
ABSTRACT
Objectives: The prognostic, diagnostic, and therapeutic potential of the human gut microbiota is widely recognised. However, translation of microbiome findings to clinical practice is challenging. Here, we discuss current knowledge and applications in the field.
Methods: We revisit some recent advances in the field of faecal microbiome analyses with a focus on covariate analyses and ecological interpretation.
Results: Population-level characterization of gut microbiota variation among healthy volunteers has allowed identifying microbiome covariates required for clinical studies. Currently, microbiome research is moving from relative to quantitative approaches that will shed a new light on microbiota–host interactions in health and disease.
Conclusions: Covariate characterization and technical advances increase reproducibility of microbiome research. Targeted in vitro/in vivo intervention studies will accelerate clinical implementation of microbiota findings.
Introduction
Due to its accessibility and clear association with host health, the faecal microbiota has been intriguing microbiologists for centuries. Even the Father of Microbiology, Antoni van Leeuwenhoek, could not resist the temptation of studying his faeces under one of his revolutionary microscopes, providing the first description of what is now believed to be Giardia intestinalis. Notwithstanding this continuing fascination, our knowledge and understanding of the composition and metabolic potential of the microbial communities in the colon has been – until recently – fairly limited. In the early days of modern microbiology, the traditional identification and characterization of faecal microorganisms was hampered substantially by what is commonly referred to as a ‘cultivation bias’. While some researchers considered this bias to result from a lack of dedication and creativity in isolation efforts rather than true unculturability of colon bacteria, an estimated 80% of faecal microorganisms was at some point considered uncultivable by classic microbiology [Citation1]. It was not until the introduction of molecular techniques (including DNA amplification/separation through gradient electrophoresis and nucleotide probing) that the black box of the colon ecosystem was opened and scientists finally caught a glimpse of its overwhelming microbial diversity. Recently, advances in sequencing technologies have accelerated microbial ecosystem research even further, allowing the study of human-body-associated microbial communities at a previously unseen scale and resolution [Citation2]. Fuelling interest and investment in microbiome research is the putative contributing role of human-associated microbial communities to emergent diseases permeating western societies, including inflammatory bowel disease, type II diabetes, and autoimmune diseases such as rheumatoid arthritis and multiple sclerosis [Citation3].
Currently, two approaches shape the narrative of gut microbiome research, most frequently using faecal samples as proxies for the colon ecosystem [Citation4]. The first – amplicon sequencing – allows the study of bacterial, archaeal, and even micro-eukaryotic composition of the faecal microbiota up to species level. The technique relies on sequencing of taxonomic marker genes (usually 16S rRNA genes) after initial targeted PCR amplification. The benefits of well-established analytical pipelines and fairly cheap sample processing come with a few limitations. Most notably, amplicon sequencing provides no information on metabolic potential, and library preparation biases exaggerate the relative abundance of dominant taxa. The second approach, metagenomic shotgun sequencing, enables the assessment of not only the composition but also functionality of the gut microbiota. In contrast with amplicon sequencing, shotgun procedures provide sequence information on a randomly picked set of DNA fragments extracted from a faecal sample while attempting to restrict amplification biases to a minimum. In spite of decreasing sequencing costs, the latter approach remains relatively expensive and significant expertise is required for bioinformatic processing and data analysis. The wealth of information generated by metagenomic sequencing of faecal microbiomes is far from trivial to tackle; therefore, untargeted, data-driven studies have tended to confirm obvious findings. However, published metagenomic datasets provide a rich reference for future studies to revisit. The design of analytical frameworks that allows distilling hypothesis-relevant information out of metagenomics datasets in a timely and user-friendly way is one of the key challenges in future microbiome research [Citation5,Citation6].
The colon ecosystem
Of the estimated total of 100 trillion microbes living in and on the human host, a vast majority resides in the large intestine [Citation7]. One gram of human faeces contains an impressive total of 100 billion microorganisms. It should be noted that the human colon is an open environment, subject to a regimen of intermittent nutrient fluxes composed of (partially) undigested food ingredients, comparable to a plug flow reactor. In such reactor system, contents travel in (semi-)discrete entities in the axial direction of a tubular system, allowing for each plug to have a different composition than the previous or next one. Together with the strict surveillance imposed by the host immune system, this plug flow regimen largely defines the composition of the colon ecosystem [Citation7,Citation8]. First, to avoid wash-out through egestion, colon microorganisms are required to either maintain a sufficient duplication rate or to adhere to/colonize host intestinal tissues [Citation9,Citation10]. Second, both the gradients of nutrient composition and availability combined with matrix heterogeneity structurally define the colon environment. One of the major intestinal gradients is linked to the influx of undigested carbohydrates exploited by microorganisms in the proximal colon for saccharolytic fermentation, high replication rates, and acidification of the luminal environment. Upon depletion of readily fermentable substrates, the colon microbiota turns toward a more proteolytic metabolism [Citation9,Citation11], with slower growing taxa taking over from initial colonizers [Citation9,Citation10]. Alongside these metabolic processes of qualitative and quantitative changes in nutrient availability, water (re-)absorption results in incomplete mixing of the luminal content. This induces the formation of substrate as well as end-product gradients in the colon ecosystem, creating micro-niches and promoting microbial diversification [Citation8].
Immune system surveillance of the gut microbiota is only one aspect of the complex bidirectional communication processes taking place between both partners. The gut ecosystem plays an important role in both the development and maturation of the gut-associated lymphoid tissue (GALT) and conditions many immune-associated actors such as regulatory T-cells (Treg) [Citation12–Citation15]. The GALT system has unique elements that promote tolerance of commensal microorganisms and keep pathogenic microbes in check. Like the primary digestive functions of the intestinal system, the GALT structure differs along the continuum of the gastrointestinal tract [Citation16]. The small intestine has an increased abundance of Paneth cells, Peyer’s patches, and dendritic cells, which sample luminal antigens to survey, signal, and interact with many regulatory actors to maintain the integrity of the epithelial barrier during nutrient absorption [Citation17]. Some offensive strategies of the gastrointestinal tract include the production of antimicrobial peptides, defensins, and immunoglobulin A (IgA) which target invading pathogens [Citation18,Citation19]. In the colon, where the highest concentration of gut microorganisms resides and water reabsorption primarily occurs, the immunological strategy consists of increased production of antimicrobial mucous by goblet cells [Citation16]. Recently, IgA in mucous was seen to contribute to the microbiome composition, as commensal microorganisms like Bacteroides fragilis attach to the peptide [Citation20]. Such processes would provide a basis for stable colonization and prevent pathogenic microorganisms from embedding into the mucosal barrier. Additionally, commensal microorganisms produce metabolites, like butyrate, to ensure that the constitutively active Treg cells do not trigger an adaptive immune response and subsequent chronic inflammation [Citation21,Citation22].
Given the transition from the aerobic exterior to the anaerobic intestinal environment, microbial nutritional specificities, continuous interactions with the human host’s (innate) immune system, and digestive processes, the phylogenetic architecture of the colon microbiota unsurprisingly demonstrates characteristics of microorganisms that have undergone bottleneck selection prior to habitat colonization [Citation23]. Such selection processes result in a limited and isolated community of initial colonizers that subsequently spread and diversify extensively whenever colonic conditions are in their favour. Indeed, while total bacterial/archaeal richness of the gut microbiota in a western population has been estimated to comprise up to 800 genera, the phylogenetic diversity of the overall composition is fairly shallow as most taxa belong to only five phyla [Citation24]. Population-level relative microbiome profiling efforts have established two main axis of phylogenetic and functional diversification of the colon microbiota, shared among both healthy and diseased individuals [Citation24,Citation25]. The first axis represents phylum-level differences in the relative abundances of Gram-positive Firmicutes and Gram-negative Bacteroidetes. Variation along a second axis is defined by a genus-level shift in the Bacteroides:Prevotella ratio – the two dominant genera composing the Bacteroidetes phylum in the gut ecosystem. Defining associations between microbiome variation and host health status have been shown to be far from straightforward, as conflicting findings resulting from technical variation between studies, cohort size limitations, and inadequate confounder control in study design muddle interpretations. As a result, while the phylogenetic landscape of microbiome variation appears well-characterized, current understanding of host and environmental covariates, drivers, and responders of colon microbial ecosystem variation is lagging behind.
Covariates of faecal microbiota variation
For many years, disease association studies have dominated faecal microbiome research. While such studies incited technical and computational developments that are currently facilitating metagenome analyses, they failed to meet their primary objective – being the identification of microbiome-based diagnostic markers or clinical targets for intervention. Although microbiome-based signatures have been put forward for a broad range of pathologies (for an overview of microbe disease associations, we refer to online resources such as Disbiome [Citation26] and the Human Pan-Microbe Communities Database [Citation27]), a staggering lack of reproducibility hampered research translation into clinical practice. The apparent lack of consistency in faecal microbiome findings revived the field’s interest in defining the boundaries of gut ecosystem variation in healthy hosts [Citation3]. This resulted in targeted efforts to characterize specific confounder effects as well as explorative population-wide cohort studies, extensively investing in broad phenotypic characterization of their participants in order to identify potential microbiome covariates. Nevertheless, the cumulative, non-redundant explanatory power of identified health and lifestyle variables on inter-individual microbiota diversification has recently been estimated to be less than 8% – suggesting that the forces shaping the gut ecosystem remain elusive despite numerous large-scale efforts worldwide [Citation24]. Among those established variables with a significant correlation with microbiome variation are some usual suspects such as age, gender, and body mass index (BMI; ) [Citation24,Citation25]. As expected, diet impacts microbiome variation, with for example fruit consumption frequency and bread-type preference listed among top covariates. Population-wide microbiome analyses also re-emphasized the need to keep track of drug intake when studying microbiome composition and metabolic potential – a finding that was recently exemplified by metformin-related findings in type II diabetes studies [Citation28]. Compared to lifestyle and anthropometric covariates, the impact of host genetics on microbiota composition has been reported to be only minor [Citation29]. Somewhat surprising was the identification of stool consistency, described using the Bristol Stool Score (BSS), as the single covariate with largest effect size in microbiome composition variation among healthy individuals [Citation10,Citation24].
Figure 1. Lifestyle and clinical variables correlated to faecal microbiota composition in the average Flemish population. (a) Multivariate model best explaining the faecal microbiota composition of the first 1106 participants [Citation24] of the Flemish Gut Flora Project (FGFP; dbRDA on genus-level Aitchison distance). The eleven variables significantly contributing to the model are listed from top to bottom until the AIC cut-off (dashed line), with their cumulative explanatory power in dark barplots (dbRDA R2). The total cumulative explanatory power of the best model sums to 4.8% (red stacked barplot). The variables’ individual explanatory power is also provided (lighter colour barplot). (b) Principal coordinate analysis depiction of FGFP faecal microbiota variation, with individuals coloured according to the samples’ enterotypes (DMM method), and arrows representing the effect size and direction of the three highest microbiome covariates in the FGFP: stool consistency (Bristol Stool Score), age, and gender.
![Figure 1. Lifestyle and clinical variables correlated to faecal microbiota composition in the average Flemish population. (a) Multivariate model best explaining the faecal microbiota composition of the first 1106 participants [Citation24] of the Flemish Gut Flora Project (FGFP; dbRDA on genus-level Aitchison distance). The eleven variables significantly contributing to the model are listed from top to bottom until the AIC cut-off (dashed line), with their cumulative explanatory power in dark barplots (dbRDA R2). The total cumulative explanatory power of the best model sums to 4.8% (red stacked barplot). The variables’ individual explanatory power is also provided (lighter colour barplot). (b) Principal coordinate analysis depiction of FGFP faecal microbiota variation, with individuals coloured according to the samples’ enterotypes (DMM method), and arrows representing the effect size and direction of the three highest microbiome covariates in the FGFP: stool consistency (Bristol Stool Score), age, and gender.](/cms/asset/49fd15fb-3e9f-4386-8bb9-fcff29f2de6d/yacb_a_1583782_f0001_oc.jpg)
The BSS scale was developed as an instrument to categorize stool consistency by visual description, as a proxy to study large intestinal transit time [Citation30]. Previous research had firmly linked stool consistency to several of microbiome covariates like age, gender, BMI, and diet [Citation31]. Moreover, the use of certain medication – like metformin [Citation32] – has been known to affect stool firmness. In fact, transit time as estimated through faecal consistency reflects a bidirectional relationship between the microbiota and the host. While host health and medication can impact transit time, individuals also actively respond to the alterations in stool consistency by taking medication or adjusting their diet and lifestyle habits to modulate their intestinal passage rates. Reversely, the microbiota has been shown to influence transit time, through its metabolic action (production of gasses and short chain fatty acids) and communication with the human host, while co-varying with host health status, lifestyle, and medication (). The impact of transit time on microbiota composition and activity can again be understood by analogy to the plug flow reactor [Citation8]. In healthy individuals, the faecal microbial community can be considered as an ecosystem under development along its transit – a development process initiated at ingestion, but dominated by microbial transformations from the moment the chime passes the ileocaecal valve. Within the colon habitat, the ecosystem is subject to a maturation process, eventually abruptly halted by egestion. The duration of this maturation process is dependent on transit time, which determines the degree of nutrient depletion, water content, and microbial end-product concentration in the resulting faecal matrix. Moreover, colon residence time determines which bacteria will be able to proliferate in the large intestinal lumen in function of their growth rates [Citation9,Citation10].
Figure 2. Transit time is at the intersection of human health, lifestyle, and gastrointestinal microbiota. Transit time, and its proxy stool consistency, can be shaped by disease and medication, diet and physical activity, and microbial metabolism. Reciprocally, transit time is a key parameter reflecting the maturation of the colon ecosystem in healthy individuals, along which water is reabsorbed and nutrients are depleted, leading to rearrangements in microbial metabolism and the fitness of different microbiota fractions.
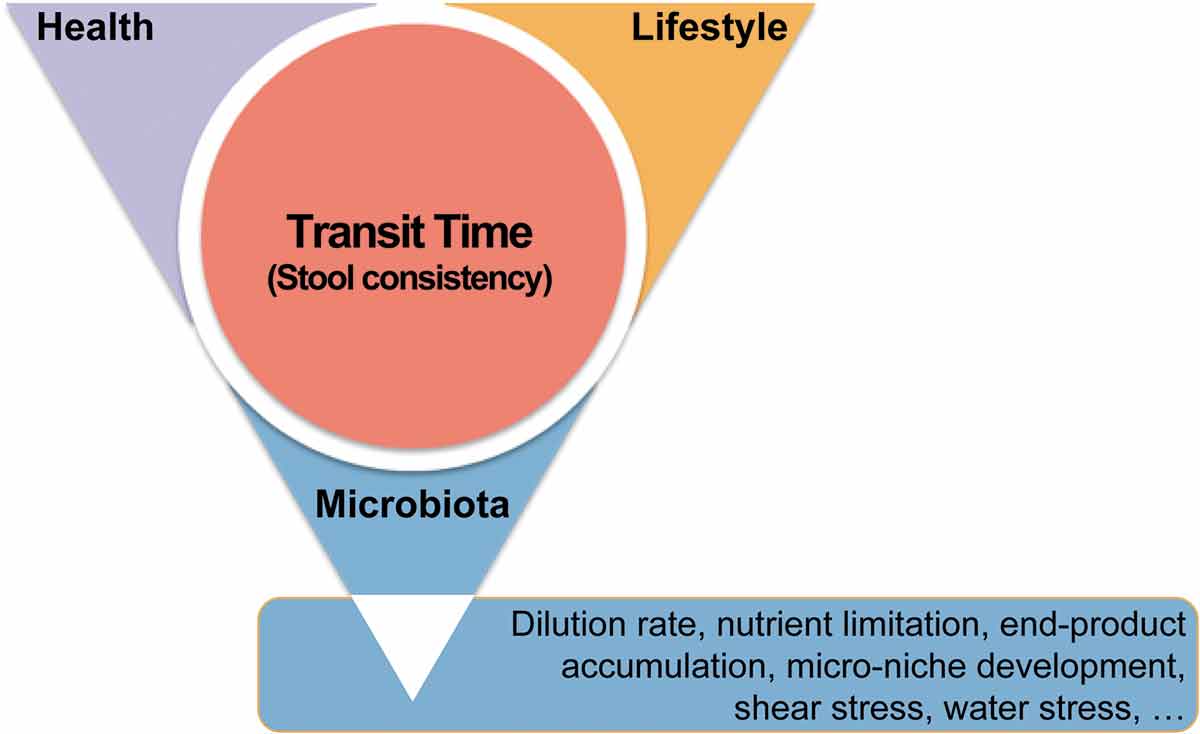
Overall, while population-wide analyses have identified several covariates of microbiome variation that need to be taken into account when setting up studies in a clinical context, they also provided a transit-time-dependent ecological framework that dominates microbiota variation among healthy individuals. Any indication of disease-associated microbiota alteration should be contrasted against this ecological background, to verify whether the fluctuations observed are not merely resulting from (coincidental or symptomatic) variation in transit time.
Enterotypes of the human colon ecosystem
Enterotypes have been described as more densely populated areas in a multidimensional space of faecal microbiota community composition [Citation33,Citation34]. In light of the interindividual differences observed in taxonomic and functional microbiota variation over time, the enterotype concept is built on the observation that several statistical clustering methods reproducibly established a limited number of prevalent community configurations in microbiome space. Approaches such as enterotyping aim at reducing the complexity of microbiome space into strata or categories, thus facilitating analyses aiming to link overall community variation to host health status or environmental variables. Immediately after the concept was launched, the existence of enterotypes was the subject of intense scientific debate. Unfortunately, the scientific enterotype argument rather quickly narrowed down to an off-topic discussion regarding the discrete or continuous nature of microbiota variation [Citation35] – although no claim on discreteness of enterotype configurations was expressed in the original publication [Citation33]. In light of the heated controversy, the concept of community-type microbiome stratification was somewhat abandoned by a part of the gut microbiome research community. More recently however, alternative strategies in community-type clustering of stool samples have been proposed and the research community has reached a consensus about the value of such ‘reductionist’ models. Not relying on between-sample distance measures, a Dirichlet Multinomial Mixtures (DMM) modelling approach has been shown to allow robust identification of enterotypes across datasets [Citation36]. Rather consistently, it allows identifying four enterotypes (), not only aligning with the main axes of gut microbiota described above, but also reflecting community richness variation and corresponding with different stages of ecosystem maturation [Citation24,Citation37]. Labelled according to their principal taxonomic identifiers, DMM community typing allows to distinguish a Ruminococcaceae (R), Prevotella (P), Bacteroides1 (B1), and Bacteroides2 (B2) enterotype. Dominated by Firmicutes species, the R enterotype is prevalent in hard stools, displaying high rates of proteolytic fermentation and populated by a relatively large fraction of slower growing bacteria. The enigmatic P microbiota constellation is associated with loose stools and an agrarian diet [Citation38] but has also been reported to be more prevalent among for example individuals suffering from rheumatoid arthritis [Citation38], while the B2 enterotype has been found to have an 80% prevalence in patients suffering from inflammatory pathology like Crohn’s disease [Citation37]. B1 is the most common enterotype in healthy populations adopting a western diet. It not only harbours a large fraction Bacteroides spp., but also contains a relatively high proportion of Faecalibacterium – a genus that has been the focus of intensive research efforts given its anti-inflammatory properties [Citation39]. The decreased relative abundance of Faecalibacterium distinguishes B2 samples from their B1 counterparts, in agreement with the depletion of Faecalibacterium in faecal samples of Crohn’s Disease patients. Regardless of any ongoing remaining debate on the nature of enterotypes [Citation34], these observations not only highlight the value of categorizing samples as a powerful tool in explorative microbiome analysis and emphasize its potential in cohort stratification in (clinical) study design, but also demonstrate how the enterotyping concept evolved from rather arbitrary classifications to biologically relevant community characterization.
A practical approach to microbiome research
A key aspect in studying the gut microbial ecosystem is the acquisition of representative samples. Meal frequency and peristaltic propulsion subdivide bowel contents in several semi-discrete entities all representing a microbial ecosystem at a different stage of maturation, distributed longitudinally over the large intestine [Citation8]. In practice, only the final phases of this ecosystem maturation process – the faecal material egested – can be obtained in a truly non-invasive manner. More invasive methods such as collecting gut lining biopsies remain mostly reserved for diagnosing or monitoring medical conditions and are less commonly applied in healthy individuals. For explorative purposes, the faecal microbiota is considered an adequate surrogate to study microbial maturation processes taking place within the large intestine.
With the development of the gut microbiomics research field, both research teams and commercial providers have globally presented a plethora of sampling methods and preservation protocols for faecal material [Citation40]. Setting up a microbiome monitoring project thus warrants careful evaluation of the available options, especially in light of the fact that not all protocols developed ensure a similar degree of reproducibility. Researchers can choose to obtain faecal samples from study participants using a variety of collection devices, including specially designed toilet collection devices, storage tubes, swabs, transport vials with stabilizing buffer, paper matrices laced with cell lysing and nucleic acid stabilizing buffers, or recipients that pre-aliquot faecal material to facilitate subsequent analyses. Each of these collection methods has its particular strengths and limitations. Differences in user friendliness, price, storage space, downstream processing requirements, and the amount of collected sample impact the degree of applicability to study objectives. However, of the aforementioned features of each sampling method, the most important to keep in mind when determining which one to use is the preservation method, since the manner in which the sample is preserved has the largest impact on the quality of the resulting microbiome profiles. The microbial signature of a sample needs to be preserved to such extent that relevant conclusions can be drawn regarding the research question. Given the small effect sizes of environmental parameters on gut microbiota composition, there is almost no room for concessions regarding quality aspects in explorative research [Citation41]. Based on protocols developed in tissue and culture preservation, the current gold standard is to freeze faecal material at –20°C immediately after egestion, with posterior storage at −80°C. Several studies have shown that microbiota composition as evaluated by 16S RNA gene sequencing does not change significantly when samples are stored at –80°C over the course of months or years. Besides amplicon sequencing, frozen faecal material can be used for metagenomic (DNA), metatranscriptomic (RNA), proteomic (protein), and metabolomic (metabolite) analyses [Citation40]. However, as −80°C freezing protocols are rarely compatible with home sampling protocols commonly applied in large-scale microbiome research, several alternative conservation strategies have been explored [Citation42]. Refrigeration for a shorter period of time (up to 24 h) seems to induce limited variation. The lower temperature slows down bacterial growth and thus limits deviations from the original composition. As refrigeration slows down fermentation reactions, it allows subsequent metabolomic analyses. Yet, storage time should be restricted to a few hours and it should be noted that volatile components might be lost during storage [Citation43]. Several stabilisation buffers have been explored with varying degrees of success, in the hope of reducing the variation in microbiota composition induced by room temperature (RT) storage. In first instance, solutions used in other bio-medical applications, such as Tris-EDTA, RNAlater, Carry Blair medium, and ethanol in various ranges of concentration were tested or directly applied to studies. Later, specifically designed transport vials entered the market and quickly found their way into research projects. While some stabilisation buffers do succeed in reducing variation in microbiome composition, none of them completely addresses the issue. Nonetheless, some can be useful, as a certain degree of storage-induced deviation from the original microbiota composition can be allowed, depending on the estimated effect size of the parameter investigated and the study sample size. It is however important to keep in mind that specific deviations can be attributable to the applied protocols. Indeed, the statistical implications of a procedure generating random noise are far more manageable than procedures inducing specific alterations in taxa abundances or diversity estimates. An altered detection of key species might for example lead to misinterpretation in disease research. As a case in point, an increased abundance of Proteobacteria has been reported for Tris-EDTA buffer, RT transport vials, or faecal swabs, and is thought to be a result of the aerobic storage conditions. This is particularly worrisome given the resemblance of these changes with reports of diet-induced variation or disease-associated microbiome signatures. Faecalibacterium, Bifidobacterium, and Verrucomicrobia (comprising Akkermansia) are other important taxa related to health and disease that are regularly reported to differ from the gold standard as a result of inadequate choice of sample preservation. The use of stabilization buffers further limits the amount of applicable techniques, such as proteomics, metabolomics, and culture-based approaches, which are rapidly gaining importance for follow-up functional studies. As a result, researchers must carefully consider the preservation methods used in order to obtain valuable results and make the most of their sampling effort [Citation40].
The issue of compositionality
The apparent lack of explanatory power of rather obvious contributors to microbiome variation such as diet has been speculated to arise from the impact of currently unknown factors, stochastic effects, and/or biotic interactions [Citation24]. However, a more straightforward explanation could just be found in the inadequacy of matching relative microbiome data to quantitative host read-outs on diet, lifestyle, physiology, and health. Although the term ‘quantitative metagenomics’ has been appropriated repeatedly to describe sequencing protocols implementing less-biased library preparation procedures, most – if not all –metagenomics finding are based on relative microbiome profiles. Current gold standard sequencing procedures indeed do not permit the determination of taxa abundances per gram of stool, but only provide proportional information on the fraction of the microbiota belonging to a particular genus in the sequenced library. The proportional nature of sequencing data does not facilitate interpretation of microbiome findings. In case of substantial variation of microbial load between faecal samples, changes in proportional abundances do not provide any information on the directionality of observed shifts in terms of absolute bacterial numbers. When studying microbial co-occurrence, positive correlations are often overlooked in proportional datasets while the relative nature of the data induces false co-exclusion patterns (if the proportion of one fraction increases, other fractions are bound to decrease – although their absolute numbers might be maintained). Similarly, linking variation in relative microbial abundances to quantitative data on faecal, urine, or serum metabolite concentrations has been shown to be far from trivial. Above all, relative sequencing approaches simply ignore the fact that changes in total microbial density in faecal samples might be a key feature of a dysbiotic ecosystem and therefore a crucial element in microbiome-based diagnostics. Hence, the development of quantitative methods was considered one of the major challenges in microbiome research.
The issues related to dealing with relative or compositional datasets are widely recognised among microbiome scientists. A variety of statistical methods have been proposed and implemented to reduce the risk of identifying false positive signals in metagenomics datasets [Citation44]. However, none of these techniques recover the quantitative information lost during the various technical steps that make up most of current DNA-extraction, library preparations, and (pooled) sequencing protocols. In order to trade in ratios for counts and enable genuine characterization of host–microbiota interactions, we have recently developed a quantitative microbiome profiling (QMP) protocol that allows bypassing the issue [Citation37]. Combining microbiome sequencing with flow cytometry enumeration of microbial cells, this novel workflow presents a straightforward and readily implementable method to quantitatively assess microbiota variation across individuals, over time, associated to a diseased or healthy state, or resulting from disease treatment. Initial QMP results demonstrate the important extent to which relative approaches have distorted our current perception of gut ecosystem composition. The observed up to tenfold inter-individual variation in faecal microbial loads seen among healthy controls was shown to substantially drive gut ecosystem variation: while the R, P, and B1 enterotypes are characterized by relatively high microbial loads, the B2 community type mostly comprises low-density samples. Of note, the integration of microbiota cell counts affects taxa correlations with physiological metadata. As the current scientific consensus on host–microbiome associations is largely based on the analysis of such correlations, the introduction of QMP can be expected to lead to a paradigm shift in clinical microbiome research. As a case study, we re-analysed faecal samples collected in a Crohn’s Disease cohort and found disease-associated microbiota dysbiosis to be far more striking in a quantitative setting than generally assumed based on relative read-outs. QMP not only showed that about half of the genera composing a healthy gut ecosystem were suppressed in CD patients, but also showed that lowered microbiota richness – considered a key microbiome signal in inflammatory bowel diseases – mainly resulted from the up to 50-fold (!) reduction in microbial cell count observed in diseased individuals. Overall, these preliminary QMP results clearly indicate that microbial load is a key feature of microbiota (dysbiosis) signatures, with implications for microbiome-based prognostic, diagnostic, and therapeutic applications.
Microbiome-based diagnostics
Detailed monitoring of the communities present in faecal material has linked alterations in the gut microbiota composition to pathological states as well as chronic suboptimal health and well-being. The establishment of these associations has caused tremendous excitement in the fields of both microbiology and clinical diagnostics. A previously unwitnessed gold-rush for specific microbiome-based biomarkers or signatures linked to a wide range of pathologies is ongoing and a plethora of disease-focused, medium-scale studies, attempting to link particular taxa, bacterial genes, and metabolic pathways to a targeted health condition, have already been published [Citation26,Citation27]. Differences in faecal microbial community diversity, composition, and functionality have been associated with Crohn’s disease, ulcerative colitis, obesity, type 2 diabetes, cardiovascular disease, depression, and colon cancer, among others. These findings have clearly demonstrated the potential of faecal microbiome-based biomarker discovery.
While biomarker discovery would not necessarily be affected by the compositional nature of microbiome datasets, pathology-associated fluctuations in transit time and faecal microbial load might affect diagnostic specificity and reproducibility. As stool consistency has been identified as the top microbiome covariate among healthy individuals, inadequate tracking of consistency fluctuations between cases and controls could yield signatures purely driven by transit time. Given the correlation between stool consistency and microbial loads [Citation10,Citation45] – with loose stool containing less bacteria than their firm counterparts in healthy controls – care needs to be taken regarding detection of less abundant taxa. Relative microbiome profiling analytical methods generally include a downsizing or rarefaction step. During these procedures, all samples are trimmed down to a similar sequencing depth, independently of the number of bacteria present in the samples analysed [Citation37]. While rarefying is commonplace in ecology, it is considered inadequate when samples are drawn from ecosystems with markedly different population densities and species abundance distributions such as the faecal microbiota. In disease association studies, rarefying microbiome profiles to even sequencing depth results in markedly different sampling depths, with loose stools being sequenced much more deeply than harder ones (). These differences in sampling depth affect the probability of detection of less abundant taxa, which end up detected more frequently in samples with lower microbial loads. Reproducibility of diagnostic signatures based on the presence or relative abundances of such taxa will thus be highly dependent of technical aspects of the protocols applied. In contrast, QMP analyses suggest to downsize microbial profiles to even sampling depth, as calculated based on the number of sequencing reads generated and the microbial load present in the original sample. Hence, we expect biomarker development – particularly their reproducibility and validation across datasets – to benefit substantially from consistent application of QMP in disease association studies, accelerating the translation of microbiome-based diagnostic signatures to a clinical setting.
Figure 3. Comparison of faecal microbiota relative and quantitative microbiota profiling. The middle panel depicts the relative faecal microbiota profiles (RMP) of 40 healthy individuals [Citation37], while the right panel depicts the corresponding quantitative microbiota profiles (QMP), as obtained by combining 16S RNA amplicon sequencing with total microbial load quantification by flow cytometry. Substantial differences in microbial cell density in faecal samples translate in uneven sampling depth after equimolar pooling and sequencing in RMP protocols (left panel), with oversampling of low microbial load samples.
![Figure 3. Comparison of faecal microbiota relative and quantitative microbiota profiling. The middle panel depicts the relative faecal microbiota profiles (RMP) of 40 healthy individuals [Citation37], while the right panel depicts the corresponding quantitative microbiota profiles (QMP), as obtained by combining 16S RNA amplicon sequencing with total microbial load quantification by flow cytometry. Substantial differences in microbial cell density in faecal samples translate in uneven sampling depth after equimolar pooling and sequencing in RMP protocols (left panel), with oversampling of low microbial load samples.](/cms/asset/1c8e57a6-d9eb-43ad-bbad-09395fc77faf/yacb_a_1583782_f0003_oc.jpg)
Microbiota modulation strategies
Beside its diagnostic potential, the intestinal microbiota has been put forward as a target for both preventive and therapeutic interventions to maintain or restore host health. In the clinical field, the rationale behind such modulation strategies can mostly be traced back to the concept of a dysbiotic ecosystem state that would fail to perform its commensal role and contribute to disease development or activity. This (stable) microbiota configuration has been proposed to increase the host’s susceptibility to disease development, (co-)initiate or sustain inflammatory processes affecting host health, or create an environment allowing (opportunistic) pathogens to thrive in an otherwise unwelcoming habitat. However, moving away from disease prevention or treatment, the gut microbiota has also been speculated to contribute to host suboptimal health or well-being. In this case, interventions do not aim to tilt the gut ecosystem equilibrium away from a dysbiotic state, but attempt to enhance host physical or mental fitness through fine-tuning of gut microbial composition and metabolism. Of note, any intervention in gut microbiota composition or metabolic activity to affect host health would require a thorough mechanistic understanding of underlying host–microbiota interactions promoting the effects envisaged. While several metabolic, endocrine, immune, and neural communication lines have been shown to be implicated in bidirectional host–microbiota interactions [Citation46], current understanding of these processes remains largely insufficient to allow development of novel designer modulation strategies.
Among the currently available microbiota modulation strategies, drug-based interventions could appear to be the most straightforward and deterministic. However, while the impact of medication on the gut microbiota has been established beyond doubt, knowledge on the specific alterations induced on the ecosystem by specific drugs is still limited. The current knowledge on the impact of drugs on microbiota composition and metabolic activity mostly originate from observed microbial ‘side-effects’ in patient cohort studies, making it hard to distinguish between the direct effect of the active ingredient on the large intestinal microbiota and its impact on host physiology associated to disease treatment or secondary effects. However, both the research community’s and the pharmaceutical industry’s interest in drug–microbiota interaction are growing, with specific intervention studies in healthy volunteers being set up [Citation47]. In parallel, several research teams are engaging in large scale in vitro studies to elucidate the effect of therapeutic molecules on intestinal isolates [Citation48].
Non-drug-based intervention strategies aim at modulating the gut microbial ecosystem by introducing or promoting growth of beneficial bacteria or bacterial consortia. Such approaches range from dietary interventions to the substitution of a dysbiotic bacterial community by a health-associated microbiota through faecal transplantation. A long-standing dietary strategy include the use of probiotics – live microorganisms that, when administered in adequate amounts, confer a health benefit to the host [Citation49]. Although probiotic research was previously mainly – although not exclusively – focused on bifidobacteria and lactic acid bacteria, more recently, a revived interest has led to the development of what has been termed ‘next-generation probiotics’. Rather broadly defined as probiotics that have not been coined as health-promoting to date [Citation50], this category comprises several taxa that are currently being evaluated as potential agents to counter obesity-associated comorbidities (Akkermansia) or intestinal inflammation (butyrate producers such as Faecalibacterium and Butyricicoccus). Another traditional intervention avenue was the use of prebiotics – substrates that are selectively utilized by host microorganisms conferring a health benefit [Citation51]. Recent sequencing-based research has been applied to confirm their impact on the colon microbiota, as exemplified by the confirmation of the bifidogenic nature of inulin [Citation52]. While health-claims on fermentable fibres nowadays mostly emphasize host responses – such as their contribution to normal bowel function by modulating stool frequency – ecological insights in gut ecosystem development as described above might just well prelude a return to phrasings that assert beneficial effects on the intestinal microbiota.
Faecal microbiota transplantation
Faecal microbiota transplantation (FMT, also known as faecal infusion, faecal microbiota transfer, or bacteriotherapy) is a treatment whereby faeces from a healthy donor are transferred/transplanted to an unhealthy person with curative intent. The rationale behind FMT stems from the principle of engrafting the microbiota from a healthy donor into a patient to re-introduce or re-establish a stable environment that promotes the growth of both endogenous microorganisms of the recipient and newly introduced donor species. The first reference to FMT can be found in traditional Chinese medicine, were faecal enemas were used for treating enteric pathology [Citation53]. Following the recent advance in gut microbiome research, a revived interest in FMT can be observed and several administration procedures have been developed. While the optimal FMT delivery protocol remains unknown, administration through colonoscopy or enema has become the most widely applied methods. FMT can also be delivered through the working channel of a gastroscope, or through nasogastric, -jejunal, or gastrostomy tube, though it is not preferred due to safety reasons. Recent reports describe a novel FMT application in the form of frozen capsules that can be administered orally with no apparent side effects and with success rates equivalent to those observed after delivery by colonoscopy. Further development of such methods would allow to avoid invasive gastrointestinal procedures [Citation54]. Other trending topics in current FMT research include the exploration of novel target diseases as well as the formulation of alternative therapies based on defined cocktails of gut micro-organisms – which would not only increase treatment safety, but also facilitate regulatory harmonization [Citation55].
Despite a revived interest in FMT and the development of several administration procedures, the only approved indication for FMT today is refractory Clostridium difficile infection (rCDI) – severe antibiotic-associated diarrhoea, in which FMT treatment has been shown superior to vancomycin administration. This is particularly exciting as 20–35% of patients with CDI will fail initial antibiotic treatment [Citation56–Citation59]. Of those who initially respond, 40–60% will have a recurrence after antibiotic treatment [Citation60,Citation61]. FMT thus fills an important role in the CDI treatment algorithm. In the case of rCDI FMT, the mode of administration seems to have little impact on the overall efficacy. Oral FMT capsules, FMT enemas, and FMT duodenal infusions all produced success rates in the 70–90% range [Citation62–Citation64]. The donor choice also appears of minor importance for efficacy, although potential donors should be screened according to the international consensus guidelines (similar to those for blood donors) to increase safety [Citation65]. This implies that only healthy adults without acute or chronic diseases qualify as stool donors, but no specific microbial profile or enterotype is required. Until now, FMT has shown to be a safe and cost-effective treatment for rCDI, but it should be noted that long-term follow-up data of interventions are currently lacking.
Beyond the treatment of rCDI, FMT has also been investigated in other disorders associated with the alterations of the gut microbiota. In particular, several studies have been set up in patients with ulcerative colitis (UC), Crohn’s Disease (CD), irritable bowel syndrome (IBS), and metabolic syndrome (MS). So far, four randomized controlled FMT trials in active UC patients have been performed with variable, but with overall positive success rates [Citation66–Citation69]. Unlike in rCDI, the efficacy of FMT in the treatment of IBD appears to be influenced by a number of factors, including donor microbial profiles, inflammatory burden of the recipient, microbial diversity of the recipient, FMT preparation procedures, and administration route and frequency. Overall, FMT could become a safe potential treatment for UC, but further investigation is needed to answer several pressing questions, including necessity of pre-selecting donors based on potential treatment effectiveness, of repeated administration, and of strict (anaerobic) preparation of the FMT. In contrast, researchers have been less successful in applying FMT in CD [Citation70]. Several factors have been put forward to explain the discrepancy in FMT effectiveness between both Inflammatory Bowel Diseases. First, the role of mucosa adherent bacteria such as adherent-invasive Escherichia coli is larger in CD pathology, potentially affecting FMT impact. Second, CD is characterized by transmural inflammation and therefore longer treatment cycles of FMT might be required to improve modification of the disease state. Third, CD patients tend to have a much lower baseline microbial load than UC patients, potentially indicating a more pronounced state of dysbiosis.
Regarding IBS, only two small randomized controlled studies have been published so far – with conflicting results emphasizing the need for further investigations [Citation71,Citation72]. For MS, different case reports have been published, and multiple hypotheses have been raised to explain the observed effects, though formal human studies remain scarce. A 2012 study indicated, for example, that the transfer of faeces from a lean donor to treatment-naïve male subjects with metabolic syndrome temporarily increased their peripheral insulin sensitivity. This was positively correlated with an increase in the number of butyrate-producing bacteria [Citation73]. These results are in line with results from animal models [Citation74]. The potential of FMT in management of autoimmune and allergic diseases, neurodevelopmental and neurodegenerative disorders, and chronic fatigue syndrome has been suggested but remains to be explored in more detail [Citation75–Citation80]. Some studies demonstrate encouraging results, but well-designed, randomized controlled trials are needed to establish the efficacy of FMT for all of these diseases.
Conclusions
While no longer in its infancy, gut microbiome research has yet to fulfil its expectations of revolutionizing clinical diagnostics and providing alternative preventive or therapeutic targets for dysbiosis-associated pathologies. Efforts to characterize microbiota variation in healthy individuals on population level have now provided an ecological framework that can be translated into guidelines for study design and interpretation of results. Together with the recent shift from relative microbiota profiles to genuine taxon quantifications, this framework can be expected to accelerate translation of microbiome findings from bench to bedside.
Author contributions
All authors contributed to writing the manuscript and revised and approved the final version for publication.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Lagier J-C, Armougom F, Million M, et al. Microbial culturomics: paradigm shift in the human gut microbiome study. Clin Microbiol Infect. 2012;18:1185–1193.
- Eckburg PB, Bik EM, Bernstein CN, et al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638.
- Bäckhed F, Fraser CM, Ringel Y, et al. Defining a healthy human gut microbiome: current concepts, future directions, and clinical applications. Cell Host Microbe. 2012;12:611–622.
- Valles-Colomer M, Darzi Y, Vieira-Silva S, et al. Meta-omics in inflammatory bowel disease research: applications, challenges, and guidelines. J Crohns Colitis. 2016;10:735–746.
- Falony G, Vieira-Silva S, Raes J. Microbiology meets big data: the case of gut microbiota-derived trimethylamine. Annu Rev Microbiol. 2015;69:305–321.
- Darzi Y, Falony G, Vieira-Silva S, et al. Towards biome-specific analysis of meta-omics data. ISME J. 2016;10:1025–1028.
- Falony G, De Vuyst L. Ecological interactions of bacteria in the human gut. In: Charalampopoulos D, Rastall RA, editors. Prebiotics and probiotics science and technology. New York: Springer New York; 2009. p. 639–679.
- Falony G, Vieira-Silva S, Raes J. Richness and ecosystem development across faecal snapshots of the gut microbiota. Nat Microbiol. 2018;3:526–528.
- Vieira-Silva S, Falony G, Darzi Y, et al. Species-function relationships shape ecological properties of the human gut microbiome. Nat Microbiol. 2016;1:16088.
- Vandeputte D, Falony G, Vieira-Silva S, et al. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut. 2016;65:57–62.
- Roager HM, Hansen LBS, Bahl MI, et al. Colonic transit time is related to bacterial metabolism and mucosal turnover in the gut. Nat Microbiol. 2016;1:16093.
- Durack JLynch SV 2019. The gut microbiome: relationships with disease and opportunities for therapy. J Exp Med 216 (1):20–40. doi:10.1084/jem.20180448.
- Lambrecht BN, Hammad H. The immunology of the allergy epidemic and the hygiene hypothesis. Nat Immunol. 2017;18:1076–1083.
- Brown EM, Sadarangani M, Finlay BB. The role of the immune system in governing host-microbe interactions in the intestine. Nat Immunol. 2013;14:660–667.
- Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–323.
- Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol. 2014;14:667–685.
- Bergqvist P, Stensson A, Hazanov L, et al. Re-utilization of germinal centers in multiple Peyer’s patches results in highly synchronized, oligoclonal, and affinity-matured gut IgA responses. Mucosal Immunol. 2013;6:122–135.
- Perez-Lopez A, Behnsen J, Nuccio S-P, et al. Mucosal immunity to pathogenic intestinal bacteria. Nat Rev Immunol. 2016;16:135–148.
- Chairatana P, Nolan EM. Defensins, lectins, mucins, and secretory immunoglobulin A: microbe-binding biomolecules that contribute to mucosal immunity in the human gut. Crit Rev Biochem Mol Biol. 2017;52:45–56.
- Donaldson GP, Ladinsky MS, Yu KB, et al. Gut microbiota utilize immunoglobulin A for mucosal colonization. Science. 2018;360:795–800.
- Furusawa Y, Obata Y, Fukuda S, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–450.
- Arpaia N, Campbell C, Fan X, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451–455.
- Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124:837–848.
- Falony G, Joossens M, Vieira-Silva S, et al. Population-level analysis of gut microbiome variation. Science. 2016;352:560–564.
- Zhernakova A, Kurilshikov A, Bonder MJ, et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science. 2016;352:565–569.
- Janssens Y, Nielandt J, Bronselaer A, et al. Disbiome database: linking the microbiome to disease. BMC Microbiol. 2018;18:50.
- Forster SC, Browne HP, Kumar N, et al. HPMCD: the database of human microbial communities from metagenomic datasets and microbial reference genomes. Nucleic Acids Res. 2016;44:D604–D609.
- Forslund K, Hildebrand F, Nielsen T, et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature. 2015;528:262–266.
- Rothschild D, Weissbrod O, Barkan E, et al. Environment dominates over host genetics in shaping human gut microbiota. Nature. 2018;555:210–215.
- Lewis SJ, Heaton KW. Stool form scale as a useful guide to intestinal transit time. Scand J Gastroenterol. 1997;32:920–924.
- Probert CS, Emmett PM, Heaton KW. Some determinants of whole-gut transit time: a population-based study. Qjm. 1995;88:311–315.
- Florez H, Luo J, Castillo-Florez S, et al. Impact of metformin-induced gastrointestinal symptoms on quality of life and adherence in patients with type 2 diabetes. Postgrad Med. 2010;122:112–120.
- Arumugam M, Raes J, Pelletier E, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180.
- Costea PI, Hildebrand F, Arumugam M, et al. Enterotypes in the landscape of gut microbial community composition. Nat Microbiol. 2018;3:8–16.
- Knights D, Ward TL, McKinlay CE, et al. Rethinking “enterotypes”. Cell Host Microbe. 2014;16:433–437.
- Holmes I, Harris K, Quince C. Dirichlet multinomial mixtures: generative models for microbial metagenomics. PLoS One. 2012;7:e30126.
- Vandeputte D, Kathagen G, D’hoe K, et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature. 2017;551:507–511.
- Wu GD, Chen J, Hoffmann C, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–108.
- Sokol H, Pigneur B, Watterlot L, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A. 2008;105:16731–16736.
- Vandeputte D, Tito RY, Vanleeuwen R, et al. Practical considerations for large-scale gut microbiome studies. FEMS Microbiol Rev. 2017;41:S154–S167.
- Debelius J, Song SJ, Vazquez-Baeza Y, et al. Tiny microbes, enormous impacts: what matters in gut microbiome studies? Genome Biol. 2016;17:217.
- Sinha R, Chen J, Amir A, et al. Collecting fecal samples for microbiome analyses in epidemiology studies. Cancer Epidemiol Biomarkers Prev. 2016;25:407–416.
- Loftfield E, Vogtmann E, Sampson JN, et al. Comparison of collection methods for fecal samples for discovery metabolomics in epidemiologic studies. Cancer Epidemiol Biomarkers Prev. 2016;25:1483–1490.
- Gloor GB, Macklaim JM, Pawlowsky-Glahn V, et al. Microbiome datasets are compositional: and this is not optional. Front Microbiol. 2017;8:2224.
- Vandeputte D, Falony G, D’hoe K, et al. Water activity does not shape the microbiota in the human colon. Gut. 2017;66:1865–1866.
- Cani PD, Knauf C. How gut microbes talk to organs: the role of endocrine and nervous routes. Mol Metab. 2016;5:743–752.
- Elbere I, Kalnina I, Silamikelis I, et al. Association of metformin administration with gut microbiome dysbiosis in healthy volunteers. PLoS One. 2018;13:e0204317.
- Maier L, Pruteanu M, Kuhn M, et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature. 2018;555:623–628.
- Hill C, Guarner F, Reid G, et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat Rev Gastroenterol Hepatol. 2014;11:506–514.
- O’Toole PW, Marchesi JR, Hill C. Next-generation probiotics: the spectrum from probiotics to live biotherapeutics. Nat Microbiol. 2017;2:17057.
- Gibson GR, Hutkins R, Sanders ME, et al. Expert consensus document: The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of prebiotics. Nat Rev Gastroenterol Hepatol. 2017;14:491–502.
- Vandeputte D, Falony G, Vieira-Silva S, et al. Prebiotic inulin-type fructans induce specific changes in the human gut microbiota. Gut. 2017;66:1968–1974.
- Zhang F, Luo W, Shi Y, et al. Should we standardize the 1,700-year-old fecal microbiota transplantation? Am J Gastroenterol. 2012;107:1755–1756.
- Youngster I, Mahabamunuge J, Systrom HK, et al. Oral, frozen fecal microbiota transplant (FMT) capsules for recurrent Clostridium difficile infection. BMC Med. 2016;14:134.
- Verbeke F, Janssens Y, Wynendaele E, et al. Faecal microbiota transplantation: a regulatory hurdle? BMC Gastroenterol. 2017;17:128.
- Aslam S, Hamill RJ, Musher DM. Treatment of clostridium difficile-associated disease: old therapies and new strategies. Lancet Infect Dis. 2005;5:549–557.
- Kelly CP, LaMont JT. Clostridium difficile – more difficult than ever. N Engl J Med. 2008;359:1932–1940.
- Cornely OA, Miller MA, Louie TJ, et al. Treatment of first recurrence of clostridium difficile infection: fidaxomicin versus vancomycin. Clin Infect Dis. 2012;55(Suppl 2):S154–S161.
- Lowy I, Molrine DC, Leav BA, et al. Treatment with monoclonal antibodies against clostridium difficile toxins. N Engl J Med. 2010;362:197–205.
- McFarland LV, Elmer GW, Surawicz CM. Breaking the cycle: treatment strategies for 163 cases of recurrent clostridium difficile disease. Am J Gastroenterol. 2002;97:1769–1775.
- McFarland LV, Surawicz CM, Greenberg RN, et al. A randomized placebo-controlled trial of saccharomyces boulardii in combination with standard antibiotics for clostridium difficile disease. JAMA. 1994;271:1913–1918.
- Kassam Z, Lee CH, Yuan Y, et al. Fecal microbiota transplantation for clostridium difficile infection: systematic review and meta-analysis. Am J Gastroenterol. 2013;108:500–508.
- Furuya-Kanamori L, Paterson DL, Helms SK, et al. Upper versus lower gastrointestinal delivery for transplantation of fecal microbiota in recurrent or refractory clostridium difficile infection: a collaborative analysis of individual patient data from 14 studies. J Clin Gastroenterol. 2017;51:145–150.
- Hagel S, Fischer A, Ehlermann P, et al. Fecal microbiota transplant in patients with recurrent clostridium difficile infection. Dtsch Arztebl Int. 2016;113:583–589.
- Cammarota G, Ianiro G, Tilg H, et al. European consensus conference on faecal microbiota transplantation in clinical practice. Gut. 2017;66:569–580.
- Costello SP, Waters O, Bryant RV, et al. Short duration, low intensity, pooled fecal microbiota transplantation induces remission in patients with mild-moderately active ulcerative colitis: a randomised controlled trial. Gastroenterology. 2017;152:S198–S199.
- Paramsothy S, Kamm MA, Kaakoush NO, et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: a randomised placebo-controlled trial. Lancet. 2017;389:1218–1228.
- Rossen NG, Fuentes S, van der Spek MJ, et al. Findings from a randomized controlled trial of fecal transplantation for patients with ulcerative colitis. Gastroenterology. 2015;149:110–118.e4.
- Moayyedi P, Surette MG, Kim PT, et al. Fecal microbiota transplantation induces remission in patients with active ulcerative colitis in a randomized controlled trial. Gastroenterology. 2015;149:102–109.e6.
- Colman RJ, Rubin DT. Fecal microbiota transplantation as therapy for inflammatory bowel disease: a systematic review and meta-analysis. J Crohns Colitis. 2014;8:1569–1581.
- Holvoet T, Joossens M, Wang J, et al. Assessment of faecal microbial transfer in irritable bowel syndrome with severe bloating. Gut. 2017;66:980–982.
- Halkjær SI, Christensen AH, Lo BZS, et al. Faecal microbiota transplantation alters gut microbiota in patients with irritable bowel syndrome: results from a randomised, double-blind placebo-controlled study. Gut. 2018;67:2107–2115.
- Vrieze A, Van Nood E, Holleman F, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012;143:913–6.e7.
- Turnbaugh PJ, Ley RE, Mahowald MA, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031.
- He Z, Cui B-T, Zhang T, et al. Fecal microbiota transplantation cured epilepsy in a case with Crohn’s disease: the first report. World J Gastroenterol. 2017;23:3565–3568.
- Frémont M, Coomans D, Massart S, et al. High-throughput 16S rRNA gene sequencing reveals alterations of intestinal microbiota in myalgic encephalomyelitis/chronic fatigue syndrome patients. Anaerobe. 2013;22:50–56.
- Finegold SM, Molitoris D, Song Y, et al. Gastrointestinal microflora studies in late-onset autism. Clin Infect Dis. 2002;35:S6–S16.
- Song Y, Liu C, Finegold SM. Real-time PCR quantitation of clostridia in feces of autistic children. Appl Environ Microbiol. 2004;70:6459–6465.
- Kang D-W, Adams JB, Gregory AC, et al. Microbiota transfer therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: an open-label study. Microbiome. 2017;5:10.
- Kurokawa S, Kishimoto T, Mizuno S, et al. The effect of fecal microbiota transplantation on psychiatric symptoms among patients with irritable bowel syndrome, functional diarrhea and functional constipation: an open-label observational study. J Affect Disord. 2018;235:506–512.