097/01.aids.0000183629.20041.bb
- Calza L, Manfredi R, Chiodo F. Dyslipidaemia associated with antiretroviral therapy in HIV-infected patients. J Antimicrob Chemother. 2004;53:10–14.
- Hruz PW, Murata H, Mueckler M. Adverse metabolic consequences of HIV protease inhibitor therapy: the search for a central mechanism. Am J Physiol Endocrinol Metab. 2001;280:E549–E553.
- Fauvel J, Bonnet E, Ruidavets JB, et al. An interaction between apo C-III variants and protease inhibitors contributes to high triglyceride/low HDL levels in treated HIV patients. AIDS. 2001;15:2397–2406.10.1097/00002030-200112070-00007
- Vigano A, Aldrovandi GM, Giacomet V, et al. Improvement in dyslipidaemia after switching stavudine to tenofovir and replacing protease inhibitors with efavirenz in HIV-infected children. Antivir Ther. 2005;10:917–924.
- Sabin CA, Worm SW, Weber R, et al. Use of nucleoside reverse transcriptase inhibitors and risk of myocardial infarction in HIV-infected patients enrolled in the D:A:D study: a multi-cohort collaboration. Lancet 2008;371:1417–1426.
- Bernal E, Masiá M, Padilla S, et al. High-density lipoprotein cholesterol in HIV-infected patients: evidence for an association with HIV-1 viral load, antiretroviral therapy status, and regimen composition. AIDS Patient Care STDs. 2008;22:569–575.10.1089/apc.2007.0186
- Lo J, Abbara S, Shturman L, et al. Increased prevalence of subclinical coronary atherosclerosis detected by coronary computed tomography angiography in HIV-infected men. AIDS 2010;24:243–253.10.1097/QAD.0b013e328333ea9e
- Friis-Moller N, Sabin CA, Weber R, et al. Combination antiretroviral therapy and the risk of myocardial infarction. N Engl J Med. 2003;349:1993–2003.
- Saves M, Chene G, Ducimetiere P, et al. Risk factors for coronary heart disease in patients treated for human immunodeficiency virus infection compared with the general population. Clin Infect Dis. 2003;37:292–298.10.1086/375844
- Lohse N, Hansen AB, Pedersen G, et al. Survival of persons with and without HIV infection in Denmark, 1995–2005. Ann Intern Med. 2007;146:87–95.10.7326/0003-4819-146-2-200701160-00003
- Grunfeld C, Pang M, Doerrler W, et al. Lipids, lipoproteins, triglyceride clearance, and cytokines in human immunodeficiency virus infection and the acquired immunodeficiency syndrome. J Clin Endocrinol Metab. 1992;74:1045–1052.
- Hadigan C, Meigs JB, Corcoran C, et al. Metabolic abnormalities and cardiovascular disease risk factors in adults with human immunodeficiency virus infection and lipodystrophy. Clin Infect Dis. 2001;32:130–139.10.1086/317541
- Triant VA, Lee H, Hadigan C, et al. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. J Clin Endocrinol Metab. 2007;92:2506–2512.10.1210/jc.2006-2190
- Grinspoon SK, Grunfeld C, Kotler DP, et al. State of the science conference: initiative to decrease cardiovascular risk and increase quality of care for patients living with HIV/AIDS: executive summary. Circulation 2008;118:198–210.10.1161/CIRCULATIONAHA.107.189622
- Palella FJ Jr, Baker RK, Moorman AC, et al. Mortality in the highly active antiretroviral therapy era. J Acquir Immune Defic Syndr. 2006;43:27–34.10.1097/01.qai.0000233310.90484.16
- Mothe B, Perez I, Domingo P, et al. HIV-1 infection in subjects older than 70: a multicenter cross-sectional assessment in Catalonia, Spain. Curr HIV Res. 2009;7:597–600.10.2174/157016209789973691
- Hejazi N, Rajikan R, Kwok Choong CL, et al. Metabolic abnormalities in adult HIV infected population on antiretroviral medication in Malaysia: a cross-sectional survey. BMC Public Health. 2013;13:358.10.1186/1471-2458-13-758
- Julius H, Basu D, Ricci E, et al. The burden of metabolic diseases amongst HIV positive patients on HAART attending The Johannesburg Hospital. Curr HIV Res. 2011;9:247–252.10.2174/157016211796320360
- Kamin D, Hadigan C. Hyperlipidemia in children with HIV infection: an emerging problem. Expert Rev Cardiovasc Ther. 2003;1:143–150.10.1586/14779072.1.1.143
- Miller TI, Borkowsky W, DiMeglio LA, et al. Metabolic abnormalities and viral replication are associated with biomarkers of vascular dysfunction in HIV-infected children. HIV Med. 2012;13:264–275.10.1111/j.1468-1293.2011.00970.x
- Riddler SA, Smit E, Cole SR, et al. Impact of HIV infection and HAART on serum lipids in men. JAMA 2003;289:2978–2982.10.1001/jama.289.22.2978
- Periard D, Telenti A, Sudre P, et al. Atherogenic dyslipidemia in HIV-infected individuals treated with protease inhibitors. The Swiss HIV Cohort Study. Circulation 1999;100:700–705.
- El-Sadr WM, Lundgren J, Neaton JD, et al. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med. 2006;355:2283–2296.
- Duprez DA, Kuller LH, Tracy R, et al. Lipoprotein particle subclasses, cardiovascular disease and HIV infection. Atherosclerosis 2009;207:524–529.10.1016/j.atherosclerosis.2009.05.001
- Lang S, Boccara F, Mary-Krause M, et al. Epidemiology of coronary heart disease in HIV-infected versus uninfected individuals in developed countries. Arch Cardiovasc Dis. 2015;108:206–215.10.1016/j.acvd.2015.01.004
- Fishbein GA, Micheletti RG, Currier JS, et al. Atherosclerotic oxalosis in coronary arteries. Cardiovasc Pathol. 2008;17:117–123.10.1016/j.carpath.2007.07.002
- Hulten E, Mitchell J, Scally J, et al. HIV positivity, protease inhibitor exposure and subclinical atherosclerosis: a systematic review and meta-analysis of observational studies. Heart 2009;95:1826–1835.10.1136/hrt.2009.177774
- Stein JH, Currier JS, Hsue PY. Arterial disease in patients with human immunodeficiency virus infection JACC: Cardiovasc Imaging. 2014;7:515–525.10.1016/j.jcmg.2013.08.019
- Kuller LH, Tracy R, Belloso W, et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 2008;5:e203.10.1371/journal.pmed.0050203
- Tenorio AR, Zheng Y, Bosch RJ, et al. Soluble markers of inflammation and coagulation but not t-cell activation predict non-AIDS-defining morbid events during suppressive antiretroviral treatment. J Infect Dis. 2014;210:1248–1259.10.1093/infdis/jiu254
- Torriani FJ, Komarow L, Parker RA, et al. Endothelial function in human immunodeficiency virus-infected antiretroviral-naive subjects before and after starting potent antiretroviral therapy. J Am Coll Cardiol. 2008;52:569–576.10.1016/j.jacc.2008.04.049
- Karmochkine M, Ankri A, Calvez V, et al. Plasma hypercoagulability is correlated to plasma HIV load. Thromb Haemost. 1998;80:208–209.
- Dubé MP, Stein JH, Aberg JA, et al. Guidelines for the evaluation and management of dyslipidemia in human immunodeficiency virus (HIV)-infected adults receiving antiretroviral therapy: recommendations of the HIV Medicine Association of the Infectious Disease Society of America and the Adult AIDS Clinical Trials Group. Clin Infect Dis. 2003;37:613–627.10.1086/378131
- Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III). JAMA 2001;285:2486–2497.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004;110:227–239.10.1161/01.CIR.0000133317.49796.0E
- Silverberg MJ, Leyden W, Hurley L, et al. Response to newly prescribed lipid-lowering therapy in patients with and without HIV infection. Ann Intern Med. 2009;150:301–313.10.7326/0003-4819-150-5-200903030-00006
- Law MG, Friis-Moller N, El-Sadr WM, et al. The use of the Framingham equation to predict myocardial infarctions in HIV-infected patients: comparison with observed events in the D:A:D study. HIV Med. 2006;7:218–230.10.1111/hiv.2006.7.issue-4
- Silverberg MJ, Leyden WA, Xu L, et al. Immunodeficiency and risk of myocardial infarction among HIV-positive individuals with access to care. J Acquir Immune Defic Syndr. 2014;65:160–166.10.1097/QAI.0000000000000009
- Friis-Møller N, Thiébaut R, Reiss P, et al. Predicting the risk of cardiovascular disease in HIV-infected patients: the data collection on adverse effects of anti-HIV drugs study. Eur J Cardiovasc Prev Rehabil. 2010;17:491–501.10.1097/HJR.0b013e328336a150
- Sitbon O, Gressin V, Speich R, et al. Bosentan for the treatment of human immunodeficiency virus-associated pulmonary arterial hypertension. Am J Respir Crit Care Med. 2004;170:1212–1217.10.1164/rccm.200404-445OC
- Reuter H, Burgess LJ, Doubell AF. Epidemiology of pericardial effusions at a large academic hospital in South Africa. Epidemiol Infect. 1999;133:393–399.10.1017/S0950268804003577
- Ghofrani HA, Friese G, Discher T, et al. Inhaled iloprost is a potent acute pulmonary vasodilator in HIV-related severe pulmonary hypertension. Eur Respir J. 2004;23:321–326.10.1183/09031936.03.00057703
- Third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation 2002;106:3143–3421.
- Marzilli M. Pleiotropic effects of statins. Am J Cardiovasc Drugs. 2010;10(10):3–9.10.2165/1153644-S0-000000000-00000
- Gili S, Grosso Marra W, D’Ascenzo F, et al. Comparative safety and efficacy of statins for primary prevention in human immunodeficiency virus-positive patients: a systematic review and meta-analysis. Eur Heart J. 2016;37:3600–3609.10.1093/eurheartj/ehv734
- Singh S, Willig JH, Mugavero MJ, et al. Comparative effectiveness and toxicity of statins among HIV-infected patients. Clin Infect Dis. 2011;52:387–395.10.1093/cid/ciq111
- Chow D, Chen H, Glesby MJ, et al. Short-term ezetimibe is well tolerated and effective in combination with statin therapy to treat elevated LDL cholesterol in HIV-infected patients. AIDS 2009;23:2133–2141.10.1097/QAD.0b013e32833068e3
- Wohl DA, Waters D, Simpson RJ Jr, et al. Ezetimibe alone reduces low‐density lipoprotein cholesterol in HIV‐infected patients receiving combination antiretroviral therapy. Clin Infect Dis. 2008;47:1105–1108.10.1086/593299
- Digby JE, Ruparelia N, Choudhury RP. Niacin in cardiovascular disease: recent preclinical and clinical developments. Arterioscler Thromb Vasc Biol. 2012;32:582–588.10.1161/ATVBAHA.111.236315
- Brunzell JD. Hypertriglyceridemia. N Engl J Med. 2007;357:1009–1017.10.1056/NEJMcp070061
- Calza L, Manfredi R, Chiodo F. Use of fibrates in the management of hyperlipidemia in HIV-infected patients receiving HAART. Infection 2002;30:26–31.10.1007/s15010-001-2052-3
- Bays HE, Tighe AP, Sadovsky R, et al. Prescription omega-3 fatty acids and their lipid effects: physiologic mechanisms of action and clinical implications. Expert Rev Cardiovasc Ther. 2008;6:391–409.10.1586/14779072.6.3.391
- Harris WS, Kris-Etherton PM, Harris KA. Intakes of long-chain omega-3 fatty acid associated with reduced risk for death from coronary heart disease in healthy adults. Curr Atheroscler Rep. 2008;10:503–509.10.1007/s11883-008-0078-z
- Bartlett JG. New guidelines for management of dyslipidemia from IDSA and the ACTG. Hopkins HIV Rep. 2004;16:4–5.
- Gerber JG, Kitch DW, Fichtenbaum CJ, et al. Fish oil and fenofibrate for the treatment of hypertriglyceridemia in HIV-infected subjects on antiretroviral therapy. J Acquir Immune Defic Syndr. 2008;47:459–466.10.1097/QAI.0b013e31815bace2
- Peters BS, Wierzbicki AS, Moyle G, et al. The effect of a 12-week course of omega-3 polyunsaturated fatty acids on lipid parameters in hypertriglyceridemic adult HIV-infected patients undergoing haart: a randomized, placebo-controlled pilot trial. Clin Ther. 2012;34:67–76.10.1016/j.clinthera.2011.12.001
- Muñoz MA, Liu W, Delaney JA, et al. Comparative effectiveness of fish oil versus fenofibrate, gemfibrozil, and atorvastatin on lowering triglyceride levels among HIV-infected patients in routine clinical care. J Acquir Immune Defic Syndr. 2013;64:254–260.10.1097/QAI.0b013e3182a60e82
Specialty-Related List of the Expert Panel Members
- (In alphabetical order)
- Cardiologists
- AnilBHAT, Max Super Speciality Hospital, New Delhi, Delhi
- Anupam GOEL, Max Hospital, Saket, New Delhi, Delhi
- Arnab DUTTA, Medical College, Kolkata, West Bengal
- Asha MAHILMARAN, Apollo Hospitals, Chennai, Tamil Nadu
- Ashwani MEHTA, Sir Ganga Ram Hospital, New Delhi, Delhi
- Asit KHANNA, Max Hospital, New Delhi, Delhi
- Bharat SHIVDASANI, Jaslok Hospital, Mumbai, Maharashtra
- Bhashar SHAH, Jupiter Hospital, Mumbai, Maharashtra
- Bipin DAXINI, Fortis Hospital, Mumbai, Maharashtra
- Brian PINTO, Holy Family Hospital, Mumbai, Maharashtra
- CV HARI NARAYAN, Sakra World Hospitals, Bengaluru, Karnataka
- Chetan SHAH, Zynova Hospital, Mumbai, Maharashtra
- DS GAMBHIR, Kailash Hospital& Research Institute, Noida, Uttar Pradesh
- HS ISSER, Safdarjung Hospital, New Delhi, Delhi
- H WARDHAN, Primus Super Speciality Hospital, New Delhi, Delhi
- Hygriv RAO, KIMS Hospital, Hyderabad, Andhra Pradesh
- JS MAKKAR, Eternal Hospital, Jaipur, Rajasthan
- Jamal YUSUF, GB Pant Hospital, New Delhi, Delhi
- Joy M THOMAS, Electrophysiologist, Frontier Life Line Hospital, Dr K M Cherian Heart Foundation, Chennai, Tamil Nadu
- Justin PAUL, Madras Medical College, Chennai, Tamil Nadu
- K JAISHANKER, Madras Medical College, Chennai, Tamil Nadu
- M VISHWANATHAN, Jaslok Hospital, Mumbai, Maharashtra
- Mahesh SHAH, Nanavati Hospital, Mumbai, Maharashtra
- Manoj MASHRU, Reliance Hospital, Mumbai, Maharashtra
- Navneet KUMAR, P.D. Hinduja Hospital, Mumbai, Maharashtra
- Neeraj BHALLA, B L Kapoor Hospital, New Delhi, Delhi
- Neeraj PANDIT, DR RML Hospital and PGIMER, New Delhi, Delhi
- Nishith CHANDRA, Fortis, Escorts Hospital, New Delhi, Delhi
- Partik SONI, Wockhardt Hospital, Mumbai, Maharashtra
- Pramod KUMAR, Paras HOSPITAL, Patna, Bihar
- RD YADAV, Batra Hospital, New Delhi, Delhi
- RR MANTRI, Sir Ganga RamHospital, New Delhi, Delhi
- Rajeev AGARWAL, Jaswant Rai Speciality Hospital, Meerut, Uttar Pradesh
- Rajiv KARNIK, Fortis Hospital, Mumbai, Maharashtra
- Rajneesh KAPOOR, Medanta The Medicity Hospital, Gurugram, Haryana
- Rajneesh SARDANA, Jaypee Hospital, Noida, Uttar Pradesh
- Rohit KAPOOR, Carewell Hospital & Superspeciality Hospital, Amritsar, Punjab
- SK GUPTA, Indraprastha Apollo Hospitals, New Delhi, Delhi
- S MUKHOPADHYAY, G B PANT Hospital, New Delhi, Delhi
- Sadanand SHETTY, D Y Patil, Mumbai, Maharashtra
- Sameer KUBA, Narinder Mohan Hospital& Heart Center, Ghaziabad, Uttar Pradesh
- Sameer MEHROTRA, B L Kapoor Hospital, New Delhi, Delhi
- Sameer SHRIVASTAVA, Fortis Escorts Hospital, New Delhi, Delhi
- Samir PAGAD, Bombay Hospital, Mumbai, Maharashtra
- Sandeep BANSAL, Safdarjung Hospital, New Delhi, Delhi
- Sanjeev ROY, Fortis Escorts Hospital, Jaipur, Rajasthan
- Santosh Kumar AGARWAL, Kailash Hospital& Research Institute, Noida, Uttar Pradesh
- Satish VAIDYA, JupiterHospital, Mumbai, Maharashtra
- Sunil MODI, Indraprastha Apollo Hospitals, New Delhi, Delhi
- TR MURLIDHARAN, Sri Ramachandra University, Chennai, Tamil Nadu
- UC SAMAL, PMCH, Patna, Bihar
- Ulhas M PANDURANGI, The Madras Medical Mission, Chennai, Tamil Nadu
- VK CHOPRA, MedantaTheMedicity Hospital, Gurugram, Haryana
- Venkatesh TEKUR, Apollo Hospitals, Bengaluru, Karnataka
- Vijaya Chandra REDDY, Apollo Hospitals, Chennai, Tamil Nadu
- Vikas KOHLI, Paedriatic Cardiologist, Indraprastha Apollo Hospitals, New Delhi, Delhi
- Vinayak AGGARWAL, MedantaTheMedicity Hospital, Gurugram, Haryana
- Vivek MITTAL, Max Super Speciality Hospital, New Delhi, Delhi
- Zakia KHAN, FortisHospital, Mumbai, Maharashtra
- Physicians
- AS ISRANI, Private Practitioner, New Delhi, Delhi
- A MURUGUNATHAN, Shristi A. G. Hospital, Tirupur, Chennai, Tamil Nadu
- Abha GUPTA, LLRM Medical College, Meerut, Uttar Pradesh
- Ajay KANTHARIA, Saifee Hospital, Mumbai, Maharashtra
- Alka CHADHA, Hope N Care Hospital, Mumbai, Maharashtra
- Anand MOSES, The Tamil Nadu DR MGR Medical University, Chennai, Tamil Nadu
- Anil CHATURVEDI, Private Practitioner, New Delhi, Delhi
- Anil DAXINI, Jyotirmay Heart Care, Mumbai, Maharashtra
- Arvind GUPTA, Jaipur Diabetes Research Centre, Jaipur, Rajasthan
- Asim KATYAL, Fortis Hospital Vasant Kunj, New Delhi, Delhi
- Avishkar SABHARWAL, Maulana Azad Medical College, New Delhi, Delhi
- BMS LAMBA, RML Hospital and PGIMER, New Delhi, Delhi
- Benny NEGALUR, JupiterHospital, Mumbai, Maharashtra
- Bharat SHAH, Jaslok Hospital, Mumbai, Maharashtra
- Deepak SARMA, Private Practitioner, Kaziranga, Assam
- Dilip KUMAR, Safdarjung Hospital, New Delhi, Delhi
- Girish Chandra VERMA, Kota Medical College, Kota, Rajasthan
- Girish RAJADHYAKSHA, Nair Hospital, Mumbai, Maharashtra
- GovindSHARMA, Private Practitioner, Jaipur, Rajasthan
- Hemant THACKER, Bhatia Hospital, Mumbai, Maharashtra
- Keshav CHATURVEDI, Private Practitioner, Jaipur, Rajasthan
- MPS CHAWLA, RML Hospital and PGIMER, New Delhi, Delhi
- Madhur YADAV, Lady Hardinge Medical College, New Delhi, Delhi
- Meena CHABRA, Private Practitioner, New Delhi, Delhi
- Mukesh JAIN, Private Practitioner, Jaipur, Rajasthan
- NPS OBEROI, CGHS, New Delhi, Delhi
- Om SRIVASTAVA, Jaslok Hospital, Mumbai, Maharashtra
- RD LELE, Jaslok Hospital, Mumbai, Maharashtra
- RR VYAS, Private Practitioner, Jaipur, Rajasthan
- Rajbeer BENIWAL, Private Practitioner, Jaipur, Rajasthan
- Rajesh Kumar MARYA, Sukhmani Hospital, New Delhi, Delhi
- Ram SINGH, Govt. Medical College, Chandigarh, Punjab
- Ramesh DARGAD, Lilavati Hospital, Mumbai, Maharashtra
- Rashmi NANDA, Indraprastha Apollo Hospitals, New Delhi
- Richa GIRI, CSVM Medical College, Kanpur, Uttar Pradesh
- SK AGGARWAL, Indraprastha Apollo Hospitals, New Delhi, Delhi
- SS LAKSHMANAN, Priya Nursing Home, Chennai, Tamil Nadu
- Sanjay GODBOLE, Saifee Hospital, Mumbai, Maharashtra
- Sanjay GUPTA, Private Practitioner, New Delhi, Delhi
- Sanjeev BHALLA, Private Practitioner, New Delhi, Delhi
- Shahid BARMARE, Kohinoor Hospital, Mumbai, Maharashtra
- Sharat KOLKE, Kohinoor Hospital, Mumbai, Maharashtra
- Shoab ZAHEER, Jawaharlal Nehru Medical College, Aligarh, Uttar Pradesh
- Sudhir BALDWA, Private Practitioner, Jaipur, Rajasthan
- Sudhir BHANDARI, S M S Medical College, Jaipur, Rajasthan
- Sunil K MAHAVAR, S M S Medical College, Jaipur, Rajasthan
- Uma KALIA, SBI, Jaipur, Rajasthan
- VD SHARMA, Private Practitioner, Jaipur, Rajasthan
- VS BALDWA, Private Practitioner, Jaipur, Rajasthan
- Vijay NEGALUR, JupiterHospital, Mumbai, Maharashtra
- VIkas AHULWALIA, Max Super Speciality Hospital, New Delhi, Delhi
- Vinod MITTAL, Delhi Heart and Lung Institute, New Delhi, Delhi
- Zoojer RANGWALA, Narayan Hrudyalay, Mumbai, Maharashtra
- Endocrinology
- Ameya JOSHI, Bhakti Vedant Hospital, Mumbai, Maharashtra
- Bhuma SRINIVASAN, Vijaya Hospital, Chennai, Tamil Nadu
- Bindu KULSHRESTHA, RML Hospitaland PGIMER, New Delhi, Delhi
- Deepak KHANDELWA, Maharaja Agarsen Hospital, New Delhi, Delhi
- Girish PARMAR, Kolilaben Ambani Hospital, Mumbai, Maharashtra
- JS WASIR, Medanta The Medicity Hospital, Gurugram, Haryana
- Jayshree GOPAL, Apollo Hospital, Chennai, Tamil Nadu
- Krishna BISWAS, Safdarjung Hospital, New Delhi, Delhi
- Krishna SESHADRI, SRI Ramachandra University, Chennai, Tamil Nadu
- Manoj CHADHA, Hinduja Hospital, Mumbai, Maharashtra
- Pramod GANDHI, Gandhi Hospital, Mumbai, Maharashtra
- Richa CHATURVEDI, Indraprastha Apollo Hospitals, New Delhi, Delhi
- S MURTHY, Endocrine Diagnostic& Research Centre, Chennai, Tamil Nadu
- Sachin JAIN, Lady Hardinge Medical College, New Delhi, Delhi
- SachinMITTAL, Fortis Hospital, Mohali, Punjab
- Sarad KUMAR, Eras Medical College, Lucknow, Uttar Pradesh
- Shalini JAGGI, Private Practitioner, New Delhi, Delhi
- Surinder KUMAR, Sir Ganga RamHospital, New Delhi, Delhi
- Suresh DAMODRAN, Ramakrishna Hospital, Coimbatore, Tamil Nadu
- Gastroentrologists
- Abha NAGRAL, Jaslok Hospital, Mumbai, Maharashtra
- Deepak LOHATI, Max Super Speciality Hospital, Patparganj, New Delhi, Delhi
- Dipak AMRAPURAKAR, Bombay Hospital, Mumbai, Maharashtra
- P BASUMANI, Fortis Malar Hospital, Chennai, Tamil Nadu
- Sanjay JAIN, Indraprastha Apollo Hospitals, New Delhi, Delhi
- Sanjay SIKKA, Indraprastha Apollo Hospitals, New Delhi, Delhi
- Vijay PRAKASH, PMCH, Patna, Bihar
- Nephrology
- Arun SHAH, Arogyanidhi Hospital, Mumbai, Maharashtra
- Ashok KRIPLANI, Bombay Hospital, Mumbai, Maharashtra
- Ashok SARIN, Indraprastha Apollo Hospitals, New Delhi, Delhi
- B Ramesh RAO, Raheja Hospital, Mumbai, Maharashtra
- Edwin FERNANDO, Govt. Stanley Medical College, Chennai, Tamil Nadu
- Hardik SHAH, Bombay Hospital, Mumbai, Maharashtra
- Himanshu MAHAPATRA, RML Hospitaland PGIMER, New Delhi, Delhi
- Jayant Kumar HOTA, Indraprastha Apollo Hospitals, New Delhi, Delhi
- MS AMARESAN, Madras Medical College, Chennai, Tamil Nadu
- N Gopal KRISHNAN, Madras Medical College, Chennai, Tamil Nadu
- Om KUMAR, IGIMS, Patna, Bihar
- Rushi DESHPANDE, Saifee Hospital, Mumbai, Maharashtra
- SC CHHABRA, Max Super Speciality Hospital, Patparganj, New Delhi, Delhi
- Vijay KHER, EscortsHospital, New Delhi, Delhi
- Neurology
- AK SINGH, IGIMS Patna, Patna, Bihar
- AV SRINIVASAN, TheTamil Nadu DR MGR Medical University, Chennai, Tamil Nadu
- Dinesh KHANDELWAL, S M S Medical College, Jaipur, Rajasthan
- KS ANAND, DR RML Hospital and PGIMER, New Delhi, Delhi
- U MEENAKSHISUNDARAM, Sri RamachandraUniversity, Chennai, Tamil Nadu
- Pankaj HANS, PMCH Patna, Patna, Bihar
- Sanjay SAXENA, Max Super Speciality Hospital, New Delhi, Delhi
- Lipidologists
- Anvay MULE, CVTS, Heart Transplant Wockhardt Hospital, Mumbai, Maharashtra
- Krishan KEWAL, CTVS Max Super Speciality Hospital, Saket, New Delhi, Delhi
- Vascular Surgery
- Rakesh MAHAJAN, Indraprastha Apollo Hospitals, New Delhi, Delhi
- N Shastri, Indraprastha Apollo Hospitals, New Delhi, Delhi
- Rheumatologists
- V KRISHNAMOORTHY, Eenakshi Mission Hospital, Chennai, Tamil Nadu
- Infectious Diseases (HIV)
- D Suresh KUMAR, Apollo Hospitals, Chennai, Tamil Nadu
- Nalin NAG, Indraprastha Apollo Hospitals, New Delhi, Delhi
- Genetic Diseases
- IC VERMA, Sir Ganga Ram Hospital, New Delhi, Delhi