
Abstract
The aim of this research was to screen candidate genes affecting growth traits in pigs by studying the difference expression genes (DEGs) between the longissimus dorsi of Large White (LW) and Mashen (MS) pigs. RNA-sequencing (RNA-seq) technology was applied to analyse the transcriptomes from six longissimus dorsi in two pig breeds (LW and MS). Based on sequencing, the DEGs were screened, and functional annotation and enrichment analysis were performed. The results showed that 697 different significant genes were obtained (p < 0.05, FDR <0.05 and |FC| ≥ 2). The results of Gene Ontology (GO) functional annotation showed that DEGs were mainly related to the growth and development of skeletal muscle, the formation of organelles and the function of protein binding. DEGs were mainly involved in the pathways of Phagosome, Viral myocarditis, Antigen processing and presentation, Allograft rejection, Graft-versus-host disease, Cardiac muscle contraction by Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. Combined with other livestock genomics studies, SFRP2, KDM6A, OGT may be candidate genes involved in pig growth traits. The results provided a theoretical basis for the improvement of pig growth traits and the study of candidate genes.
In this study, three candidate genes (SFRP2, KDM6A, OGT) which affecting growth traits were screened.
The experimental animals selected in this study were the local breed Mashen pig and the Western commercial breed Large White pig with obvious growth traits difference.
In this study, RNA-Seq technology was applied to analysis the transcriptomes of pig muscle.
Highlights
Introduction
Pork is the main source of animal protein in the human diet, and research on pork yield and meat quality has been a focus of animal husbandry. Studies have focused on growth and development regulation mechanisms of pigs to improve their growth rate and quality of meat. Many factors affect the growth and development of pigs, among which the growth and development of skeletal muscle is one of the main factors determining production performance, which directly affects the yield and quality of pork.
RNA high-throughput sequencing technology (RNA-seq) was used to analyse DEGs, functional genes, allele-specific expression, RNA splicing and fusion gene transcripts, etc. (Mitelman et al. Citation2007; Wang et al. Citation2008; Trapnell et al. Citation2010). RNA-seq technology can accurately describe the regulation of gene expression in tissue cells of transcript sequences and has been widely used in animal and plant breeding, including genetic screening in pig growth performance. Genes expressing particular growth traits can be selected to obtain gene expression profiles using DEGs with RNA-seq technology. The key genes can be determined by analysing the DEGs, to accelerate the understanding of the metabolic regulation of pig growth performance and other aspects (Shang Citation2017).
Using RNA-seq technology, Sodhi et al. (Citation2014) found that there were 169 DEGs in liver and 39 in the longissimus dorsi in Jeju and Bucklebury pigs. In the identification of the DEGs, GO and KEGG analysis revealed 41 genes in liver and 9 genes in muscle, which played a role in the gene regulation of meat quality. These genes were mainly related to metabolism, cellular immune response, regulation of protein binding sites, inflammation. Liu et al. (Citation2014) studied the effects of piglet birth weight on muscle growth by analysing skeletal muscle protein. They found 46 differentially expressed proteins, including heat shock protein, and a protein regulating fat metabolism and cell apoptosis. Cai (Citation2011) screened DEGs in the subcutaneous adipose tissue of castrated and non-castrated boars using RNA-seq technology. A total of 1919 genes that differentially expressed more than twice were screened in the subcutaneous adipose tissue of castrated boars, and the selective splicing and transcripts were identified. Zhu et al. (Citation2016) analysed the transcriptome of extensor digitorum longus and soleus muscles of Large White pig using RNA-seq technology, obtaining 561 DEGs and four genes associated with muscle CRSP3, ACTN2, MYL1 and MYH6. Li, Liu, et al. (Citation2016) constructed two RNA libraries from pooled samples of longissimus dorsi muscles with the highest and lowest drip loss pig longissimus dorsi and obtained 150 different genes using RNA-seq technology, screening two candidate genes, TRDN and MSTN. TRDN has roles in muscle contraction and fat deposition, whereas MSTN has a role in muscle growth. Li, Qian, et al. (Citation2016) analysed the transcriptome of longissimus dorsi muscles in Wannanhua and Large Yorkshire pigs using RNA-seq. The obtained sequences were subject to GO and Pathway significant enrichment analysis, which produced gene expression profiles of pig longissimus dorsi muscle and identified candidate genes associated with meat quality and growth traits. Although RNA-seq technology has been widely used in the study of DEGs and candidate genes, the study of candidate genes for pig growth traits in longissimus dorsi muscle has not been reported.
The MS pig is a local breed in north China, which has the advantages of resistance to crude feed, strong adaptability and good meat quality. However, the growth rate and feed conversion ratio are much lower than those in Western commercial pig breeds such as LW and Landrace (Zhang et al. Citation2001; Yang et al. Citation2005; Zhao et al. Citation2015). The significant difference in the growth rate of MS and LW pigs makes them suitable for comparative studies of pig growth traits. In this study, RNA-seq technology was used for sequencing analysis of longissimus dorsi muscle in 90-day old MS and LW pigs and to screen candidate genes of pig growth traits.
Materials and methods
The Code of Ethics of the World Medical Association was strictly followed for the animal experiments (http://ec.europa.eu/environment/chemicals/lab_animals/legislation_en.htm). The methods used in our study conformed to the Good Experimental Practices adopted by the College of Animal Science and Veterinary Medicine, Shanxi Agricultural University (Shanxi, China). Local animal welfare laws, guidelines and policies regarding the feeding and use of experimental animals were strictly adhered to.
Animals and sample collection
Three healthy MS male pigs (mean body weight: 20.11 kg) and three healthy LW male pigs (mean body weight: 39.56 kg) were used in this study. These pigs were provided by the Datong Pig Breeding Farm (Datong, Shanxi, China), an animal research facility of Shanxi Agricultural University. All pigs selected for the experiment were kept under the same feeding and environmental conditions. All pigs were slaughtered at 90 days after birth. Longissimus dorsi tissues were collected and immediately frozen in liquid nitrogen, and stored at –80 °C for subsequent use.
RNA extraction, library preparation and illumina sequencing
Total RNA was extracted using TRIZOL Reagent (Life Technologies, Carlsbad, USA) following manufacturer instructions. The total RNA was further purified using RNeasy Kits (Qiagen, GmbH, Germany). RNA concentration, quality and integrity were measured with Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, USA). A cDNA library was constructed according to the specifications of the TruSeqRapid Duo cBot Sample Loading Kit (Illumina, San Diego, California), and 2 × 100 bp double-end sequencing was performed using the IlluminaHiSeq 2500 platform (Illumina, San Diego, California).
Quality control for paired-end reads
Quality control of raw data was processed using Perl scripts. Clean data were obtained from the raw data by removal of reads containing more than 10% unknown nucleotides, and low-quality reads (more than 50% of the bases had quality scores ≤10). Q20 (the 1% base error rate), Q30 (the 0.1% base error rate) and GC (the percentage of guanine and cytosine) content, and sequence duplication level of the clean data were calculated.
Screening of RNA transcripts
Clean reads were mapped to the pig genome (UCSC susScr3, from the UCSC Genome Browser [http://hgdownload.soe.ucsc.edu/goldenPath/susScr3/bigZips/susScr3.fa.gz; accessed 24 August 2017) with TopHat (v2.0.14). Known transcripts were named with ensembl ID, and unknown transcriptions with TCONS ID. Transcripts were reconstructed with cufflinks (Trapnell et al. Citation2012; Ghosh and Chan Citation2016) and cuffmerge and assembled into new annotation files. Finally, the transcriptional information of differential expression in the two different pig groups at 90 d was obtained with cuffdiff.
Identification of DEGs
DESeq R package (1.10.1) (Bioconductor, Buffalo, USA) was used to analyse differential expression in the two groups (Anders and Huber Citation2010). The parameter FPKM (Fragments Per Kilobase of exon per Million fragments mapped) was used to quantify the expression of DEGs. The data were filtered by p < .05, false discovery rate (FDR) < 0.05 and absolute value of Fold Change (|FC|) ≥ 2.
GO and KEGG enrichment analysis of DEGs
Cytoscape was used to implement GO enrichment analysis and KEGG pathway analysis of DEGs.
Validation of differentially expressed genes with quantitative real-time PCR (qPCR)
To validate the repeatability and reproducibility of gene expression data obtained by RNA sequencing in MS and LW pigs, qPCR was carried out for 10 randomly selected DEGs. 18S RNA and GAPDH were used as the internal control. Primer sequences are shown in Supplementary material (Table S1). qPCR was performed using an SYBR® PrimeScriptTMII RT-PCR Kit (Takara, China) implemented on an ABI-7500 (Applied Biosystems, Foster City, CA). The reaction conditions were as follows: pre-denaturation at 95 °C for 30 s, 45 cycles of 95 °C for 30 s and 60 °C for 34 s, one cycle of 95 °C for 30 s, 60 °C for 1 min, 95 °C for 30 s. Each sample was tested in triplicate for robustness. The relative expressions were quantified by 2–ΔΔCt.
Results
Sequencing and mapping of the pig longissimus dorsi transcriptome
In this study, as shown in Table , the sequencing generated 913 million raw reads from three MS and three LW pigs, i.e. six longissimus dorsi tissues. After quality control, the sequencing results generated 892 million clean reads, with Q20 ≥ 96.00% and Q30 ≥ 87.00% for LW pigs, with Q20 ≥ 95.80% and Q30 ≥ 86.70% for MS pigs. These clean reads were used for further analysis.
Table 1. Number and Q Value of RNA-seq Reads.
Alignment of the sequence reads against the pig genome UCSC susScr3 yielded 69.41%–73.04% of uniquely aligned reads across the six samples (Table ). Unmapped or multi-position matched reads (1.51%–28.83%) were excluded from further analysis (Table ). Mapping Ratios were 71.17% to 74.54% in six longissimus dorsi tissues from three MS and three LW pigs.
Table 2. Mapping results of RNA-seq reads.
Identification of DEGs
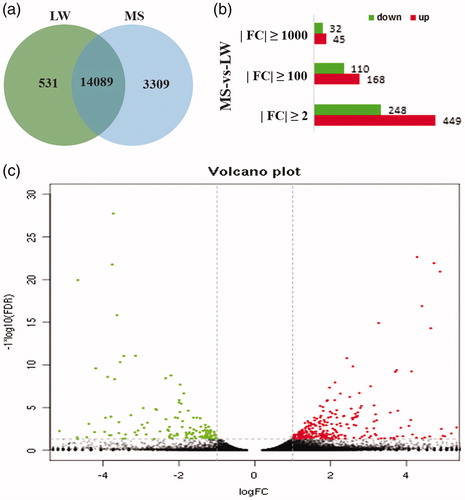
Sequencing results show that there were 14,089 genes in both LW and MS pigs, 531 unique genes in LW pigs and 3309 unique genes in MS pigs (Figure ). A total of 697 different significant genes were obtained with the condition of p < 0.05, FDR <0.05 and |FC| ≥ 2. In comparison with LW pigs, there were 449 up-regulated differentially expressed genes and 248 down-regulated differential genes (Figure ). Of the DEGs, 35.58% (248 genes)had higher expression in LW pigs (log2(FC) from 1.07 to 27.89), whereas 64.42% (449 genes) DEGs had higher expression in the MS pigs (log2(FC) from 1.18 to 23.04) (Figure ). When |FC| ≥100, there were 278 significant genes, 168 increases and 110 downgrades. When |FC| ≥ 1000, 77 significant genes were obtained, 45 were up-regulated and 32 were down-regulated (Figure ). There are 271 known genes and 426 unknown genes in the 697 genes that control |FC| ≥ 2.
Figure 1. Number of DEGs between LW and MS pigs. (a) Number of genes between LW and MS pigs; (b) Number of DEGs under different FC; (c) Scatter plot for DEGs and p-value volcano plot. The points below of the “---” denotes genes with no significant changes; The points on the left of the “|” denotes up-regulated genes; The points on the right of the “|” denotes down-regulated genes.LW: Large White; MS: Mashen; FC: Fold Change.
GO enrichment and KEGG pathway analysis
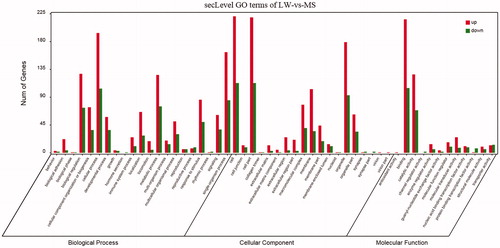
GO analysis revealed that DEGs are associated with biological processes such as biological adhesion, cellular process, metabolic process and single-organism process; and play roles in molecular functions such as catalytic activity and binding; and are related to cellular components such as macromolecular complex, membrane and organelle (Figure ).
Figure 2. GO analysis of DEGs. LW: Large White; MS: Mashen; GO: Gene Ontology; DEGs: difference expression genes.
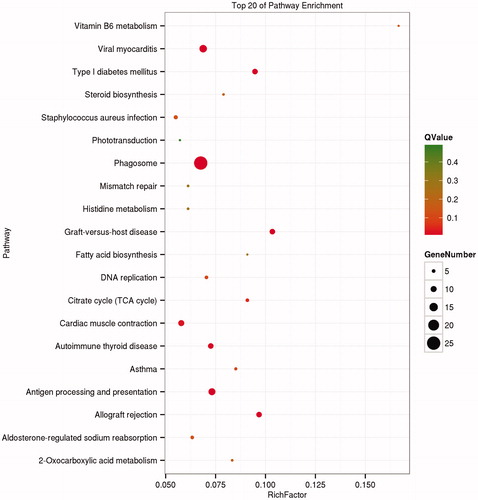
Using the KEGG database, the top 20 enrichment pathways are shown in Figure . The pathways are mainly involved in Phagosome, Viral myocarditis, Antigen processing and presentation, Allograft rejection, Graft-versus-host disease, Cardiac muscle contraction and Autoimmune thyroid disease.
Figure 3. KEGG pathway analysis of DEGs. KEGG: Kyoto Encyclopedia of Genes and Genomes; DEGs: difference expression genes.
Validation of DEGs between LW and MS pigs
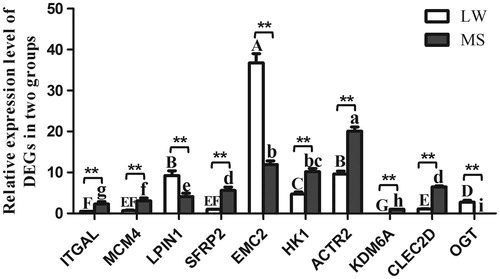
qPCR was used to validate selected DEGs identified from the RNA-seq data. Ten DEGs (ITGAL, MCM4, LPIN1, SFRP2, EMC2, HK1, ACTR2, KDM6A, CLEC2D, OGT) were selected from the DEGs, which included up- and down-regulated genes between two groups. The results from qPCR confirmed the expression pattern of DEGs at two breeds (Figure ).
Figure 4. The expression level of DEGs validated by qPCR. Large White pig (LW), Mashen pig (MS). Values in the bar diagram indicate ‘mean ± standard errors’. In the same breed, means with no common letter differ significantly. In the same tissue, ‘*’ indicates that the difference between LW and MS had a significance level of .05 level, ‘**’ indicates that the difference between LW and MS had a significance level of 0.01. LW: Large White; MS: Mashen.
Identification of candidate genes
On the basis of screening of DEGs, GO and KEGG analysis, we according to previous studies, three candidate genes related to pig growth traits, namely SFRP2, KDM64 and OGT, were preliminarily identified.
Discussion
Zhao et al. (Citation2015) found that there was a significant difference in the growth rate between 90-day old LW and MS pigs. The growth rate of LW pig is significantly higher than that of MS pig. At the age of 90 days, the average weight of LW pigs is 39.63 kg, and that of MS pigs is 20.09 kg. Therefore, it is of great significance to study the candidate genes related to the pig growth traits use LW and MS pigs. In addition, the growth and development of skeletal muscle is one of the main factors determining production performance. Gao et al. (Citation2017) found that the fibre diameter significantly increased, whereas fibre density significantly decreased with age. The myofibre diameters of MS at the same age were significantly smaller than those of LW pigs, whereas the fibre density were much greater than those of LW pigs. These further demonstrate the reliability of this study. Moreover, as the results relies on theoretical approach rather than functional data, hence it provided a theoretical basis for the improvement of pig growth traits and the study of candidate genes and its specific action mechanism needs further study.
After sequencing the longissimus dorsi tissue of LW and MS we obtained 271 candidate genes affecting the growth traits of these pigs. SFRP2 belongs to the Secreted Frizzled-Related Proteins (SFRPs) family. SFRPs are a regulator of extracellular WNT signalling and play an important role in development and carcinogenesis (Ladher et al. Citation2000). Previous research found that SFRP2 plays important roles in muscle development in mice, and the proliferation and differentiation of muscle satellite cells (Levin et al. Citation2001; Descamps et al. Citation2008). MicroRNA-1 and microRNA-206 were specifically expressed in pig skeletal muscle. Sun (Citation2013) found SFRP2 was the target gene of microRNA-1 and microRNA-206 by establishing the mutation carrier and expression vector and speculated that SFRP2 was significant to the development of skeletal muscle during the pig embryogenesis.
KDM6A is a histone demethylase, which was discovered in 1998 by Geenfield et al. (1998) and located in the Xp11. 3-p11. 23 regions of the human chromosome. Previous studies have shown that Homeobox (HOX) family genes were downstream target genes of KDM6A and that their promoter regions were easily methylated by KDM6A and involved in the regulation of anterior and posterior body axis development (Lan et al. Citation2007; Lederer et al. Citation2012). In addition, KDM6A can also participate in the formation of the myeloid/lymphoid or mixed-lineage leukaemia 2/3 (MLL2/3) complex and plays a role in the regulation of methylation for histone H3 lysine-4 sites (Lee et al. Citation2007). Mansour et al. (Citation2012) identified KDM6A as a new mediator gene involved in the reconstruction and transformation of pluripotent embryonic cell development. Gao et al. made KDM6A stable overexpression in the slow virus, which enhanced the activity of BMSCs early osteogenesis index-ALP, the ability of mineralisation in vitro and the expression OPN and OCN that are the related genes in osteogenesis differentiation manifest. The results confirmed that histone demethylase KDM6A had the potential to promote BMSCs in vitro osteogenic differentiation (Gao and Fan Citation2014).
O-GlcNAc modification has been shown to affect protein degradation, protein–protein interaction and transcriptional activation. In the process of O-GlcNAc modification, only needs these two proteins, the O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA) proteins, OGT was used to catalyse add and OGA was used to catalyse removal (Shafi et al. Citation2000; Nolte and Muller Citation2002). N-acetylglucosamine from the activated glycogen donor UDP-GlcNAc can be transferred to the Serine or Threonine of the protein by OGT (Lubas and Hanover Citation2000; Wells et al. Citation2002) and catalysed the O-GlcNAc (Copeland et al. Citation2008). At present, many kinds of proteins with O-GlcNAc modification occur. In multiple signalling pathways, the corresponding proteins occurred O-GlcNAc modification to ensure that the signals were transmitted correctly in the downstream pathways. Sequence analysis shows that the OGT can be divided into two structural domains, the Teratricopetide Repeats (TPR) of the amino terminal and the catalytic structure domain of the carboxyl terminal (Lubas et al. Citation1997). TPRs are a serial duplication of 34 amino acids and are often involved in protein–protein interactions (Golovan et al. Citation2008). The removal of OGT from the ES cells of mice can lead to cell death, suggesting that OGT is necessary in cell functioning (Shafi et al. Citation2000). In addition, OGT can closely combine with PP1α and PP1β, which suggests that this combined enzyme agent can cause the removal of phosphate groups and add O-GlcNAc group simultaneously in many cases (Wells et al. Citation2004). Therefore, OGT can be an important candidate gene for pig growth traits.
In biology, the completion of various biological functions is based on interactions between various genes to enable coordinated expression. Biological functions of genes and their interaction with other genes can be further understood by gene KEGG pathway enrichment analysis. The KEGG pathway database stores information about gene function, and represents biological processes in cells, such as metabolism, membrane transport, signal transduction and cell growth cycle. In this study, the difference genes between LW and MS pigs were mainly involved in Phagosome, Viral myocarditis. Most of the DEGs are enriched in immune-related pathways, such as Phagosome, Viral myocarditis, Antigen processing and presentation, Allograft rejection, Graft-versus-host disease and Staphylococcus aureus infection. Related genes contain FCGR1A, ELMOD3, RFX1 and so on. These results are consistent with the stronger resistance of MS pigs compared with that of LW pigs, and provides a basis for the screening of candidate genes related to swine immunisation in Immune organs. In addition, the pathways of cardiac muscle contraction, 2-carboxylic acid metabolism, Aldosterone-regulated sodium reabsorption, vitamin B6 metabolism, type I diabetes mellitus, steroid biosynthesis, histidine metabolism, fatty acid biosynthesis and the citrate cycle (TCA cycle) were also enriched; these are related to growth and development. These results provide a new perspective and theoretical basis for the study of pig growth traits.
Conclusions
RNA-seq technology was used to sequence and analyse the longissimus dorsi of LW and MS pigs. Nine hundred and fourteen million raw reads and 892 million clean reads were generated from the sequencing results, and 697 DEGs were further screened. GO function annotation and KEGG enrichment analysis revealed that these genes were mainly involved in the process of immunisation and growth. As suggested by other livestock genomics studies, SFRP2, KDM6A and OGT may be used as candidate genes for growth traits. RNA-seq can thus provide comprehensive information on the pig longissimus dorsi transcriptome, providing basic data and reference for the improvement of pig growth traits and functional genomics.
Ethical approval
The methods were performed in accordance with the Good Experimental Practices adopted by the College of Animal Science and Veterinary Medicine, Shanxi Agricultural University (Taigu, P.R. China), and the experimental protocols were approved by it.
Acknowledgements
The authors thank Editage (www.editage.cn) for its linguistic assistance during the preparation of this manuscript. They also thank the staff of the Datong pig farm.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol. 11:R106.
- Cai ZW. 2011. Study on molecular mechanisms of the effect of castration on fat deposition in male pigs. Zhejiang University, Hangzhou, Zhejiang, China.
- Copeland RJ, Bullen JW, Hart GW. 2008. Cross-talk between GlcNAcylation and phosphorylation: roles in insulin resistance and glucose toxicity. Am J Physiol Endocrinol Metab. 295:E17–E28.
- Descamps S, Arzouk H, Bacou F, Bernardi H, Fedon Y, Gay S, Reyne Y, Rossano B, Levin J. 2008. Inhibition of myoblast differentiation by Sfrp1 and Sfrp2. Cell Tissue Res. 332:299–306.
- Gao PF, Guo XH, Du M, Cao GQ, Yang QC, Pu ZD, Wang ZY, Zhang Q, Li M, Jin YS, et al. 2017. LncRNA profiling of skeletal muscles in large white pigs and Mashen pigs during development. J Anim Sci. 95:4239–4250.
- Gao RT, Fan ZP. 2014. Histone demethylase KDM6A enhanced the ostegenic differentiation potential of bone marrow mesechymal stem cells. Beijing J Stomatol. 22:5–8.
- Ghosh S, Chan CK. 2016. Analysis of RNA-Seq data using TopHat and Cufflinks. Methods Mol Biol. 1374:339–361.
- Golovan SP, Hakimov HA, Verschoor CP, Walters S, Gadish M, Elsik C, Schenkel F, Chiu DKY, Forsberg CW. 2008. Analysis of Sus scrofa liver proteome and identification of proteins differentially expressed between genders, and conventional and genetically enhanced lines. Comp Biochem Physiol Part D Genomics Proteomics. 3:234–242.
- Greenfield A, Carrel L, Pennisi D, Philippe C, Quaderi N, Siggers P, Steiner K, Tam PP, Monaco AP, Willard HF, et al. 1998. The UTX gene escapes X inactivation in mice and humans. Hum Mol Genet. 7:737–742.
- Ladher RK, Church VL, Allen S, Robson L, Abdelfattah A, Brown NA, Hattersley G, Rosen V, Luyten FP, Dale L, et al. 2000. Cloning and expression of the Wnt antagonists Sfrp-2 and Frzb during chick development. Dev Biol. 218:183–198.
- Lan F, Bayliss PE, Rinn JL, Whetstine JR, Wang JK, Chen S, Iwase S, Alpatov R, Issaeva I, Canaani E, et al. 2007. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature. 449:689–694.
- Lederer D, Grisart B, Digilio MC, Benoit V, Crespin M, Ghariani SC, Maystadt I, Dallapiccola B, Verellen-Dumoulin C. 2012. Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am J Hum Genet. 90:119–124.
- Lee MG, Villa R, Trojer P, Norman J, Yan K-P, Reinberg D, Di Croce L, Shiekhattar R. 2007. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science. 318:447–450.
- Levin JM, El Andalousi RA, Dainat J, Reyne Y, Bacou F. 2001. SFRP2 expression in rabbit myogenic progenitor cells and in adult skeletal muscles. J Muscle Res Cell Motil. 22:361–369.
- Li B, Liu K, Weng Q, Li P, Wei W, Li Q, Chen J, Huang R, Wu W, Liu H, et al. 2016. RNA-seq analysis reveals new candidate genes for drip loss in a Pietrain × Duroc × Landrace × Yorkshire population. Anim Genet. 47:192–199.
- Li XJ, Qian KQ, Liu LQ, Wang CL, Zhou J. 2016. Screening and annotation of differentially expressed genes in longissimus muscle of different pig breeds based on RNA-seq technology. J Northwest A & F University. 44:1–8.
- Liu J, He J, Yu J, Mao X, Zheng P, Huang Z, Yu B, Chen D. 2014. Birth weight alters the response to postnatal high-fat diet-induced changes in meat quality traits and skeletal muscle proteome of pigs. Br J Nutr. 111:1738–1747.
- Lubas WA, Frank DW, Krause M, Hanover JA. 1997. O-Linked GlcNAc transferase is a conserved nucleocytoplasmic protein containing tetratricopeptide repeats. J Biol Chem. 272:9316–9324.
- Lubas WA, Hanover JA. 2000. Functional expression of O-linked GlcNAc transferase. Domain structure and substrate specificity. J Biol Chem. 275:10983–10988.
- Mansour A. AFatah, Gafni O, Weinberger L, Zviran A, Ayyash M, Rais Y, Krupalnik V, Zerbib M, Amann-Zalcenstein D, Maza I, et al. 2012. The H3K27 demethylase Utx regulates somatic and germ cell epigenetic reprogramming. Nature. 488:409–413.
- Mitelman F, Johansson B, Mertens F. 2007. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 7:233–245.
- Nolte D, Muller U. 2002. Human O-GlcNAc transferase (OGT): genomic structure, analysis of splice variants, fine mapping in Xq13.1. Mamm Genome. 13:62–64.
- Shafi R, Iyer SP, Ellies LG, O'Donnell N, Marek KW, Chui D, Hart GW, Marth JD. 2000. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc Natl Acad Sci USA. 97:5735–5739.
- Shang P. 2017. Identification of candidate genes on growth traits intergrating transcriptome and proteome of embrynic muscle in pigs. China Agricultural University, Beijing, China.
- Sodhi SS, Song K-D, Ghosh M, Sharma N, Lee SJ, Kim JH, Kim N, Mongre RK, Adhikari P, Kim JY, et al. 2014. Comparative transcriptomic analysis by RNA-seq to discern differential expression of genes in liver and muscle tissues of adult Berkshire and Jeju Native Pig. Gene. 546:233–242.
- Sun W. 2013. Identification of target genes of MicroRNA-1 and MicroRNA-206 in muscle development of pig. Northwest A & F University, Yangling, Shanxi, China.
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 28:511–515.
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L, et al. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 7:562–578.
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. 2008. Alternative isoform regulation in human tissue transcriptomes. Nature. 456:470–476.
- Wells L, Gao Y, Mahoney JA, Vosseller K, Chen C, Rosen A, Hart GW. 2002. Dynamic O-glycosylation of nuclear and cytosolic proteins: further characterization of the nucleocytoplasmic beta-N-acetylglucosaminidase, O-GlcNAcase. J Biol Chem. 277:1755–1761.
- Wells L, Kreppel LK, Comer FI, Wadzinski BE, Hart GW. 2004. O-GlcNAc transferase is in a functional complex with protein phosphatase 1 catalytic subunits. J Biol Chem. 279:38466–38470.
- Yang WP, Cao GQ, Shi JZ, Liu JH, Zhou ZX. 2005. Study on the finishing ability of different cross combination in pig. Chinese J Anim Sci. 41:48–49.
- Zhang JG, Wang X, Du MH, Zhou ZX. 2001. Species diversity and the way to protect Ma shen zhu. J Shanxi Agric Univ. 21:188–191.
- Zhao Y, Gao P, Li W, Zhang Y, Xu K, Guo X, Li B, Cao G. 2015. Study on the developmental expression of Lbx1 Gene in longissimus dorsi of Mashen and large white pigs. Ital J Anim Sci. 14:3720–2087.
- Zhu JY, Shi XE, Lu HZ, Xia B, Li YF, Li X, et al. 2016. RNA-seq transcriptome analysis of extensor digitorum longus and soleus muscles in large white pigs. Mol Genet Genomics. 291:687–701.