
Abstract
A growing number of studies in the last decade described the microbial populations in different niches of the organism thank to High-throughput DNA sequencing techniques, that are easily accessible to researchers. Furthermore, network analysis allows characterisation of bacteria that are indirectly linked to outcomes such as disease, diet and host sex through their association with other taxa. The present work on the gut microbiome follows previous research in which data were collected from 8 in-house dietary intervention studies in healthy dogs. Animals were divided according to diet and sex. The process of sample collection, storage, DNA extraction and sequencing, bioinformatic and statistical analysis followed a tailored, internally defined pipeline. The extracted DNA was prepared for sequencing of the V3 and V4 regions of the 16 rRNA gene. Correlation network analysis was performed by calculating pairwise relationships between taxa using the Sparse Correlations for Compositional Data (SparCC) algorithm. First, we identified candidate bacteria that were highly abundant in the microbial community, and second, we examined taxa that were directly related to diet and sex factors. In summary, this study underpins the network structure of the gut microbiome of dogs categorised by diet and sex and provides a better explanation for the interactions between bacteria that led to the grouping of dogs based on environmental or genetic factors. Specifically, this study has provided the basis for understanding how the bacterial community in the gut is interconnected and functions as a function of diet composition and sex.
Network analysis could allow indirect association between bacteria composing the gut microbiome and outcomes, such as disease, diet and sex of the host.
This research put the basis on the definition of enterotypes also in dogs, a concept already applied in humans.
Positively correlated bacteria, belonging to modules could help to modulate diets and therapies for the treatment of enteropathies or gut-related pathologies linked to a dysbiosis status of the host.
Highlights
Introduction
The human and non-human gut harbours a large community of microorganisms that are extremely different from each other in terms of microbial diversity but have the ability to interact closely with the host. The field of the microbiome in relation to health status and environmental factors continues to expand in companion animals and livestock (Sandri et al. Citation2014; Deng and Swanson Citation2015). In these years, researchers have focussed on the role of gut microbiota involved in metabolic activities, protection against pathogens, the transmission of signals to the immune system, and direct and indirect influence on most physiological functions (Pilla and Suchodolski Citation2019).
Several associations have been discovered between the composition of the gut microbiome and the co-occurrence of diseases, which may or may not be related to the gastrointestinal tract but correlate with dysbiosis in the animal. The definition of intestinal dysbiosis refers to a change in the composition of the microbial population living in the gastrointestinal tract. The change in the balance between the abundance of bacteria inhabiting the gut can lead to functional changes in the microbial transcriptome, proteome, or metabolome (Zeng et al. Citation2017). These changes can have important consequences for the host, such as the onset of diseases like obesity, cardiovascular disease, inflammatory bowel disease, and diabetes (Muegge et al. Citation2011; David et al. Citation2014; Thorburn et al. Citation2014). These functional changes may not always occur due to processes such as functional redundancy.
The composition of the gut microbiome is influenced by several factors, both genetic and environmental. Improving host health, especially in dogs, through nutritional management or selecting the best therapy based on a response to a genetic factor are the goals of recent studies. Several experiments have shown that dietary composition is reflected in different profiles of the intestinal microbiome of dogs (Pilla and Suchodolski Citation2019; Vastolo et al. Citation2022), although the composition of the microbial population inhabiting the gut can be considered an individual fingerprint (Garcia-Mazcorro et al. Citation2017). Even though there are a growing number of studies on the intestinal microbiome of dogs, most of them focus on characterising the microbial community in individuals with different gastroenteropathies (Aggiungere citazioni). It is of utmost importance to study and characterise the gut microbiome of healthy dogs in all aspects, as it is not possible to fully understand all the variations and interactions in a microbial community of a diseased host without knowing what is happening in a healthy subject. In the previously published research on healthy dogs (Scarsella et al. Citation2020b) the role that diet and sex have on the gastrointestinal microbiota has been reported. The results suggested that the gut microbiome of dogs can be divided into enterotypes, as is the case in humans. An enterotype is a group of individuals with the same abundance of certain gut microbial taxa (Arumugam et al. Citation2011). This categorisation has not yet been applied to dogs, mainly because of the conflicting results found in the literature, which is due to the small sample size of the experiments. Although a definition of enterotypes was not yet defined for companion animals, an attempt has been done by Alessandri et al. (Citation2020), where the authors were able to perform a reconstruction of the core gut microbiota of dogs and cats with their classification into distinct community state types at both genus and species levels, identifying Bacteroides, Fusobacterium, and Prevotella 9 as the main bacterial components of the canine and feline gut microbiota.
Recently, studies on the complexity of the microbiome have made significant progress, especially thanks to high-throughput DNA sequencing technologies. These powerful techniques enabled the identification of the non-culturable bacteria in the gastrointestinal tract (Suchodolski et al. Citation2008). The sequences obtained can be analysed using a variety of data techniques to describe microbial composition and diversity and to determine how bacterial populations may change in response to various factors. To date, most of these techniques have been better suited to study individual taxa and highlight one property per time (Barberán et al. Citation2012). Thus, one of the big questions related to generating large datasets of sequences is, for example, how these microbial communities are organised. Deciphering these microbial interactions has become one of the main goals of most researchers, as it could lead to the detection of key bacteria for health and disease (Bäumler and Sperandio Citation2016; Schirmer et al. Citation2016; Eickhoff and Bassler Citation2018). The construction of co-occurrence networks based on correlations is one of the preferred methods to study these microbial interactions. Accordingly, the microorganisms are connected with an edge and the bacteria that are strongly correlated can be grouped into groups called modules. Combining taxa into modules can be useful to fully understand reciprocal relationships, such as cross-feeding or shared ecological niches (Lozupone et al. Citation2012a; Ban et al. Citation2015).
Here we present a network analysis for bacterial species in 340 faecal samples from 132 healthy dogs. This study is an evolution of a previously published work (Scarsella et al. Citation2020b) and presents unprecedented interpretations. The aim is to analyse co-occurrence patterns of microorganisms in dogs depending on their diet and sex. The proposed method is useful to study the gut microbiome of healthy dogs and to better highlight the interactions between bacteria and between bacteria and host. In particular, this study has provided the basis for understanding how the intestinal community of bacteria is interconnected and functions as a function of diet composition and sex.
Materials and methods
Sample population
The dataset is composed of individual records of dogs obtained from 8 dietary intervention studies (DIS) conducted in the past 5 years, for a total of 340 samples. All the dogs were recruited with the same inclusion criteria, which consisted of health conditions, as ascertained by a clinical examination, freedom from external and internal parasites, no pharmacological treatments for at least 3 months. A summary of the studies is reported in the S1 Table. Briefly, dogs were recruited from the different living environments for every DIS and they were undergone through diet modulation. The description of the different DIS and the collection and analytical procedures was already reported (Scarsella et al. Citation2020b). The factors considered for this study were diet and sex, which were already discussed in this research. Briefly, the diets considered in this database were four, called commercial extruded complete diet (KIBBLE), commercial moist complete diet (MOIST), home-made diet (HOME) and a raw meat diet with the addition of complementary food, called BASETM (BASE) (www.nutrigenefood.com). Moreover, dogs were grouped based on their sex, in males (M), castrated males (MC), females (F) and spayed females (FC). All protocols, procedures, and care of the animals complied with the Italian legislation on animal care and were approved by the ethical committee of the University of Udine (OBPA, Prot. N. 2/2017, Prot. N. 7/2019, Prot. N. 3/2020).
Faecal DNA extraction, sequencing and bioinformatic analysis
The entire procedure, starting from the microbial DNA extraction method and ending with taxonomic annotation with bioinformatic analysis, was standardised and utilised for all the samples. The protocols used from the DNA extraction to MiSeq sequencing (Illumina; San Diego, CA, USA) are described in the previous research from Scarsella et al. (Citation2020a).
The resulting raw sequences (FASTQ) were processed using the bioinformatic tool called Quantitative Insights Into Microbial Ecology 2 (QIIME2) (Bolyen et al. Citation2019). After demultiplexing, sequenced reads that passed the quality check (Phred score ≥30) were annotated for 16S rRNA against the green genes database. Chimaeras were also detected and then filtered from the reads and the remaining sequences were clustered into Amplicon Sequence Variants (ASVs) by using an open reference approach in QIIME 2. Sequences can be found in NCBI, and uploaded to Sequence Read Archive (Supplementary Table 1).
We used Sparse Cooccurrence Network Investigation for Compositional data (SCNIC) (Shaffer et al. Citation2020) in QIIME2 (q2-SCNIC) to perform the network analysis. The correlation network was built using the Sparse Correlations for Compositional data (SparCC) algorithm; the network was built using edges with the default correlation coefficient of at least 0.35 using SparCC method (Friedman and Alm Citation2012), and the network was visualised by Cytoscape.
Computation and statistical analysis
Annotated ASVs were imputed on a spreadsheet together with age, sex, breed and the number of the study to allow and facilitate further statistical analysis. The annotates sequences from each sample and each taxonomic level was normalised to ‰ abundance profiles, already known as Relative Abundance (RA). Taxa were attributed to the corresponding module, found with SCNIC. To confirm the results of the SparCC correlation analysis regarding taxa belonging to the same module and groups of diets and sex, a non-parametric Kruskal-Wallis test was applied at the genus level, with Bonferroni correction for multiple comparisons. A p-value < 0.05 was considered statistically significant (Addinsoft Citation2020). ASVs and modules that differed in terms of diet and sex were also identified using Analysis of Composition of Microbiomes (ANCOM) (Mandal et al. Citation2015).
Results
Microbial Co-Abundance network modules
Before the building of the microbial network and the differentiation of the taxa based on environmental and genetic factors such as diet and sex, the gut microbiome has been analysed through the SparCC algorithm, to find an existing correlation between bacteria. Afterwards, a graphic network has been constructed with only taxa that have an R-value below 0.35, and modules were created from the aggregation of bacteria based on the network analysis. This threshold has been chosen based on results from a comparative analysis of Shaffer et al. (Citation2020). Eighteen modules were detected, with a total bacterial taxa number of 55 (Supplementary Table 2). Module 0 contains 8 taxa, 4 belonging to the phylum Firmicutes, 2 bacteria from the Bacteroidetes phylum, one belonging to the phylum Proteobacteria and the last one from Fusobacteria. Module 1 has 5 taxa, all of them belonging to the Firmicutes phylum. These two modules are the largest in terms of taxa number.
Taxa and modules associated to diet
Microbial composition analysis (ANCOM) was performed to study the effect of factor Diet on the gut microbiome network. Fifty taxa were found significant to Diet, and they are shown in Figure . 28 bacteria out of 50 were aggregated into modules through SCNIC analysis. Of the 18 modules obtained from the correlation analysis, 11 were found statistically significant for the factor Diet. From a graphic appraisal of the network (Figure ), taxa belonging to modules 5, 18, 7, 8 and 11 were grouped, moreover, clusters were possible to be observed also for bacteria belonging to modules 10, 6 and 15, and for bacteria belonging to modules 9, 12 and 3. The list of bacteria belonging to the significant modules related to the Diet factor is summarised in Table . Regarding the first group of modules, most bacteria were part of the phylum Bacteroidetes, although in module 18 there were bacteria belonging to the Order of Clostridiales (phylum Firmicutes), and in module 7 present Sporobacter termitidis (phylum Firmicutes). In the second group, in modules 6 and 15 there were bacteria of the phylum Firmicutes, whilst taxa in module 10 belonged to the phylum Actinobacteria. The last group of modules showed only taxa belonging to phylum Firmicutes.
Figure 1. Volcano plot of the significative taxa and modules on factor Diet after the ANCOM analysis.
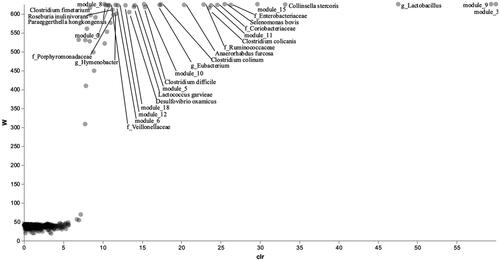
Figure 2. A graphic view of the gut microbiome network resulted from SCNIC analysis; highlighted with different colours are bacteria from significative modules for factor Diet after ANCOM analysis.
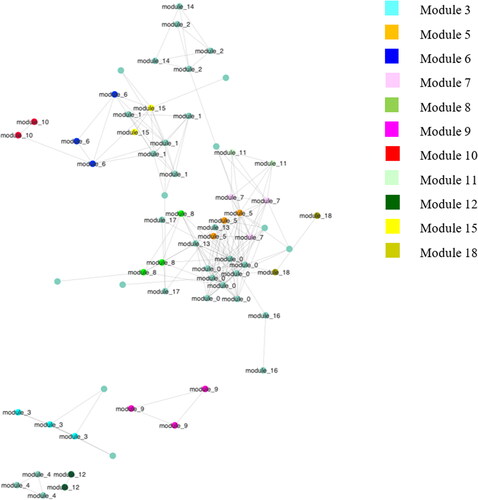
Table 1. Taxa belonging to modules significative to factor Diet at the ANCOM analysis.
The relative abundances of each of the significant modules for Diet factors were analysed with a non-parametric Kruskal-Wallis test, in order to verify the difference between the categories analysed. Modules 5 and 9 contained bacteria with higher significative RAs in the BASE diet than the other three diets. Modules 18 and 11 showed higher RAs in the MOIST diet whilst modules 8 and 6 were significantly higher in the KIBBLE diet than in the other classes. Interestingly, RAs in module 3 was almost at the same level in the BASE, HOME and MOIST diets, and were significantly higher than in the KIBBLE diet (Table ).
Table 2. Results of the Kruskal-Wallis non parametric test applied to the sum of RAs of the taxa belonging to significative modules to factor Diet after ANCOM analysis.
Taxa and modules associated to sex
ANCOM was also performed to study the effect of the factor Sex on the gut microbiome network. 43 taxa were found significant for the factor Sex (Figure ). Twenty-five bacteria of these 43 belonged to modules found through SCNIC network analysis. 9 out of 18 modules discriminated the microbiome for the factor Sex. Figure shows the network with the taxa highlighted that belonged to the modules significant for Sex. As in the case of Diet, modules 5, 7 and 11 were close together. Also, the taxa belonging to the remaining significant modules appeared to be close, but it was still possible to distinguish a group of bacteria from modules 1, 15, 6 and 10, and a second group with bacteria from modules 14 and 2. Taxa from significant modules for the factor Sex are summarised in Table . In the first group, there were almost all taxa belonging to the Bacteroidetes phylum, apart from module 7, where Sporobacter termitidis (Firmicutes phylum) was also present. In the second group, all bacteria belonged to the Firmicutes phylum, while in the last group there were taxa belonging to Firmicutes in module 14, while in module 2 there was the genus Pseudoramibacteri_Eubacterium (Firmicutes phylum), Bifidobacterium breve (Actinobacteria phylum) and S24-7 family (Bacteroidetes phylum).
Figure 3. Volcano plot of the significative taxa and modules on factor Sex after the ANCOM analysis.
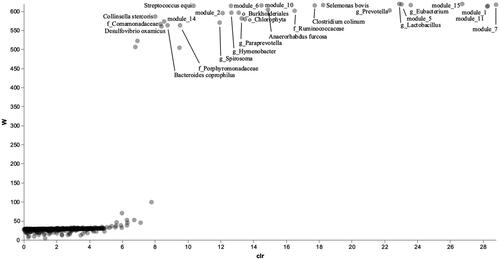
Figure 4. A graphic view of the gut microbiome network resulted from SCNIC analysis; highlighted with different colours are bacteria from significative modules for factor Sex after ANCOM analysis.
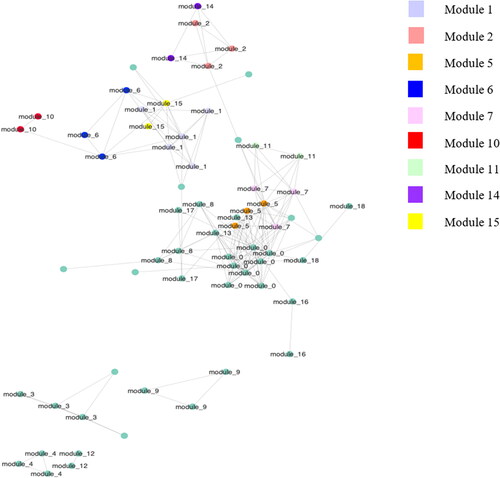
Table 3. Taxa belonging to modules significative to factor Sex at the ANCOM analysis.
Table 4. Results of the Kruskal-Wallis non-parametric test applied to the sum of RAs of the taxa belonging to significative modules to factor Sex after ANCOM analysis.
The RAs of the modules were subjected to a non-parametric Kruskal-Wallis test, to verify the significance of the factor Sex. The modules belonging to the first group contained taxa with significantly higher abundances in the entire female category, compared to entire male dogs and the remaining neutered dogs. The second group of modules appeared to have higher relative abundances in castrated subjects, both male and female, compared to entire male and female dogs.
Discussion
Network analysis could contribute to a better understanding of the canine gut microbiome by providing an integrative view of gut microbial ecology based on microbial modules. To the best of our knowledge, this is the first network analysis approach that has been applied to the canine gut microbiome.
As noted in other studies, analysis of co-occurring microorganisms along with correlation network analysis allows for the summary visualisation of much information (Chaffron et al. Citation2010). This method has been successfully applied to distinguish the links between marine microorganisms and their environment (Ruan et al. Citation2006). For this study, most of the bacteria that make up the gut microbiome of dogs were grouped into modules. The graphic representation of the network shows that significant modules in the categories of diet and sex were tightly grouped based on the degree of significance with each category of diet and sex. These structural features allow for easy comparison between gut microbiomes derived from a complex dataset to demonstrate how environmental and genetic factors such as diet and gender can influence microbial community composition and function.
Only positive associations are calculated with the SCNIC bioinformatics tool, which uses the SparCC algorithm. It may seem unnatural at first glance, but these results are logical when research focuses on the gut microbiome. In the anoxic environment of the gut, microbial energy production is limited, so positive associations, such as cross-feeding, would make energy production and utilisation more efficient (Pacheco et al. Citation2019). In addition, positive microbial associations mitigate potential stresses on the ecosystem, further increasing the diversity of a healthy gut microbiome (Stachowicz Citation2001; Lozupone et al. Citation2012b). Moreover, it has been demonstrated that predator-prey interactions decrease co-occurrence network performance. This is due mainly to the existing limitations related to the observed species abundances, which should be considered as cross-sectional data rather than longitudinal data since the time resolutions study are still low. The detection of predator-prey interactions is more difficult than the detection of other types of interactions (Hirano and Takemoto Citation2019). In the end, another reason to explain why we choose to calculate only positive associations is that identifying clusters within a biological network, which can be called modules as well, is a key issue in network biology, that will likely grow in the future. These clusters could be, for example, groups of co-existing or coevolving microbes contributing toward a disease (Layeghifard et al. Citation2017).
After network analysis with SCNIC, 18 modules were found, of which 11 were significant for the factor Diet. For the factor sex, 9 of 18 modules were significant. Diet is known to alter microbial population composition and activity (Sandri et al. Citation2020; Scarsella et al. Citation2020a) through the introduction of new bacteria, provision of nutrients, selection for enrichment or depletion of certain taxa through nutrient excess or starvation, and finally through shifts in the expression profiles of some bacteria (David et al. Citation2014). These studies have shown that diet has a greater influence on the microbiome than host genotype, but it is not clear whether the differences in the microbiome are due to the influence of diet on the subjects or to the influence that some bacteria exert on other bacteria and on the host itself. Other studies have shown the major influence of existing bacterial interactions on the composition of the gut microbiome. Network analysis is relatively new and a limited number of studies involve animals and none involve dogs. However, the principles of co-occurrence can be applied to other environments. In a study by Mandakovic et al. (Citation2018), the authors focussed on comparing two microbial co-occurrence networks representing soil bacterial communities from two different sections of a pH, temperature, and moisture gradient running along a western slope of the Andes in the Atacama Desert. They found that despite the large physicochemical and nutritional differences affecting bacterial community structure along the slope, both patterns of association of the network were quite consistent. They slightly modified the traditional notion of community resilience by suggesting that relationships among OTUs can also be resilient. In another paper, the authors analysed the co-occurrence of bacteria in either Ixodes ricinus ticks or the spleen of one of their primary hosts, the vole Myodes glareolus. To assess the effects of habitat on the ecological communities of bacteria associated with ticks and voles, two different biotopes were included in the study. The authors discovered that the networks exhibited a structure called a "small world." This was a scale-free property that strongly correlated with the robustness of the network. Thus, the networks that emerged from the study were quite resilient, meaning that the deletion of one node did not significantly affect the connectivity of the other nodes. This observation suggests the absence of trophic dependence or competition between co-occurring taxa (Estrada-Peña et al. Citation2018). From studies on bacterial dynamics, microorganisms related to bacteria already present in the gut are more readily accepted and included in the microbial community. This is the so-called phylogenetic under-dispersion (nepotism) hypothesis (Loftus et al. Citation2021). All these observations are consistent with the results reported in this study. Indeed, the bacteria that make up the gut microbiome form interactions that are characteristic of macro groups of healthy subjects. These interactions are dependent on and simultaneously influence subjects categorised by environmental and genetic factors such as diet and sex.
Several studies have shown that the composition of the gut microbiome varies widely between subjects (Forster et al. Citation2018; Cintio et al. Citation2020; Sandri et al. Citation2020). It is likely that each dog has a unique faecal microbiome that responds to even slight changes in diet. Cintio et al. (Citation2020) found that the gut microbiome varies in arthritic dogs, although there is great intervariability between dogs. They suggested that different bacterial strains might contribute in their own way to alter the inflammatory status of the subjects. Thus, it was not possible to attribute a role in arthritic disease to each taxa that make up the microbiome, but the hypothesis is that variation in the overall composition of the gut microbiome could reshape the entire physiology of the host.
The most informative modules related to factor Diet were module 3, module 9, and module 15. All bacteria included in these modules belong to the Firmicutes phylum. Firmicutes are thought to play a role in modulating the immune system (Ling et al. Citation2014). In addition, obesity was shown to be associated with an increase in Firmicutes and a decrease in Bacteroidetes in humans and mice (Turnbaugh et al. Citation2006; Bruni et al. Citation2020). Module 3 consists only of Firmicutes bacteria, particularly taxa of the genus Clostridium, both from the families Clostridiceae and Peptostreptococcaceae, and Clostridium baratii. This module has shown that the sum of RAs of the bacteria belonging to it is significantly higher in all diets that have a "wet" form, i.e. BASE, HOME and commercial wet diets. This evidence would confirm that the presence of raw meat and the physical form of these diets have a similar influence on shaping the gut microbiome (Scarsella et al. Citation2020b). The increase in Clostridium in dogs fed the diets BASE and HOME is consistent with another study in which these taxa decreased in dogs fed a commercial extruded diet compared to dogs fed a raw meat diet (Bermingham et al. Citation2017).
Very little information is available on the variation of the gut microbiome as a function of sex in dogs. In mice, the gut microbiota of males and females differs after puberty. The mechanism of sex-specific influence remains unclear, but there appears to be a bidirectional interaction between the microbiota and host endocrine status, in part because differences in microbial composition between males and females are reversed by male castration (Markle et al. Citation2013). Module 1 appeared to be the most significant for the sex factor in the ANCOM analysis. All bacteria in this module belong to the Firmicutes strain and the sum of RAs showed a higher significant value for the castrated male and female categories than for the entire male and female subjects. This is consistent with the results of the previous study (Scarsella et al. Citation2020b). Also included in this module are Dorea longicatena and Ruminococcus lactaris. The genera of these two taxa were found to discriminate against whole subjects compared to castrated subjects in the Scarsella et al. (Citation2020b) study.
The clustering of bacteria in these modules could be indicative of factors affecting the healthy gut microbiome of dogs. However, we found modules that were not significant for either the diet factor or the sex factor. The latter can be explained by the presence of taxa that are more resistant to change, as observed in humans by Loftus et al. (Citation2021). The authors suggest that these taxa have specific compound requirements for their metabolism and then compete or cooperate with the other bacteria.
Conclusion
This study has helped to better understand the ecology of the canine gut microbiome in the context of environmental and genetic factors such as diet and sex and paves the way for the definition of enterotypes in dogs, a concept already used in humans. With these results, individuals could be clustered based on the abundance of gut microbial taxa. In addition, taxa belonging to modules could help understand how to modulate diets and therapies to treat enteropathies or gut-related pathologies associated with host dysbiosis. Understanding the microbiota from an ecological perspective could shed light on how to promote host health by targeting this microbial community in clinical treatments. Once we understand the desired composition and functional state of the gut microbiota, we can determine which features, when disrupted, are associated with the disease. However, the complexity of the microbiota and the variability within and between subjects make it difficult to define a "desired" state for a population and, consequently, for an individual. Further studies are needed to reveal new associations between bacteria and other factors that may influence the gut microbiome.
Supplemental Material
Download MS Excel (18.6 KB)Supplemental Material
Download MS Excel (11.3 KB)Disclosure statement
The authors declare that there is no conflict of interest associated with the paper. The authors alone are responsible for the content and writing of this article.
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the supplementary materials of the article.
References
- Addinsoft 2020. XLSTAT statistical and data analysis solution. Boston, USA.
- Alessandri G, Milani C, Mancabelli L, Longhi G, Anzalone R, Lugli GA, Duranti S, Turroni F, Ossiprandi MC, van Sinderen D, et al. 2020. Deciphering the bifidobacterial populations within the canine and feline guy microbiota. Appl Environ Microbiol. 86(7):e02875-19.
- Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto J-M, et al. 2011. Enterotypes of the human gut microbiome. Nature. 473(7346):174–180.
- Ban Y, An L, Jiang H. 2015. Investigating microbial co-occurrence patterns based on metagenomic compositional data. Bioinformatics. 31(20):3322–3329.
- Barberán A, Bates ST, Casamayor EO, Fierer N. 2012. Using network analysis to explore co-occurrence patterns in soil microbial communities. Isme J. 6(2):343–351.
- Bäumler AJ, Sperandio V. 2016. Interactions between the microbiota and pathogenic bacteria in the gut. Nature. 535(7610):85–93.
- Bermingham EN, Maclean P, Thomas DG, Cave NJ, Young W. 2017. Key bacterial families (clostridiaceae, erysipelotrichaceae and bacteroidaceae) are related to the digestion of protein and energy in dogs. PeerJ. 5(march):e3019.
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 37(8):852–857.
- Bruni N, Martello E, Fusi E, Meineri G, Giardini A. 2020. Study of faecal parameters and body condition in dogs with a diet supplemented with Lactobacillus acidophilus D2/CSL (CECT 4529). Ital J Anim Sci. 19(1):704–711.
- Chaffron S, Rehrauer H, Pernthaler J, von Mering C. 2010. A global network of coexisting microbes from environmental and whole-genome sequence data. Genome Res. 20(7):947–959.
- Cintio M, Scarsella E, Sgorlon S, Sandri M, Stefanon B. 2020. Gut microbiome of healthy and arthritic dogs. Veterinary Sciences. 7(3):92.
- David LA, Materna AC, Friedman J, Campos-Baptista MI, Blackburn MC, Perrotta A, Erdman SE, Alm EJ. 2014. Host lifestyle affects human microbiota on daily timescales. Genome Biol. 15(7):R89.
- Deng P, Swanson KS. 2015. Gut microbiota of humans, dogs and cats: current knowledge and future opportunities and challenges. Br J Nutr. 113(S1):S6–S17.
- Eickhoff MJ, Bassler BL. 2018. SnapShot: bacterial quorum sensing. Cell. 174(5):1328–1328.e1.
- Estrada-Peña A, Cabezas-Cruz A, Pollet T, Vayssier-Taussat M, Cosson JF. 2018. High throughput sequencing and network analysis disentangle the microbial communities of ticks and hosts within and between ecosystems. Frontiers in Cellular and Infectious Microbiology. 8:236.
- Forster GM, Stockman J, Noyes N, Heuberger AL, Broeckling CD, Bantle CM, Ryan EP. 2018. A comparative study of serum biochemistry, metabolome and microbiome parameters of clinically healthy, normal weight, overweight, and obese companion dogs. Top Companion Anim Med. 33(4):126–135.
- Friedman J, Alm EJ. 2012. Inferring correlation networks from genomic survey data. PLOS Comput Biol. 8(9):e1002687.
- Garcia-Mazcorro JF, Barcenas-Walls JR, Suchodolski JS, Steiner JM. 2017. Molecular assessment of the fecal microbiota in healthy cats and dogs before and during supplementation with fructo-oligosaccharides (FOS) and inulin using high-throughput 454-Pyrosequencing. PeerJ. 5(aprile):e3184.
- Hirano H, Takemoto K. 2019. Difficulty in inferring microbial community structure based on co-occurrence network approaches. BMC Bioinformatics. 20(1):329 doi:10.1186/s12859-019-2915-1. PMC: 31195956
- Layeghifard M, Hwang DM, Guttman DS. 2017. Disentangling interactions in the microbiome: A network perspective. Trends Microbiol. 25(3):217–228. doi:10.1016/j.tim.2016.11.008.
- Ling Z, Li Z, Liu X, Cheng Y, Luo Y, Tong X, Yuan L, Wang Y, Sun J, Li L, et al. 2014. Altered fecal microbiota composition associated with food allergy in infants. Appl Environ Microbiol. 80(8):2546–2554.
- Loftus M, Al-Deen Hassouneh S, Yooseph S. 2021. Bacterial associations in the healthy human gut microbiome across populations. Sci Rep. 11(1):2828.
- Lozupone CA, Faust K, Raes J, Faith JJ, Frank DN, Zaneveld J, Gordon JI, Knight R. 2012a. Identifying genomic and metabolic features that can underlie early successional and opportunistic lifestyles of human gut symbionts. Genome Res. 22(10):1974–1984.
- Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. 2012b. Diversity, stability and resilience of the human gut microbiota. Nature. 489(7415):220–230.
- Mandakovic D, Rojas C, Maldonado J, Latorre M, Travisany D, Delage E, Bihouée A, Jean G, Díaz FP, Fernández-Gómez B, et al. 2018. Structure and co-occurrence patterns in microbial communities under acute environmental stress reveal ecological factors fostering resilience. Sci Rep. 8(1):5875.
- Mandal S, Van Treuren W, White RA, Eggesbø M, Knight R, Peddada SD. 2015. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb Ecol Health Dis. 26(1):27663.
- Markle JGM, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, von Bergen M, McCoy KD, Macpherson AJ, Danska JS. 2013. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science. 339(6123):1084–1088.
- Muegge BD, Kuczynski J, Knights D, Clemente JC, González A, Fontana L, Henrissat B, Knight R, Gordon JI. 2011. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science. 332(6032):970–974.
- Pacheco AR, Moel M, Segrè D. 2019. Costless metabolic secretions as drivers of interspecies interactions in microbial ecosystems. Nat Commun. 10(1):103.
- Pilla R, Suchodolski JS. 2019. The role of the canine gut microbiome and metabolome in health and gastrointestinal disease. Front Vet Sci. 6:498.
- Ruan Q, Dutta D, Schwalbach MS, Steele JA, Fuhrman JA, Sun e F. 2006. Local similarity analysis reveals unique associations among marine bacterioplankton species and environmental factors. Bioinformatics. 22(20):2532–2538.
- Sandri M, Manfrin C, Pallavicini A, Stefanon B. 2014. Microbial biodiversity of the liquid fraction of rumen content from lactating cows. Animal. 8(4):572–579.
- Sandri M, Sgorlon S, Scarsella E, Stefanon B. 2020. Effect of different starch sources in a raw meat-based diet on fecal microbiome in dogs housed in a shelter. Animal Nutrition. 6(3):353–361.
- Scarsella E, Cintio M, Iacumin L, Ginaldi F, Stefanon B. 2020a. Interplay between neuroendocrine biomarkers and gut microbiota in dogs supplemented with grape proanthocyanidins: results of dietary intervention study. Animals. 10(3):531.
- Scarsella E, Stefanon B, Cintio M, Licastro D, Sgorlon S, Dal Monego S, Sandri M. 2020b. Learning machine approach reveals microbial signatures of diet and sex in Dog. PLOS One. 15(8):e0237874.
- Schirmer M, Smeekens SP, Vlamakis H, Jaeger M, Oosting M, Franzosa EA, Ter Horst R, Jansen T, Jacobs L, Bonder MJ, et al. 2016. Linking the human gut microbiome to inflammatory cytokine production capacity. Cell. 167(4):1125–1136.e8.
- Shaffer M, Thurimella K, Lozupone CA. 2020. SCNIC: sparse correlation network investigation for compositional data. 10.1101/2020.11.13.380733v1.
- Stachowicz JJ. 2001. Mutualism, facilitation, and the structure of ecological communities: positive interactions play a critical, but underappreciated, role in ecological communities by reducing physical or biotic stresses in existing habitats and by creating new habitats on which many species depend. BioScience. 51(3):235–246.
- Suchodolski JS, Camacho J, Steiner JM. 2008. Analysis of bacterial diversity in the canine duodenum, jejunum, ileum, and colon by comparative 16S rRNA gene analysis. FEMS Microbiology Ecology. 66(3):567–578.
- Thorburn AN, Macia L, Mackay CR. 2014. Diet, metabolites, and “western-lifestyle” inflammatory diseases. Immunity. 40(6):833–842.
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. 2006. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 444(7122):1027–1031.
- Vastolo A, Riedmüller J, Cutrignelli MI, Zentek J. 2022. Evaluation of the effect of different dietary lipid sourses on dogs. Faecal Microbial Population and Activities. Animals. 12(11):1368.
- Zeng MY, Inohara N, Nuñez G. 2017. Mechanisms of inflammation-driven bacterial dysbiosis in the gut. Mucosal Immunol. 10(1):18–26.