
Abstract
Here, we sequenced the faecal microbiome of a local pig breed by using whole-metagenome sequencing. Metagenomic data revealed that the faecal microbiome consists of a complex and intricate admixture of microbes belonging to all domains of life, including viruses. Most of sequencing reads were mapped to multiple reference sequences of Bacteria (99.35%) whereas the remaining reads (0.65%) were assigned to different microbes belonging to Archaea, Eukarya, and viruses. The predominant bacterial phylum was Firmicutes (∼86%), whereas Ascomycota division (fungi) was the most abundant eukaryotic phyla found. BLAST homology search showed that viral reads could be assigned to a total of 13 viral families of which 7 were present in all metagenomes. Bioinformatics analysis identified a catalogue of microbial genes encoding 231,428 unique proteins grouped in 3,169 functional groups and involved in 275 biochemical pathways. This study represents a first attempt to explore the faecal microbiome of a local pig breed in order to expand our knowledge about the taxonomic and functional profiles of microbes associated with this rare and endangered-maintained pig breed.
Sequenced the faecal microbiome of a local pig breed by using whole-metagenome sequencing.
Bioinformatics analysis identified a catalogue of microbial genes.
The predominant bacterial phylum was Firmicutes (∼86%), whereas Ascomycota division (fungi) was the most abundant eukaryotic phyla found.
Highlights
Introduction
Domestic pig is one of the most important livestock species worldwide not only for historical, and socio-economic reasons (Pappas Citation2013), but also because it plays a fundamental role in many biomedical fields as an experimental model for basic and translational research (Hryhorowicz et al. Citation2020). Knowing the taxonomic structure and functions of pig microbiomes is important for commercial breeding, as clarifying how microbial taxa influence host fitness can provide rich information to promote animal health and improve feed efficiency in swine industry (Yang et al. Citation2017; Wang et al. Citation2019). Unfortunately, although the number of metagenomic studies based on 16S rRNA gene sequencing has grown exponentially in recent years (Crespo-Piazuelo et al. Citation2018), the number of those relying on shotgun metagenomics is still low (Xiao et al. Citation2016; Yang et al. Citation2017; Wang et al. Citation2019; Fenske et al. Citation2020), especially considering the hundreds of pig breeds existing worldwide. In fact, globally, there are more than 700 pig breeds of which 97 are considered as endangered or threatened (FAO Citation2015). One of these is known as ‘Nero Siciliano’ pig, an autochthonous genetic type that lives in the woods of the Nebrodi and Madonie mountains in Sicily (Italy) (D’Alessandro et al. Citation2019a). This pig is reared in extensive and semi-extensive farming systems using pasture and other natural resources for feeding, according to the usual traditional methods employed in this geographical region. Genetically, it is well-characterised (D'Alessandro et al. Citation2019b; D'Alessandro et al. Citation2020; Sutera et al. Citation2020), and recent findings on six Chinese pig breeds suggested that host genetics could play an important role in shaping the composition of the gut microbiome (Xiao et al. Citation2016). It is clear, therefore, that determining the microbiome profile of other pig breeds, including host traits, dietary and other environmental factors that shape microbiome composition, could be of great interest given the potentially enormous impact of this relatively modern field of research on pig health (Ramayo-Caldas et al. Citation2016; Lu et al. Citation2018; González-Prendes et al. Citation2019; Le Sciellour et al. Citation2019a; Citation2019b). In this study, we explore the faecal microbiome of a local pig breed pig by using whole-metagenome shotgun sequencing (WMS) and report a detailed taxonomic and functional profiling of the microbial community associated with this rare and endangered-maintained pig breed.
Materials and methods
Animals, faecal sampling, DNA extraction and WMS
The study was carried out on three healthy Nero Siciliano pigs bred in three different farms located in the same geographical area (Messina, Sicily, Italy). All pigs were reared using a single pens with nipples waterer and stainless-steels feeder.
Pigs were homogeneous for sex (female), age (8 months) and body weight (85 kg ± 0.75), and were fed with a pelleted complete feed (Leone Mangimi SPA, Italy) (Liotta et al. Citation2015), rationed on the basis of 3% of the live weight, supplemented either with acorn, (PIG1), citrus pulp (PIG2), pasture and other natural plant resources following the traditional practices used in this area (PIG3). A total of three stool samples were collected directly from the rectal ampoule of the animals.
DNA was extracted as reported by Tang et al. (Citation2008), following the pre-treatment B. For each sample, a shotgun metagenomic library was prepared by using Illumina Nextera DNA kit (Illumina, Italy) and then paired-end sequenced (2 × 150 bp; 20 million reads per sample) on an Illumina NextSeq500 platform. Raw reads were deposited into Sequence Reads Archive (SRA) database (BioProject: PRJNA677897).
Bioinformatics analysis
Quality analysis and taxonomic assignment methods
Low-quality reads (Phred-score: ≤20), including adapter sequences, were filtered out using the BBDuk tool (https://sourceforge.net/projects/bbmap). Cleaned reads were subsequently merged using the PEAR software (https://bio.tools/pear). Taxonomic assignment of the reads was performed by BLASTn, searching the non-redundant NCBI nucleotide database (filter used: in animal identity >60%; p-value ≤1e−06). Using the BLAST results, the lowest common ancestor was calculated using the Krona software (Ondov et al. Citation2011), and all classified bacterial reads showing a relative abundance <0.01% were filtered out. Unclassified reads were assembled with metaSPAdes software (Nurk et al. Citation2017) and longer contigs (>1kb) were mapped against 154,884 non-chordata genomes retrieved from the RefSeq database (Pasolli et al., Citation2019). Results were filtered for p-value <0.05, and Mash distance was used to perform a hierarchical cluster analysis including a phylogenetic tree that was built with hclust, limma (Ritchie et al. Citation2015) and ape (Paradis and Schliep Citation2019) programs. Mash distance values ≤0.05 and ≤0.35 were applied to classify the bins at species and genus level respectively (Pasolli et al. Citation2019; Ondov et al. 2016; Nayfach et al. Citation2019).
Functioning profiling of pigs microbiome
The functional potential of microbial communities was evaluated by aligning all cleaned reads against the UniRef90 database (www.uniprot.org/uniref) by DIAMOND (Buchfink et al. Citation2015), with post-alignment filtering using FAMLI algorithm (Golob and Minot Citation2018). The resulting UnirRef90 protein IDs were used to retrieve the Gene Ontology (GO) and Kegg Orthology (KO) terms from the respective databases (http://geneontology.org; www.genome.jp/kegg/ko.html). Microbial resistance genes were identified from assembled reads by using metaSPAdes, and resulting contigs were processed by the RGI software (https://github.com/arpcard/rgi) and the CARD database (Alcock et al. Citation2020).
Results
Illumina WMS produced a total of 76,489,348 raw reads, of which 64,536,618 (∼84.4%) passed quality analysis used in this study. After BLAST-based taxonomic assignment, a total of 12,604,688 (19.53%) reads had at least one hit in Genbank database with a minimum identity score of 60% and p-value ≤1e−06. Using two different sequence identity thresholds (97% and 99%) we identified several unique taxonomic groups (Table ).
Table 1. Overall and individual taxonomic groups detected at two identity thresholds (97 and 99%) in pig metagenomic faecal samples.
Notably, almost all the reads mapped to multiple reference sequences belonging to Bacteria (99.35%) whereas the remaining reads (0.65%) were assigned to different Archaea (∼0.18%) and Eukarya (∼0.12%) species and to a much lesser extent to viruses (0.02%). However, a large fraction of reads (∼0.33%) was unclassifiable and therefore their taxonomic origin remained undefined. In all sequenced samples, the predominant bacterial phylum was Firmicutes (averaged relative abundance 86%), followed by Actinobacteria (3.0%), Proteobacteria (∼3.0%), and Bacteroidetes (1.3%) (Figure ).
Figure 1. Relative abundances and diversity of bacterial phyla and genera detected in Nero Siciliano pig faecal samples.
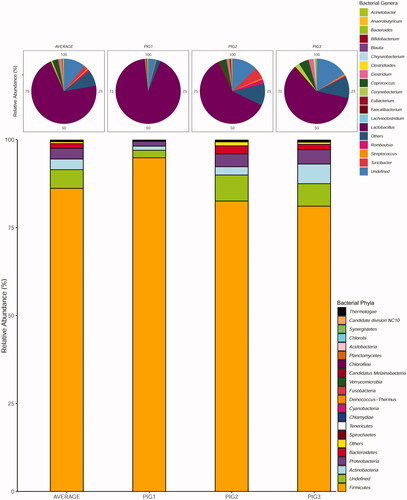
Proteobacteria were the second most abundant phylum in both PIG1 (∼1.35%) and PIG2 (3.65%) samples followed by Actinobacteria which reached ∼1.18% and ∼2.35% respectively. In contrast, in PIG3 sample, Actinobacteria (∼5.60%) and Proteobacteria (∼4.10%) were the second and third most abundant phyla, whereas Bacteroidetes was the fourth most abundant bacterial phylum in all pig metagenomes. Among non-bacterial phyla, the Ascomycota division of the fungal Kingdom was the most abundant (∼0.05%), followed by Apicomplexa (∼0.04%), a large phylum of parasitic protists belonging to the Alveolata group, and Basidiomycota (∼0.02%), another large and diverse group of eukaryotes (fungi).
Taxonomically, the PIG2 sample showed the highest number of microbial genera and species at both 97% and 99% identity, followed by PIG3 and PIG1 samples respectively (Table ). Lactobacillus was the most abundant genus across all samples (Figure ). Concerning the high number of clean reads (51,931,930/64,536,618; ∼80.5%) which did not display any match in the reference database used, we assembled this data set with metaSPAdes in 1,906,960 contigs, of which only 70,108 (∼3.7%) were ≥1kb in length. The binning produced a total of 46 bins, but only 20 (∼43.5%) showed an acceptable completeness rate >50% associated with a low contamination level (<5%). At phylum level, most of the high-quality taxonomic bins formed bacterial clusters belonging to Firmicutes (7/20; 35%), Bacteroidetes (5/20; 25%), Proteobacteria (3/20; 15%), Actinobacteria and Tenericutes (1/20; 5% each), while the remaining three bins (3/20; 15%) composed a cluster containing the filamentous cyanobacteria Trichormus sp., and two Eukaryotic (non-microbial) organisms. Moreover, we found that two Prevotella reference genomes (Genbank: GCA_002251365.1 and GCA_002251435.1) clustered with 2 of the 5 Bacteroidetes bins found in this study whereas other bins, with Mash distance values ranging from ∼0.1 to ∼0.26, clustered with different bacterial genera including Clostridium, Helicobacter, Muricauda, Bacillus, Mycoplasma, Butyrivibrio and Parvibacter.
Pig faecal mycobiome and virome
Three major divisions of fungi (Ascomycota, Basidiomycota and Mucoromycota) were identified in all metagenomic samples (Figure ).
Figure 2. Relative abundances and diversity of fungal community (phyla and genera) detected in Nero Siciliano pig faecal samples.
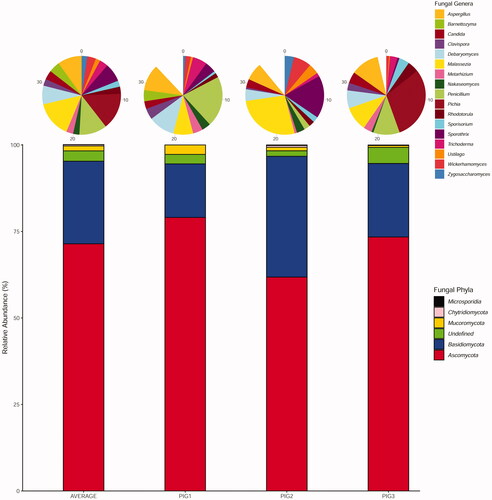
No evident variations were found in taxonomic profiles defined by the two identity thresholds used, and although fungal abundance was in general significantly lower (≤0.07%), compared to bacteria, the diversity of genera and species appeared to be remarkably high (Figure ).
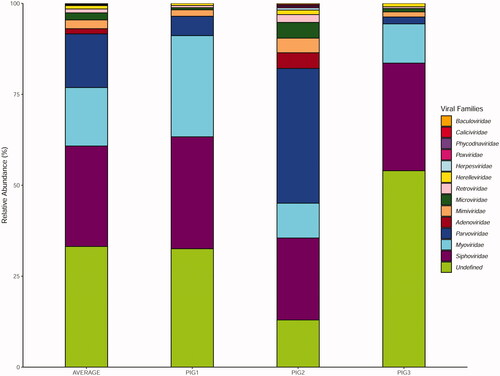
WMS analysis of pig faecal virome revealed a multifaceted viral community consisting mainly of viruses infecting Bacteria and Archaea. BLAST homology search showed that viral reads could be assigned to a total of 13 viral families of which 7 (Siphoviridae, Myoviridae, Parvoviridae, Herelleviridae, Retroviridae, Microviridae and Mimiviridae) were present in all samples, and 3 (Siphoviridae, Myoviridae and Parvoviridae) predominated each faecal virome. However, a large fraction of viral community remained unknown (Figure ).
Figure 3. Relative abundances of viral families identified in Nero Siciliano pig metagenomes.
Functional annotation of pig metagenomes
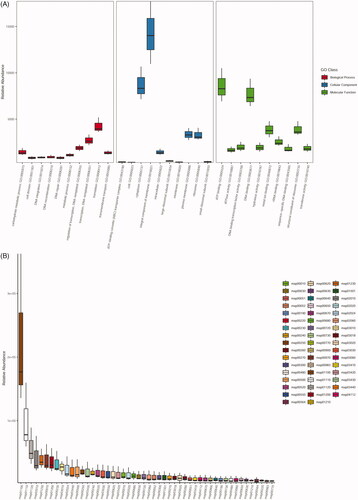
To investigate the functional role and diversity of proteins encoded by microbial genes associated with pig faecal microbiome, the Uniref90 database was used to identify 231,428 unique proteins from all sequencing reads employed by the FAMLI algorithm. Approximately 28.5% (65,898) of these proteins were shared among at least two metagenomes, but only ∼8.6% (19,863) were shared by all. Exploratory gene ontology analysis identified a total of 3,169 GO and 2,817 KO terms. The KEGG pathway related to ‘biosynthesis of secondary metabolites’ (map01110) was overrepresented in all three pig metagenomes (Figure ).
Figure 4. Relative abundances of significant gene ontology (GO) terms and KEGG maps identified in this study. (A) Top-10 ranking GO terms belonging to the biological process, cellular component and molecular function categories; (B) Top-50 ranking KEGG maps detected in pig metagenomes.
Multiple antibiotics resistance genes, in particular those conferring resistance to aminoglycoside, macrolide, and lincosamide were detected with high abundance across all metagenomes. A total of 180 antibiotic resistant ontology (ARO) terms were shared among our pigs.
Discussion
In recent years, hundreds and hundreds of metagenomic studies have appeared in the literature showing convincing evidence that gut microbiome changes play a crucial role in animal physiology, health and disease (Pascoe et al. Citation2017; Patil et al. Citation2020). In pigs, gut microbiome evaluation studies revealed a core microbial signature resembling that found in humans where Firmicutes and Bacteroidetes are generally the most dominant phyla (Isaacson and Kim Citation2012; Roura et al. Citation2016). This high level of identity, also highlights a clear functional overlap that has been firmly confirmed by recent faecal xenograft experiments showing that human microbiomes can be efficiently transplanted into pigs (Zhang et al. Citation2013). Moreover, the recent publication of the reference gene catalogue of the pig gut microbiome (Xiao et al. Citation2016) confirmed the high level of functional relationships shared by these two omnivorous species. Nevertheless, the global microbiome gene catalogue was established by investigating only 17 of the 709 (∼2.4%) pig breeds currently known (Xiao et al. Citation2016; FAO Citation2015), and therefore further investigations are needed.
In this study, we provide the first metagenomic analysis of faecal samples collected from Sicilian pigs breed, whose meat is characterised by an average intramuscular fat content of 4.6% and high levels of ‘good’ fats (Zumbo et al. Citation2020). Our data revealed that microbial taxa associated with this local breed follow, to some extent, the taxonomic pattern observed in other pig breeds worldwide (Bergamaschi et al. Citation2020), although the prevalence of some dominant bacterial phyla (Firmicutes and Bacteroidetes) was clearly different (Patil et al. Citation2020). In fact, in pigs analysed, the phylum Firmicutes was largely over-represented, while Bacteroidetes (Figure ) was by far the least abundant bacterial phylum compared to other well-known pig breeds (Pajarillo et al. Citation2014). Members of the Lactobacillaceae family, were highly enriched in all faecal samples with L. amylovorus as the predominant Latobacillus species. These data are in perfect agreement with other studies in which L. amylovorus was reported as the most dominant Lactobacillus species in pigs (Aluthge et al. Citation2019).
Most microbiome studies to date have focussed mainly on more abundant (bacterial) species, but an increasing body of evidence suggests pointing towards a better characterisation of microbial communities that are extremely low in terms of abundance (Pascoal et al. Citation2020). This less abundant, but highly diversified, intrinsic component of the microbiome is commonly referred to as ‘rare biosphere’ and includes a large variety of microbial taxa (Bhute et al. Citation2017; Jia et al. Citation2018), including fungi (Huffnagle and Noverr Citation2013). In our study, the fungal community represented a tiny part of whole microbiome which is consistent with previous reports. Nevertheless, its taxonomic composition was rather heterogeneous and characterised by a high proportion of the species D. hanseneii as previously reported in post-wean piglet faeces (Arfken et al. Citation2020). Regarding the virome, our data showed that it was dominated by three main families (Siphoviridae, Myoviridae and Parvoviridae) which have previously been reported in pigs (Kwok et al. Citation2020). Most viruses were bacteriophages (Sachsenröder et al. Citation2014; Kwok et al. Citation2020), but our metagenomes also contained at least one BLAST-recognizable sequence belonging to a novel porcine bocavirus which was first identified in 2009 in Swedish pigs (Blomström et al. Citation2009).
This study is a first attempt to explore the whole faecal microbiome of an ancient autochthonous pig which genome has also been recently sequenced and therefore future studies on the interaction between gut microbiome and host genome will be useful in order to understand the contribution of the genetic background of this breed to the enrichment of beneficial microbial phyla that promote intestinal health, growth performance and feed efficiency.
Ethical approval
All the experimental procedures used in this study were carried out according to the European guidelines for the care and use of animals in research (Directive 2010/63/EU 2010).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in Sequence Read Archive (SRA) databases under the study accession number: SRP 292297 and in Figshare under the project ‘Whole-metagenome shotgun sequencing of the Nero Siciliano pig faecal microbiome’ at https://doi.org/10.6084/m9.figshare.14899866
References
- Alcock BP, Raphenya AR, Lau TTY, Tsang KK, Bouchard M, Edalatmand A, Huynh W, Nguyen AV, Cheng AA, Liu S, et al. 2020. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48(1):517–525.
- Aluthge ND, Van Sambeek DM, Carney-Hinkle EE, Li YS, Fernando SC, Burkey E. 2019. BOARD INVITED REVIEW: The pig microbiota and the potential for harnessing the power of the microbiome to improve growth and health1. J Anim Sci. 97 (9):3741–3757.
- Arfken AM, Frey JF, Summers KL. 2020. Temporal dynamics of the gut bacteriome and mycobiome in the weanling pig. Microorganisms. 8 (6):868.
- Bergamaschi M, Tiezzi F, Howard J, Huang YJ, Gray KA, Schillebeeckx C, McNulty NP, Maltecca C. 2020. Gut microbiome composition differences among breeds impact feed efficiency in swine. Microbiome. 8 (1):110.
- Bhute SS, Ghaskadbi SS, Shouche YS. 2017. Rare biosphere in human gut: a less explored component of human gut microbiota and its association with human health. In: Kalia V, Shouche Y, Purohit H, Rahi P, editors. Mining of Microbial Wealth and MetaGenomics. Springer, p. 133–142.
- Blomström AL, Belák S, Fossum C, McKillen J, Allan G, Wallgren P, Berg M. 2009. Detection of a novel porcine boca-like virus in the background of porcine circovirus type 2 induced postweaning multisystemic wasting syndrome. Virus Res. 146(1-2):125–129.
- Buchfink B, Xie C, Huson DH. 2015. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 12(1):59–60.
- Crespo-Piazuelo D, Estellé J, Revilla M, Criado-Mesas L, Ramayo-Caldas Y, Óvilo C, Fernández AI, Ballester M, Folch JM. 2018. Characterization of bacterial microbiota compositions along the intestinal tract in pigs and their interactions and functions. Sci Rep. 8(1):12727.
- D’Alessandro E, Giosa D, Sapienza I, Giuffrè L, Aiese CR, Romeo O, Zumbo A. 2019a. Whole genome SNPs discovery in Nero Siciliano pig. Genet Mol Biol. 42 (3):594–602.
- D'Alessandro E, Sapienza I, Giosa D, Giuffrè L, Zumbo A. 2019b. In silico analysis of meat quality candidate genes among Nero Siciliano, and Italian heavy pigs genomes. Large Anim Rev. 25(4):137–140.
- D'Alessandro E, Sottile G, Sardina MT, Criscione A, Bordonaro S, Sutera AM, Zumbo A, Portolano B, Mastrangelo S. 2020. Genome-wide analyses reveal the regions involved in the phenotypic diversity in Sicilian pigs. Anim Genet. 51(1):101–105.
- FAO 2015., The Second Report on the State of the World’s Animal Genetic Resources for Foodand Agriculture. Rome, Italy: Commission on Genetic Resources for Food and Agriculture Assessments. Available from: http://www.fao.org/3/a-i4787e.pdf.
- Fenske GJ, Ghimire S, Antony L, Christopher-Hennings J, Scaria J. 2020. Integration of culture-dependent and independent methods provides a more coherent picture of the pig gut microbiome. FEMS Microbiol Ecol. 96(3):fiaa022.
- Golob JL, Minot SS. 2018. Functional Analysis of Metagenomes by Likelihood Inference (FAMLI) successfully compensates for multi-mapping short reads from metagenomic samples. Bio Rxiv. p. 1–32.
- González-Prendes R, Pena RN, Solé E, Seradj AR, Estany J, Ramayo-Caldas Y. 2019. Modulatory effect of protein and carotene dietary levels on pig gut microbiota. Sci Rep. 9(1):14582.
- Hryhorowicz M, Lipiński D, Hryhorowicz S, Nowak-Terpiłowska A, Ryczek N, Zeyland J. 2020. Application of genetically engineered pigs in biomedical research. Genes. 11(6):670.
- Huffnagle GB, Noverr MC. 2013. The emerging world of the fungal microbiome. Trends Microbiol. 21 (7):334–341.
- Isaacson R, Kim HB. 2012. The intestinal microbiome of the pig. Anim Health Res Rev. 13(1):100–109.
- Jia X, Dini-Andreote F, Falcão Salles J. 2018. Community assembly processes of the microbial rare biosphere. Trends Microbiol. 26(9):738–747.
- Kwok KTT, Nieuwenhuijse DF, Phan MVT, Koopmans MPG. 2020. Virus metagenomics in farm animals: a systematic review. Viruses. 12(1):107.
- Le Sciellour M, Renaudeau D, Zemb O. 2019a. Longitudinal analysis of the microbiota composition and enterotypes of pigs from post-weaning to finishing. Microorganisms. Nov 287(12):622.
- Le Sciellour M, Zemb O, Hochu I, Riquet J, Gilbert H, Giorgi M, Billon Y, Gourdine JL, Renaudeau D. 2019b. Effect of chronic and acute heat challenges on fecal microbiota composition, production, and thermoregulation traits in growing pigs1,2. J Anim Sci. Sep. 3(9):3845–3858.
- Liotta L, Chiofalo V, D'Alessandro E, Lo Presti V, Chiofalo B. 2015. Supplementation of rosemary extract in the diet of Nero Siciliano pigs: evaluation of the antioxidant properties on meat quality. Animal. 9(6):1065–1072.
- Lu D, Tiezzi F, Schillebeeckx C, McNulty NP, Schwab C, Shull C, Maltecca C. 2018. Host contributes to longitudinal diversity of fecal microbiota in swine selected for lean growth. Microbiome. 6(1):4.
- Nayfach S, Shi ZJ, Seshadri R, Pollard KS, Kyrpides NC. 2019. New insights from uncultivated genomes of the global human gut microbiome. Nature. 568 (7753):505–510.
- Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. 2017. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27 (5):824–834.
- Ondov BD, Bergman NH, Phillippy AM. 2011. Interactive metagenomic visualization in a Web browser. BMC Bioinformatics. 12:385.
- Pajarillo EA, Chae JP, Balolong, MP, Kim HB, Seo KS, Kang DK. 2014. Pyrosequencing-based analysis of fecal microbial communities in three purebred pig lines. J. Microbiol. 52: 646–651.
- Pappas G. 2013. Socio-economic, industrial and cultural parameters of pig-borne infections. Clin Microbiol Infect. 19 (7):605–610.
- Paradis E, Schliep K. 2019. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics. 35 (3):526–528.
- Pascoal F, Magalhães C, Costa R. 2020. The Link Between the Ecology of the Prokaryotic Rare Biosphere and Its Biotechnological Potential. Front Microbiol. 11:231.
- Pascoe EL, Hauffe HC, Marchesi JR, Perkins SE. 2017. Network analysis of gut microbiota literature: an overview of the research landscape in non-human animal studies. Isme J. 11(12):2644–2651.
- Pasolli E, Asnicar F, Manara S, Zolfo M, Karcher N, Armanini F, Beghini F, Manghi P, Tett A, Ghensi P. 2019. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age. Geography, and Lifestyle. Cell1. 76(3):649–662.
- Patil Y, Gooneratne R, Ju XH. 2020. Interactions between host and gut microbiota in domestic pigs: a review. Gut Microbes. 11(3):310–334.
- Ramayo-Caldas Y, Mach N, Lepage P, Levenez F, Denis C, Lemonnier G, Leplat JJ, Billon Y, Berri M, Doré J, et al. 2016. Phylogenetic network analysis applied to pig gut microbiota identifies an ecosystem structure linked with growth traits. Isme J. 10(12):2973–2977.
- Ritchie EM, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43(7):e47.
- Roura E, Koopmans S-J, Lallès J-P, Le Huerou-Luron I, de Jager N, Schuurman T, Val-Laillet D. 2016. Critical review evaluating the pig as a model for human nutritional physiology. Nutr Res Rev. 29(1):60–90.
- Sachsenröder J, Twardziok SO, Scheuch M, Johne R. 2014. The general composition of the faecal virome of pigs depends on age, but not on feeding with a probiotic bacterium. PLoS One. 9(2):e88888.
- Sutera AM, Zumbo A, Tardiolo G, D’Alessandro E. 2020. SNPs discovery in RRLs from DNA pools of Nero Siciliano pigs with extreme and divergent phenotypes for the Back Fat Thickness (BFT) tract. Large Anim Rev. 26(6):301–304.
- Tang J-n, Zeng Z-g, Wang H-n, Yang T, Zhang P-j, Li Y-l, Zhang A-y, Fan W-q, Zhang Y, Yang X, et al. 2008. An effective method for isolation of DNA from pig faeces and comparison of five different methods. JMicrobiolMethods75. J Microbiol Methods. 75(3):432–436.
- Wang W, Hu H, Zijlstra RT, Zheng J, Gänzle MG. 2019. Metagenomic reconstructions of gut microbial metabolism in weanling pigs. Microbiome. 7(1):48.
- Wang C, Li P, Yan Q, Chen L, Li T, Zhang W, Li H, Chen C, Han X, Zhang S. 2019. Characterization of the Pig Gut Microbiome and Antibiotic Resistome in Industrialized Feedlots in China. M Systems. 4(6):e00206–19.
- Xiao L, Estellé J, Kiilerich P, Ramayo-Caldas Y, Xia Z, Feng Q, Liang S, Pedersen AØ, Kjeldsen NJ, Liu C, Maguin E, Doré J, , Le Chatelier E, Prifti E, Li J, Jia H, Liu X, Xu X, Ehrlich SD, Madsen L, Kristiansen K, Rogel-Gaillard C, Wang J. 2016. A reference gene catalogue of the pig gut microbiome. Nat. Microbiol. 1, 16161.
- Yang H, Huang X, Fang S, He M, Zhao Y, Wu Z, Yang M, Zhang Z, Chen C, Huang L. 2017. Unraveling the Fecal Microbiota and Metagenomic Functional Capacity Associated with Feed Efficiency in Pigs. Front Microbiol. 8:1555.
- Zhang Q, Widmer G, Tzipori SA. 2013. Pig model of the human gastrointestinal tract. Gut Microbes 4:193–200.
- Zumbo A, Sutera AM, Tardiolo G, D’Alessandro E. 2020. Sicilian Black Pig: An Overview. Animals. 10(12):2326.