
Abstract
Protein misfolding and aggregation underpin several fatal neurodegenerative diseases, including Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), and frontotemporal dementia (FTD). There are no treatments that directly antagonize the protein-misfolding events that cause these disorders. Agents that reverse protein misfolding and restore proteins to native form and function could simultaneously eliminate any deleterious loss-of-function or toxic gain-of-function caused by misfolded conformers. Moreover, a disruptive technology of this nature would eliminate self-templating conformers that spread pathology and catalyze formation of toxic, soluble oligomers. Here, we highlight our efforts to engineer Hsp104, a protein disaggregase from yeast, to more effectively disaggregate misfolded proteins connected with PD, ALS, and FTD. Remarkably subtle modifications of Hsp104 primary sequence yielded large gains in protective activity against deleterious α-synuclein, TDP-43, FUS, and TAF15 misfolding. Unusually, in many cases loss of amino acid identity at select positions in Hsp104 rather than specific mutation conferred a robust therapeutic gain-of-function. Nevertheless, the misfolding and toxicity of EWSR1, an RNA-binding protein with a prion-like domain linked to ALS and FTD, could not be buffered by potentiated Hsp104 variants, indicating that further amelioration of disaggregase activity or sharpening of substrate specificity is warranted. We suggest that neuroprotection is achievable for diverse neurodegenerative conditions via surprisingly subtle structural modifications of existing chaperones.
Abbreviations
| PD | = | Parkinson's disease |
| ALS | = | amyotrophic lateral sclerosis |
| FTD | = | frontotemporal dementia |
| AD | = | Alzheimer's disease |
| α-syn | = | α-synuclein |
| WT | = | wild-type |
| HD | = | Huntington's disease |
| NBD | = | nucleotide-binding domain |
| MD | = | middle domain |
Introduction
As population demographics shift toward older age groups, several fatal and presently incurable neurodegenerative diseases, including: Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), and frontotemporal dementia (FTD) will inevitably increase in prevalence.Citation1–5 These diseases threaten public health worldwide and present a formidable barrier to living longer, more fulfilling lives. Game-changing therapeutic solutions that attack the cause of these diseases and not merely the symptoms are urgently needed. Indeed, while ‘wars’ on cancer and heart disease have yielded myriad promising drugs, the same cannot be said for the neurodegenerative diseases where, aside from one notable exception for the treatment of familial amyloid neuropathy,Citation6–11 the drug pipelines are practically empty. Although each neurodegenerative disease is fundamentally different – some debilitating movement but preserving memory, others destroying memory but sparing movement – a recurring and unifying facet is the accumulation of misfolded protein structures in the brain.Citation1–5,12–14 These misfolded structures can even become self-templating and propagate disease from single or multiple sites of origination.Citation2,12–16
Many of the misfolded proteins found in pathological inclusions, e.g. α−synuclein (α-syn) or TDP-43, are expressed in almost all cells, yet only seem to misfold and confer toxicity in specific neurons.Citation1–5,17 Thus, motor neurons are primarily afflicted in ALS,Citation3 whereas dopaminergic neurons are selectively devastated in PD.Citation4,17 In all of these diseases, the proteostasis network collapses and fails to counter specific protein-misfolding events,Citation18–20 which ultimately overwhelm the system and can even transmit disease.Citation2,12–16 A key therapeutic advance will come with ability to rescue selectively vulnerable neurons with agents that directly antagonize or reverse the deleterious protein-misfolding events that underpin neurodegeneration.Citation21,22 Indeed, agents that reverse protein misfolding and restore proteins to native form and function could simultaneously eliminate any deleterious loss-of-function or toxic gain-of-function caused by misfolded conformers.Citation21,22 Moreover, a disruptive technology of this nature would eliminate self-templating conformers that spread pathologyCitation2,12–16 and catalyze formation of soluble toxic oligomersCitation23 via secondary nucleation.Citation12,24 In other contexts, the accumulation of misfolded proteins trapped in inclusions can incur fitness costs and likely also contributes to cancer and aging.Citation25–30 Hence, agents capable of reactivating misfolded and aggregated proteins could have therapeutic utility beyond neurodegenerative disease.
The very same type of misfolded conformers (e.g., prions or amyloids) that underpin incurable neurodegenerative diseases have, surprisingly, been appropriated during evolution for adaptive purposes elsewhere.Citation31-46 For example, the same type of prion domain that enables Sup35 and Mot3 to form beneficial prions in yeastCitation31,33,41 causes TDP-43 and FUS to misfold in ALS,Citation2,47-51 and hnRNPA1 and hnRNPA2 to misfold in multisystem proteinopathy.Citation52,53 Indeed, yeast exploit prions for beneficial purposes, including stress resistance and the evolution of new traits in fluctuating environments.Citation31–35,41,44,45,54 CPEB prions might even encode our own long-term memories.Citation41,55–60 An important mission is to establish a deep and rigorous mechanistic understanding of how nature has controlled protein misfolding (sometimes even for adaptive purposes) so that we can apply and if necessary re-engineer these natural solutions to counter protein misfolding in disease. Unlocking nature's secrets to mitigate protein misfolding could empower unparalleled opportunities to eradicate neurodegenerative disease. In this review, we highlight our efforts to engineer Hsp104, a protein disaggregase from yeast, to more effectively disaggregate misfolded proteins connected with PD, ALS, and FTD.Citation61–64
Applying Hsp104 and Engineered Variants to Deleterious Protein Misfolding
Hsp104 is a hexameric AAA+ ATPase and protein disaggregase from yeast.Citation65,66 Hsp104 forms a ring-shaped complex that disaggregates protein by coupling ATP hydrolysis to partial or complete substrate translocation across its central channel.Citation21,67–72 Optimal Hsp104 activity is typically achieved in conjunction with the Hsp110, Hsp70, and Hsp40 chaperone system.Citation65,73–76 Importantly, Hsp104 is the only cellular agent known to rapidly dissolve stable amyloid fibrils,Citation73,77,88 which can self-template their own cross-β conformation and encode transmissible phenotypes.Citation12,15,41 Hsp104 activity has enabled yeast to stably propagate prions and even exploit them for beneficial purposes.Citation32,33,41,54,89,90 Hsp104 also eradicates toxic soluble oligomers that adopt a generic conformationCitation23 formed by diverse proteins.Citation78–80 Thus, amyloid fibrils and toxic oligomers are not intractable and can be eliminated rapidly by Hsp104.Citation61,73,77–84,87,91
Inexplicably, Hsp104 has no metazoan ortholog despite being highly conserved in bacteria, plants, fungi, chromista, and protozoa.Citation39,40,52 The reason for the loss of Hsp104 from metazoan lineages is uncertain, especially as Hsp104 can be expressed safely and broadly in worm, fly, mouse, and rat.Citation61,78,92–95 However, it has been suggested that the possession of Hsp104's potent disaggregase modality might have incurred a detrimental fitness cost at the divergence of metazoa from protozoa.Citation96,97 We have hypothesized that Hsp104 could be applied as a disruptive technology to combat protein-misfolding disorders.Citation21,22,98 Can we add back the powerful disaggregase activity that animals have lost very early in their evolution to impart therapeutic benefit in situations where protein misfolding has caused disease? Applying powerful biochemical activities isolated from the microbial world to solve problems posed by human disease has strong precedent. For example, botulinum neurotoxin variants can achieve therapeutic benefit across a large range of clinical conditions including various movement, urologic, and secretory disorders,Citation99–101 due to their highly potent and selective ability to cleave SNARE proteins and prevent secretion.Citation102,103
Importantly, Hsp104 returns aggregated proteins to native structure and function,Citation65,66,85 and could simultaneously reverse toxic gain-of-function and loss-of-function phenotypes linked to protein misfolding, as well as eliminate self-templating conformers that spread disease.Citation13,21,22,98 We have established that Hsp104 disassembles toxic α-syn oligomers and amyloid,Citation78,84,98 and rescues α-syn-induced dopaminergic neurodegeneration in the mammalian substantia nigra.Citation78 Hsp104 suppresses polyglutamine toxicity in Drosophila even when expressed after the onset of polyglutamine-induced degeneration, whereas Hsp70 is ineffective.Citation92 Thus, Hsp104 is the first disaggregase or chaperone treatment administered after the onset of pathogenic protein-induced degeneration that mitigates disease progression.Citation92 Moreover, Hsp104 can disassemble amyloid and soluble toxic oligomeric forms of diverse wild-type (WT) and mutant proteins connected to AD, PD, Huntington's Disease (HD) or type II diabetes, including: Aβ42, tau, α-syn, polyglutamine, and amylin.Citation84,104
For some substrates, however, Hsp104 is not as active as it is against natural yeast prions, such as those formed by Sup35.Citation79,80,84,101 Indeed, even some Sup35 prion strains are more resistant to elimination by Hsp104, and this differential sensitivity can drive ‘protein only’ evolution.Citation91 Thus, a key goal is to potentiate Hsp104 activity against specific disease-associated substrates via engineering and evolution.Citation21,22,61,62,98 However, chaperones are difficult targets for protein engineering due to their large size, and protein disaggregases such as Hsp104 have poorly understood structures,Citation67,82,83,105–108 making rational design challenging. Hence, we empl-oyed an unbiased approach to isolate improved Hsp104 variants by screening large libraries of Hsp104 variants to isolate those that rescue yeast models that recapitulate salient features of various neurodegenerative proteinopathies, including protein mislocalization, aggregation, and toxicity.Citation61,64
We focused on yeast models of neurodegenerative proteinopathies caused by aberrant TDP-43 and FUS misfolding, e.g. ALS and FTD,Citation3,109,110 or α-syn misfolding, e.g., PD and multiple system atrophy.Citation4,17,111,112 In these yeast models, the neurodegenerative disease protein is overexpressed from the galactose-inducible promoter, which induces protein mislocalization, aggregation, and toxicity idiosyncratic to each specific neurodegenerative disease.Citation47–49,113–118 Overexpression is a key tool to study the aggregation and toxicity of human neurodegenerative disease proteins in yeast and enables protein misfolding via increasing protein concentration and overwhelming proteostatic buffers.Citation117,118 Importantly, an established cause of FTD or ALS is increased expression of TDP-43Citation119 or FUSCitation120,121 respectively, and elevated α-syn expression causes PD.Citation122-125 These yeast models have proven to be an extraordinary resource and have enabled the identification of genetic and small-molecule suppressors of TDP-43, FUS, and α-syn toxicity, which can also exhibit therapeutic efficacy against degeneration in the metazoan nervous system, human cells and neurons, and even patient-derived neurons.Citation49,114,116,126–138 In the case of TDP-43, the yeast model even enabled identification of a common genetic risk factor for ALS: intermediate polyglutamine expansions (27-33 glutamines) in ataxin 2.Citation128,139–145 Indeed, several aggregation-prone human RNA-binding proteins with prion-like domains, including TAF15, EWSR1, hnRNPA1, and hnRNPA2 have been successfully predicted as neurodegenerative disease genes based on initial studies in yeast.Citation50–53,146,147 Thus, the power of yeast to elucidate protein-misfolding events and methods to counter them relevant to neurodegenerative disease should not be doubted.Citation130,137,139
Importantly, for our purposes, the toxicity of TDP-43, FUS, and α-syn in yeast is maintained in Δhsp104 backgrounds,Citation61 indicating that, unlike polyglutamine toxicity in yeast,Citation148–150 TDP-43, FUS, and α-syn toxicity does not depend on Hsp104 or Hsp104-dependent prions. Moreover, in the Δhsp104 background, overexpression of wild-type (WT) Hsp104 did not rescue TDP-43, FUS, or α-syn toxicity.Citation61–64 Thus, we could explore Hsp104 sequence space in the absence of WT Hsp104 and be certain that any Hsp104 variants that rescued TDP-43, FUS, and α-syn toxicity were due to a novel therapeutic gain of Hsp104 function.Citation61–64
Potentiating Mutations Can be Uncovered in the Middle Domain of Hsp104
Hsp104 is comprised of 5 domains: an N-terminal domain, nucleotide-binding domain 1 (NBD1), a coiled-coil middle domain (MD), NBD2, and a short acidic C-terminal domain.Citation67,81 We focused our libraries on the coiled-coil MD, which is comprised of 4 α-helices and facilitates optimal ATPase activity, communication between NBD1 and NBD2, intrinsic disaggregase activity, and interactions with Hsp70 during disordered aggregate dissolution.Citation67,82,151–154 Importantly, the MD is less conserved than the 2 NBDs, indicating that it can withstand various missense mutations without eliminating disaggregase functionality.Citation67 Indeed, the MD can even tolerate large protein insertions (e.g., insertion of lysozyme between Asn467 and Glu468 in MD helix 2) or helix replacements and yet still maintain Hsp104 disaggregase activity.Citation106,152,153 Moreover, previous studies suggested that Hsp104 MD variants can have unexpected gain-of-function phenotypes, including rescue of polyglutamine aggregation and toxicity.Citation106,150,155
Remarkably, we uncovered a large number of MD variants that enabled Hsp104 to rescue the aggregation and toxicity of TDP-43, FUS, and α-syn in yeast.Citation61 These mutations were located throughout the MD in helix 1 (e.g., V426L), the distal loop between helix 1 and 2 (e.g., A437W), and helix 3 (e.g., A503S, Y507C)Citation61 ( and ). We also uncovered potentiating mutations in the small domain of NBD1 (e.g., N539K) immediately adjacent to the C-terminal end of the MDCitation61 (). Interestingly, we recently discovered that the N-terminal domain of Hsp104 (residues 1-157) is essential for the potentiated activity of Hsp104A503V and Hsp104A503S.Citation156
TABLE 1. Summary of potentiating Hsp104 mutations. The mutations that potentiate Hsp104 activity, their location, and their properties are listed based onCitation61,63,107
Figure 1. Potentiated Hsp104 variants suppress aggregation and mislocalization of disease proteins in yeast proteinopathy models. (A) Homology model of the MD and a portion of the small domain of NBD1 of Hsp104. Side chains of indicated residues are shown as sticks. (B) Fluorescence microscopy of yeast coexpressing fluorescently tagged TDP-43 and Hsp104WT, Hsp104A503V, or vector. Cells are stained with DAPI to visualize nuclei (blue). TDP-43 only exhibits nuclear localization upon coexpression of potentiated Hsp104A503V. (C) Fluorescence microscopy of cells coexpressing FUS-GFP and Hsp104WT, Hsp104A503V, or vector. Yeast coexpressing potentiated Hsp104A503V display fewer cytoplasmic FUS foci. (D) Fluorescence microscopy of cells coexpressing α-synuclein-YFP and Hsp104WT, Hsp104A503V, or vector. Yeast coexpressing potentiated Hsp104A503V display fewer cytoplasmic α-syn foci, and α-syn only accumulates at the plasma membrane upon coexpression of Hsp104A503V.
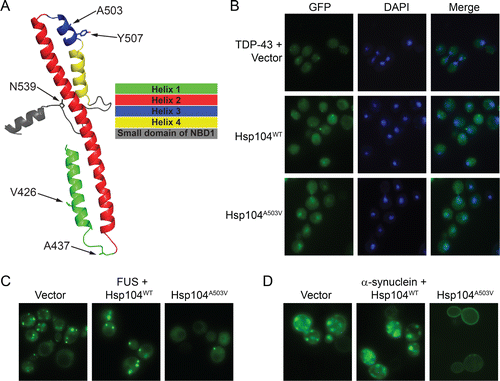
Typically, these potentiated Hsp104 variants reduced aggregation and toxicity of TDP-43, FUS, and α-syn in yeast without reducing their expression level or inducing a heat shock response.Citation61 The expression of potentiated Hsp104 variants was also similar or lower to the expression of WT Hsp104, which failed to rescue TDP-43, FUS, and α-syn aggregation or toxicity.Citation61,63 Moreover, neither the unfolded protein response nor autophagy was required for potentiated Hsp104 variants to rescue toxicity.Citation61 Remarkably, aggregates were now cleared from the majority of cells, and the correct localization of the neurodegenerative disease protein could be restored.Citation61,63 Thus, potentiated Hsp104 variants resolved cytoplasmic TDP-43 aggregates and restored TDP-43 to the nucleusCitation61,63 (). A major goal in ALS therapeutics is to achieve this phenotype in degenerating motor neurons.Citation3 For FUS, cytoplasmic aggregates were dissolved, but FUS remained cytoplasmic and did not return to the nucleus because the yeast nuclear-import machinery fails to decode the FUS PY-NLSCitation49,114 (). Several suppressors of FUS toxicity in yeast have been uncovered in genome-wide screens,Citation49,114 but none of these cleared FUS aggregates, indicating a novel mode of rescue by potentiated Hsp104 variants.Citation61,63 Potentiated Hsp104 variants cleared cytoplasmic α-syn inclusions and restored plasma membrane localization of α-synCitation61,63 (). Achieving these phenotypes in patients would provide a game-changing solution for PD. Importantly, 2 potentiated Hsp104 variants, Hsp104A503S and Hsp104DPLF-A503V (where substrate-engaging pore-loop tyrosines, Y257 and Y662 are mutated to F), rescued α-syn-induced dopaminergic neurodegeneration in a C. elegans model of PD.Citation61 Thus, potentiated Hsp104 variants confer neuroprotective phenotypes in the context of the metazoan nervous system. Next, it will be important to assess the efficacy of potentiated Hsp104 variants in mammalian models of PDCitation78 as well as TDP-43- and FUS-related ALS.Citation157,158 Nonetheless, we have established the first ever example of engineered disaggregases rescuing neurodegeneration in the metazoan nervous system. Thus, general neuroprotection via activated protein disaggregases may be achievable for a range of neurodegenerative diseases.
Several missense mutations in TDP-43 or FUS cause ALS,Citation3,159,160 whereas specific α-syn mutations cause PD.Citation17,161,162 Many of these variants cause aggressive, early-onset forms of disease.Citation3,17,159-162 Enhanced proteotoxicity often stems from an increased intrinsic propensity of the mutant protein to misfold in isolation,Citation48,163 e.g., as with TDP-43M337V and α-synA53T, or to mislocalize in vivo, e.g. as with FUSP525L and FUSR521C.Citation3,157,164–166 A subset of the potentiated Hsp104 variants, Hsp104A503V, Hsp104A503S, Hsp104A503G, Hsp104V426L, Hsp104A437W, Hsp104Y507C, Hsp104N539K, and Hsp104DPLF-A503V, were tested against yeast models expressing disease-linked: TDP-43 (TDP-43A315T, TDP-43Q331K, and TDP-43M337V Citation167,168); FUS (FUSP525L and FUSR521C Citation3,157,164-166); and α-syn (α-synA53T and α-synE46K Citation17,161,162). Remarkably, potentiated Hsp104 variants suppressed toxicity and aggregation of these disease-linked forms of TDP-43, FUS, and α-syn.Citation63
Initially, these Hsp104 variants appeared to be potentiated against all neurodegenerative disease substrates, but this was not the case.Citation63 We also tested their activity against EWSR1, another RNA-binding protein with a prion-like domain implicated in ALS and FTD, which aggregates and is toxic in yeast.Citation3,50,146,147,169 None of the potentiated Hsp104 variants suppressed EWSR1 aggregation or toxicity in yeast, and instead enhanced toxicity except for Hsp104V426L and Hsp104N539K, which had no effect like WT Hsp104.Citation63 This result was surprising as EWSR1 is closely related to FUS in terms of primary sequence and domain architecture.Citation170 Moreover, potentiated Hsp104 variants could rescue TAF15 toxicity in yeast.Citation63 TAF15 is also an RNA-binding protein with a prion-like domain implicated in ALS and FTD that is closely related to FUS and EWSR1.Citation50,146,147 We could rationalize these findings as in vitro Hsp104A503S but not WT Hsp104 could disaggregate preformed FUS and TAF15 fibrils, but not EWSR1 fibrils.Citation63 Thus, EWSR1 fibrils appear to be refractory to potentiated Hsp104 variants in vitro and in vivo.Citation63 Further engineering of Hsp104 appears to be necessary to antagonize EWSR1 aggregation and toxicity.
Potentiated Hsp104 Variants Typically Have Elevated ATPase Activity
To assess the mechanism by which potentiating mutations enhance Hsp104 activity, we explored their biochemical properties. Typically, potentiated Hsp104 variants exhibited ∼2-4-fold elevated ATPase activity than Hsp104, indicating that potentiation might stem from an ability to undergo more rapid rounds of ATP binding and hydrolysis.Citation61 However, enhanced ATPase activity was not always a feature of potentiated Hsp104 variants. For example, Hsp104D498V and Hsp104D504C exhibited ATPase activity similar to Hsp104.Citation61 Interestingly, Hsp104D498V and Hsp104D504C were more selective in their ability to rescue the yeast neurodegenerative disease models. They could both rescue FUS and α-syn toxicity in yeast, but failed to rescue TDP-43 toxicity.Citation61 These data suggest that elevated ATPase activity of potentiated Hsp104 variants is critical to rescue toxicity of a broad spectrum of neurodegenerative disease proteins, and that reducing ATPase activity in this context promotes selectivity for specific substrates.
Potentiated Hsp104 Variants Do Not Require Hsp70 and Hsp40 for Disaggregation
Rescue of toxicity by enhanced Hsp104 variants could be a consequence of an altered mechanism of disaggregation compared to Hsp104. Thus, we assessed disaggregase activity against disordered luciferase aggregates in vitro. Hsp104 failed to disaggregate luciferase unless supplemented with Hsp70 or Hsp40. In striking contrast, Hsp70 and Hsp40 were not required for any of the potentiated Hsp104 variants to reactivate aggregated luciferase.Citation61 Typically, in the absence of Hsp70 and Hsp40, potentiated Hsp104 variants were ∼3-9-fold more active than Hsp104 in the presence of Hsc70 (an Hsp70) and Hdj2 (an Hsp40).Citation61 Surprisingly, these findings suggest that absolute dependence on Hsp70 and Hsp40 hinders Hsp104 from rescuing α-syn, FUS, and TDP-43 toxicity.
This elevated disaggregase activity even in the absence of Hsp70 differentiates potentiated Hsp104 variants from hyperactive ClpB (the E. coli homolog of Hsp104) variants bearing specific MD mutations. In contrast to potentiated Hsp104 variants, hyperactive ClpB variants still require the Hsp70 chaperone system for robust disaggregase activity.Citation171 This key difference likely reflects the more stringent requirement for Hsp70 for ClpB disaggregase activity compared to Hsp104.Citation107
In most cases, potentiated Hsp104 activity could be stimulated even further by supplementation with the Hsp70 chaperone system.Citation61 Thus, potentiated Hsp104 variants can still collaborate with the Hsp70 chaperone system, but do not absolutely require it for the disaggregation of disordered aggregates. Interestingly, once again the 2 exceptions were Hsp104D498V and Hsp104D504C. The luciferase disaggregase activity of Hsp104D498V and Hsp104D504C was not stimulated further by the Hsp70 chaperone system.Citation61 These data suggest that the ability of potentiated Hsp104 variants to collaborate with Hsp70 is critical to rescue toxicity of a broad spectrum of neurodegenerative disease proteins, and that reducing collaboration with Hsp70 promotes selectivity for specific substrates.
Potentiated Hsp104 Variants Exhibit Enhanced Translocase and Unfoldase Activity
We also established that potentiated Hsp104 variants had enhanced activities against various soluble model substrates. Thus, potentiated variants translocated the intrinsically disordered soluble substrate, FITC-casein, more rapidly across their central channel.Citation61 This accelerated substrate-translocation rate likely enables potentiated Hsp104 variants to avoid kinetic traps during translocation and exert additional force to unfold stable substrates. Indeed, potentiated Hsp104 variants were also enhanced unfoldases, and unfolded GFP bearing a long (RepA1-70) or short (6-HIS-TEV) unfolded tag more rapidly than Hsp104 in the presence of ATP.Citation61,63 In fact, Hsp104 did not unfold these substrates at all in the presence of ATP, but instead required a mixture of ATP and ATPγS.Citation61,63 Even then, Hsp104 unfolded these substrates less effectively and at a slower rate than the potentiated Hsp104 variants.Citation61,63 Importantly, neither Hsp104 nor the potentiated Hsp104 variants unfolded untagged GFP, and thus the potentiated variants do not unfold any protein.Citation61,63 Collectively, these data suggest that potentiated Hsp104 variants are enhanced unfoldases that are intrinsically primed to recognize and rapidly unfold substrates bearing even short unfolded tags and unlike Hsp104 do not have to wait for regulatory events to initiate unfolding (simulated here by ATP:ATPγS mixtures).
Potentiated Hsp104 Variants Exhibit Altered Subunit Collaboration
Using a mutant doping strategy,Citation104 we also revealed that a potentiated Hsp104 variant, Hsp104A503V, promoted protein disaggregation by employing a different mechanism of intersubunit collaboration compared to Hsp104. Indeed, the Hsp104A503V hexamer possessed greater plasticity and maintained robust disaggregase activity in the presence of a wider variety of subunit-inactivating events. For example, an Hsp104A503V subunit that binds but cannot hydrolyze ATP and engages substrate will stimulate the disaggregase activity of an adjacent Hsp104A503V subunit within the hexamer. By contrast, in Hsp104, a single subunit with these properties inactivates the entire hexamer.Citation104 This increased resistance of Hsp104A503V hexamers to subunit-inactivating events likely empowers facile resolution of recalcitrant substrates. Importantly, this altered activity enabled potentiated Hsp104 variants to disaggregate diverse neurodegenerative disease substrates in vitro, including α-syn, FUS, TDP-43, and TAF15 fibrils, under conditions where Hsp104 was inactive.Citation61,63
Degeneracy of Potentiating Mutations at Specific MD Positions
Our findings establish that the Hsp104 MD plays a critical role in regulating Hsp104 function.Citation61,63 Remarkably, missense mutations to diverse residues at specific and disparate positions within the MD (e.g., A503, Y507, N539) conferred a therapeutic gain of function.Citation61 Indeed, A503 could be mutated to any residue except proline to yield potentiated Hsp104 variants capable of rescuing TDP-43, FUS, and α-syn toxicity in yeast.Citation61 Thus, potentiated disaggregase activity is enabled by loss of amino acid identity at specific positions in the MD rather than by mutation to a specific residue or class of residue. This finding indicates that Hsp104 disaggregase activity is usually tightly constrained, but can be unleashed by even very subtle changes to side chains at specific positions. The ability to attain such a wide-reaching set of gain of therapeutic functions via such minor changes in primary sequence, e.g. by adding a single methylene bridge (V426L) or by removing a single methyl group (A503G) is without precedent. It also suggests that drug-like small molecules that bind to the correct region of the MD might also enhance Hsp104 activity.Citation154 Moreover, post-translational modification of Hsp104 in specific regions of the MD could act as a switch to elicit potentiated activity in a reversible manner. Our findings suggest that the regulatory constraints placed on Hsp104 are simply too tight to counter TDP-43, FUS, and α-syn misfolding and toxicity under certain conditions. Thus, we discover an unanticipated and inimical limitation in existing disaggregase functionality. The MD can be viewed as a capacitor braced to unleash Hsp104 activity. Potentiating mutations likely destabilize autoinhibitory interactions that dampen Hsp104 activity or induce structural rearrangements that mimic or enable allosteric activation perhaps akin to the effect of Hsp70 binding the Hsp104 MD.Citation154 It remains unclear how diverse conservative and nonconservative mutations can result in this phenotype. Mutation of specific residues might subtly perturb hexamer structure, possibly promoting enhanced flexibility, altered channel properties, and stable population of the potentiated state.
Why Is WT Hsp104 Not Naturally Potentiated?
Why is WT Hsp104 not naturally potentiated? It would seem beneficial to be able to counter the excessive aggregation and toxicity caused by the overexpression of a single protein in yeast.Citation61,63 However, WT Hsp104 is unable to confer this activity.Citation61,63 Thus, it seems probable that the stress caused by the overexpression of a single, aggregation-prone, toxic protein is an unusual challenge for yeast that has not featured as a significant selective pressure sculpting Hsp104 primary sequence and activity. Rather, Hsp104 activity has likely been tuned during evolution to refold diverse aggregated proteins that accrue after a range of mild to severe environmental stresses.Citation65,66,172,173 Moreover, Hsp104 activity is also likely adapted to propagate various beneficial yeast prions.Citation31,33,34,41,46,90
With regard to these 2 important activities, the potentiated Hsp104 variants uncovered in our screen display deficits. Thus, even though Hsp104A503V can confer thermotolerance to high temperatures (e.g., 50°C) just as well or even better than Hsp104,Citation61,155 Hsp104A503V is toxic to yeast when overexpressed under conditions of very mild thermal stress (e.g. 37°C).Citation61,155 Moreover, Hsp104A503V overexpression is benign under non-stressful conditions.Citation61,155 We suggest that under mild stress conditions, many yeast proteins populate mildly destabilized or metastable states that are inappropriately recognized and unfolded by potentiated Hsp104A503V, which causes toxicity.Citation63 By contrast, WT Hsp104 is likely tuned to ignore such substrates.Citation63 Hsp104A503V also displays defects in propagation of the beneficial [PSI+] prion.Citation174 Indeed, Hsp104A503V can even display synthetic lethality with strong [PSI+] variants.Citation150 Thus, the alterations in Hsp104 activity caused by potentiating mutations might preclude stable maintenance of beneficial prions states, such as [PSI+], which would in turn inhibit revelation of cryptic variation and rapid evolution of new traits in response to environmental stress.Citation32,33,35,41,45,46 These 2 differences between WT Hsp104 and potentiated Hsp104 in yeast likely explain why the potentiated forms were not fixed during evolution.
Even so, the possibility remains that the proteomes of other organisms might present challenges and selection pressures that necessitated more potentiated versions of Hsp104. Thus, it will be of great interest to compare the activity of Hsp104 orthologues from eukaryotic and prokaryotic species with diverse proteomes. For example, a naturally occurring variant of Hsp104 could display enhanced activity able to even more effectively counter the aggregation of human neurodegenerative disease proteins.
Further Engineering of Potentiated Hsp104 Variants
Using rational design and directed evolution, it will be important to isolate potentiated Hsp104 variants that are specific for single proteins (e.g., FUS) to minimize any potential off-target effects.Citation61,62 It will also be of interest to isolate conformer-specific potentiated Hsp104 variants. Ideally, Hsp104 variants could be isolated that antagonize only toxic misfolded species, for example: Hsp104 variants that rapidly resolve toxic soluble oligomers but not amyloid fibrils, or Hsp104 variants that resolve toxic amyloid strains and not benign strains. In principle, Hsp104 could be potentiated against any protein or any misfolded conformer, which might find important applications not only in therapeutics but also in the purification of irksome recombinant proteins for valuable basic or pharmaceutical purposes.
Broader Implications for Engineering Other Chaperones or Disaggregases
Reactivation of the disease-associated proteins to their non-pathogenic states suggests that Hsp104 variants and indeed other therapeutics that achieve this goal may provide a highly promising and potentially successful strategy for halting and reversing the progression of devastating neurodegenerative diseases. Potentiation of Hsp104 activity to achieve this goal required only subtle modifications to the existing disaggregase.Citation61,63 Indeed, engineering and directed evolution of the activity of other molecular chaperones, including GroEL, Hsp70, ClpX, and Spy, has revealed that minor changes in primary sequence (often a single missense mutation) can suffice to drastically alter substrate specificity or enhance global chaperone activity.Citation175–179 Indeed, minor alterations in primary sequence in naturally occurring chaperone homologs can also radically change activity.Citation180 It will be of great interest to engineer and enhance human molecular chaperones to counter specific protein-misfolding events related to neurodegenerative disease. In particular, engineering the human Hsp110, Hsp70, and Hsp40 disaggregase system,Citation65,73–76 to more effectively disaggregate disease substrates is an important goal. Since small changes in primary sequence at specific positions can greatly enhance chaperone activityCitation61,63,175–180 it appears plausible to isolate therapeutic small molecules that elicit similar enhancements in chaperone activity against disease substrates.Citation181,182
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Korrie Mack and Mariana Torrente for feedback on the manuscript.
Funding
Our studies were supported by: an American Heart Association Post-Doctoral Fellowship and Target ALS Springboard Fellowship (MEJ); NIH Director's New Innovator Award DP2OD002177, NIH grants R21NS067354, R21HD074510, and R01GM099836, a Muscular Dystrophy Association Research Award (MDA277268), Packard Center for ALS Research at Johns Hopkins University, Target ALS, and an Ellison Medical Foundation New Scholar in Aging Award (JS).
REFERENCES
- Trojanowski JQ. PENN neurodegenerative disease research - in the spirit of Benjamin Franklin. Neurosignals 2008; 16:5-10; PMID:18097154; http://dx.doi.org/10.1159/000109753
- Cushman M, Johnson BS, King OD, Gitler AD, Shorter J. Prion-like disorders: blurring the divide between transmissibility and infectivity. J Cell Sci 2010; 123:1191-201; PMID:20356930; http://dx.doi.org/10.1242/jcs.051672
- Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci 2013; 14:248-64; PMID:23463272; http://dx.doi.org/10.1038/nrn3430
- Irwin DJ, Lee VM, Trojanowski JQ. Parkinson's disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat Rev Neurosci 2013; 14:626-36; PMID:23900411; http://dx.doi.org/10.1038/nrn3549
- Goedert M, Spillantini MG. A century of Alzheimer's disease. Science 2006; 314:777-81; PMID:17082447; http://dx.doi.org/10.1126/science.1132814
- Bulawa CE, Connelly S, Devit M, Wang L, Weigel C, Fleming JA, Packman J, Powers ET, Wiseman RL, Foss TR, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci USA 2012; 109:9629-34; PMID:22645360; http://dx.doi.org/10.1073/pnas.1121005109
- Coelho T, Maia LF, da Silva AM, Cruz MW, Plante-Bordeneuve V, Suhr OB, Conceicao I, Schmidt HH, Trigo P, Kelly JW, et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol 2013; 260(11):2802-14; PMID: 23974642; http://dx.doi.org/10.1007/s00415-013-7051-7
- Coelho T, Maia LF, Martins da Silva A, Waddington Cruz M, Plante-Bordeneuve V, Lozeron P, Suhr OB, Campistol JM, Conceicao IM, Schmidt HH, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 2012; 79:785-92; PMID:22843282; http://dx.doi.org/10.1212/WNL.0b013e3182661eb1
- de Lartigue J. Tafamidis for transthyretin amyloidosis. Drugs Today (Barc) 2012; 48:331-7; PMID:22645721; http://dx.doi.org/10.1358/dot.2012.48.5.1808486
- Nencetti S, Rossello A, Orlandini E. Tafamidis (Vyndaqel): A Light for FAP Patients. ChemMedChem 2013; 8(10):1617-9; http://dx.doi.org/10.1002/cmdc.201300245
- Said G, Grippon S, Kirkpatrick P. Tafamidis. Nat Rev Drug Discov 2012; 11:185-6; PMID:22378262; http://dx.doi.org/10.1038/nrd3675
- Knowles TP, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol 2014; 15:384-96; PMID:24854788; http://dx.doi.org/10.1038/nrm3810
- Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013; 501:45-51; PMID:24005412; http://dx.doi.org/10.1038/nature12481
- Eisenberg D, Jucker M. The amyloid state of proteins in human diseases. Cell 2012; 148:1188-203; PMID:22424229; http://dx.doi.org/10.1016/j.cell.2012.02.022
- Guo JL, Lee VM. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med 2014; 20:130-8; PMID:24504409; http://dx.doi.org/10.1038/nm.3457
- Prusiner SB. Biology and genetics of prions causing neurodegeneration. Annu Rev Genet 2013; 47:601-23; PMID:24274755; http://dx.doi.org/10.1146/annurev-genet-110711-155524
- Snead D, Eliezer D. Alpha-synuclein function and dysfunction on cellular membranes. Exp Neurobiol 2014; 23:292-313; PMID:25548530; http://dx.doi.org/10.5607/en.2014.23.4.292
- van Oosten-Hawle P, Morimoto RI. Organismal proteostasis: role of cell-nonautonomous regulation and transcellular chaperone signaling. Genes Dev 2014; 28:1533-43; PMID:25030693; http://dx.doi.org/10.1101/gad.241125.114
- Hipp MS, Park SH, Hartl FU. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol 2014; PMID:24946960
- Lindquist SL, Kelly JW. Chemical and biological approaches for adapting proteostasis to ameliorate protein misfolding and aggregation diseases: progress and prognosis. Cold Spring Harb Perspect Biol 2011; 3; PMID:21900404; http://dx.doi.org/10.1101/cshperspect.a004507
- Shorter J. Hsp104: a weapon to combat diverse neurodegenerative disorders. Neurosignals 2008; 16:63-74; PMID:18097161; http://dx.doi.org/10.1159/000109760
- Vashist S, Cushman M, Shorter J. Applying Hsp104 to protein-misfolding disorders. Biochem Cell Biol 2010; 88:1-13; PMID:20130674; http://dx.doi.org/10.1139/O09-121
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003; 300:486-9; PMID:12702875; http://dx.doi.org/10.1126/science.1079469
- Cohen SI, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, Otzen DE, Vendruscolo M, Dobson CM, Knowles TP. Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci U S A 2013; 110:9758-63; PMID:23703910; http://dx.doi.org/10.1073/pnas.1218402110
- Oromendia AB, Dodgson SE, Amon A. Aneuploidy causes proteotoxic stress in yeast. Genes Dev 2012; 26:2696-708; PMID:23222101; http://dx.doi.org/10.1101/gad.207407.112
- Xu J, Reumers J, Couceiro JR, De Smet F, Gallardo R, Rudyak S, Cornelis A, Rozenski J, Zwolinska A, Marine JC, et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat Chem Biol 2011; 7:285-95; PMID:21445056; http://dx.doi.org/10.1038/nchembio.546
- Coelho M, Dereli A, Haese A, Kuhn S, Malinovska L, DeSantis ME, Shorter J, Alberti S, Gross T, Tolic-Norrelykke IM. Fission yeast does not age under favorable conditions, but does so after stress. Curr Biol 2013; 23:1844-52; PMID:24035542; http://dx.doi.org/10.1016/j.cub.2013.07.084
- Hill SM, Hao X, Liu B, Nystrom T. Life-span extension by a metacaspase in the yeast Saccharomyces cerevisiae. Science 2014; 344:1389-92; PMID:24855027; http://dx.doi.org/10.1126/science.1252634
- David DC, Ollikainen N, Trinidad JC, Cary MP, Burlingame AL, Kenyon C. Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol 2010; 8:e1000450; PMID:20711477; http://dx.doi.org/10.1371/journal.pbio.1000450
- Geiler-Samerotte KA, Dion MF, Budnik BA, Wang SM, Hartl DL, Drummond DA. Misfolded proteins impose a dosage-dependent fitness cost and trigger a cytosolic unfolded protein response in yeast. Proc Natl Acad Sci U S A 2011; 108:680-5; PMID:21187411; http://dx.doi.org/10.1073/pnas.1017570108
- Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 2009; 137:146-58; PMID:19345193; http://dx.doi.org/10.1016/j.cell.2009.02.044
- Halfmann R, Alberti S, Lindquist S. Prions, protein homeostasis, and phenotypic diversity. Trends Cell Biol 2010; 20:125-33.; PMID:20071174; http://dx.doi.org/10.1016/j.tcb.2009.12.003
- Halfmann R, Jarosz DF, Jones SK, Chang A, Lancaster AK, Lindquist S. Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature 2012; 482:363-8; PMID:22337056; http://dx.doi.org/10.1038/nature10875
- Suzuki G, Shimazu N, Tanaka M. A yeast prion, Mod5, promotes acquired drug resistance and cell survival under environmental stress. Science 2012; 336:355-9; PMID:22517861; http://dx.doi.org/10.1126/science.1219491
- True HL, Lindquist SL. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature 2000; 407:477-83; PMID:11028992; http://dx.doi.org/10.1038/35035005
- Tyedmers J, Treusch S, Dong J, McCaffery JM, Bevis B, Lindquist S. Prion induction involves an ancient system for the sequestration of aggregated proteins and heritable changes in prion fragmentation. Proc Natl Acad Sci U S A 2010; 107:8633-8; PMID:20421488; http://dx.doi.org/10.1073/pnas.1003895107
- Cegelski L, Pinkner JS, Hammer ND, Cusumano CK, Hung CS, Chorell E, Aberg V, Walker JN, Seed PC, Almqvist F, et al. Small-molecule inhibitors target Escherichia coli amyloid biogenesis and biofilm formation. Nat Chem Biol 2009; 5:913-9; PMID:19915538; http://dx.doi.org/10.1038/nchembio.242
- Fowler DM, Koulov AV, Alory-Jost C, Marks MS, Balch WE, Kelly JW. Functional amyloid formation within mammalian tissue. PLoS Biol 2006; 4:e6; PMID:16300414; http://dx.doi.org/10.1371/journal.pbio.0040006
- Fowler DM, Koulov AV, Balch WE, Kelly JW. Functional amyloid–from bacteria to humans. Trends Biochem Sci 2007; 32:217-24; PMID:17412596; http://dx.doi.org/10.1016/j.tibs.2007.03.003
- Castellano LM, Shorter J. The Surprising Role of Amyloid Fibrils in HIV Infection. Biology 2012; 1:58-80; PMID:24832047; http://dx.doi.org/10.3390/biology1010058
- Shorter J, Lindquist S. Prions as adaptive conduits of memory and inheritance. Nat Rev Genet 2005; 6:435-50; PMID:15931169; http://dx.doi.org/10.1038/nrg1616
- Skinner JJ, Wood S, Shorter J, Englander SW, Black BE. The Mad2 partial unfolding model: regulating mitosis through Mad2 conformational switching. J Cell Biol 2008; 183:761-8; PMID:19029339; http://dx.doi.org/10.1083/jcb.200808122
- Watt B, van Niel G, Fowler DM, Hurbain I, Luk KC, Stayrook SE, Lemmon MA, Raposo G, Shorter J, Kelly JW, et al. N-terminal domains elicit formation of functional Pmel17 amyloid fibrils. J Biol Chem 2009; 284:35543-55; PMID:19840945; http://dx.doi.org/10.1074/jbc.M109.047449
- Tuite MF, Serio TR. The prion hypothesis: from biological anomaly to basic regulatory mechanism. Nat Rev Mol Cell Biol 2010; 11:823-33; PMID:21081963; http://dx.doi.org/10.1038/nrm3007
- True HL, Berlin I, Lindquist SL. Epigenetic regulation of translation reveals hidden genetic variation to produce complex traits. Nature 2004; 431:184-7; PMID:15311209; http://dx.doi.org/10.1038/nature02885
- Garcia DM, Jarosz DF. Rebels with a cause: molecular features and physiological consequences of yeast prions. FEMS Yeast Res 2014; 14:136-47; PMID:25667942; http://dx.doi.org/10.1111/1567-1364.12116
- Gitler AD, Shorter J. RNA-binding proteins with prion-like domains in ALS and FTLD-U. Prion 2011; 5:179-87; PMID:21847013; http://dx.doi.org/10.4161/pri.5.3.17230
- Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem 2009; 284:20329-39; PMID:19465477; http://dx.doi.org/10.1074/jbc.M109.010264
- Sun Z, Diaz Z, Fang X, Hart MP, Chesi A, Shorter J, Gitler AD. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol 2011; 9:e1000614; PMID:21541367; http://dx.doi.org/10.1371/journal.pbio.1000614
- King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res 2012; 1462:61-80; PMID:22445064; http://dx.doi.org/10.1016/j.brainres.2012.01.016
- Li YR, King OD, Shorter J, Gitler AD. Stress granules as crucibles of ALS pathogenesis. J Cell Biol 2013; 201:361-72; PMID:23629963; http://dx.doi.org/10.1083/jcb.201302044
- Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013; 495:467-73; PMID:23455423; http://dx.doi.org/10.1038/nature11922
- Shorter J, Taylor JP. Disease mutations in the prion-like domains of hnRNPA1 and hnRNPA2B1 introduce potent steric zippers that drive excess RNP granule assembly. . Rare Diseases 2013; 1:e25200; http://dx.doi.org/10.4161/rdis.25200
- Halfmann R, Lindquist S. Epigenetics in the extreme: prions and the inheritance of environmentally acquired traits. Science 2010; 330:629-32; PMID:21030648; http://dx.doi.org/10.1126/science.1191081
- Heinrich SU, Lindquist S. Protein-only mechanism induces self-perpetuating changes in the activity of neuronal Aplysia cytoplasmic polyadenylation element binding protein (CPEB). Proc Natl Acad Sci U S A 2011; 108:2999-3004; PMID:21270333; http://dx.doi.org/10.1073/pnas.1019368108
- Si K, Choi YB, White-Grindley E, Majumdar A, Kandel ER. Aplysia CPEB can form prion-like multimers in sensory neurons that contribute to long-term facilitation. Cell 2010; 140:421-35; PMID:20144764; http://dx.doi.org/10.1016/j.cell.2010.01.008
- Si K, Lindquist S, Kandel E. A possible epigenetic mechanism for the persistence of memory. Cold Spring Harb Symp Quant Biol 2004; 69:497-8; PMID:16117686; http://dx.doi.org/10.1101/sqb.2004.69.497
- Si K, Giustetto M, Etkin A, Hsu R, Janisiewicz AM, Miniaci MC, Kim JH, Zhu H, Kandel ER. A neuronal isoform of CPEB regulates local protein synthesis and stabilizes synapse-specific long-term facilitation in aplysia. Cell 2003; 115:893-904; PMID:14697206; http://dx.doi.org/10.1016/S0092-8674(03)01021-3
- Si K, Lindquist S, Kandel ER. A neuronal isoform of the aplysia CPEB has prion-like properties. Cell 2003; 115:879-91; PMID:14697205; http://dx.doi.org/10.1016/S0092-8674(03)01020-1
- Majumdar A, Cesario WC, White-Grindley E, Jiang H, Ren F, Khan MR, Li L, Choi EM, Kannan K, Guo F, et al. Critical role of amyloid-like oligomers of Drosophila Orb2 in the persistence of memory. Cell 2012; 148:515-29; PMID:22284910; http://dx.doi.org/10.1016/j.cell.2012.01.004
- Jackrel ME, DeSantis ME, Martinez BA, Castellano LM, Stewart RM, Caldwell KA, Caldwell GA, Shorter J. Potentiated Hsp104 variants antagonize diverse proteotoxic misfolding events. Cell 2014; 156:170-82; PMID:24439375; http://dx.doi.org/10.1016/j.cell.2013.11.047
- Jackrel ME, Shorter J. Reversing deleterious protein aggregation with re-engineered protein disaggregases. Cell Cycle 2014; 13:1379-83; PMID:24694655; http://dx.doi.org/10.4161/cc.28709
- Jackrel ME, Shorter J. Potentiated Hsp104 variants suppress toxicity of diverse neurodegenerative disease-linked proteins. Dis Model Mech 2014; 7:1175-84; PMID:25062688; http://dx.doi.org/10.1242/dmm.016113
- Jackrel ME, Tariq A, Yee K, Weitzman R, Shorter J. Isolating potentiated Hsp104 variants using yeast proteinopathy models. J Vis Exp 2014; 93:e52089; PMID:25407485
- Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell 1998; 94:73-82; PMID:9674429; http://dx.doi.org/10.1016/S0092-8674(00)81223-4
- Parsell DA, Kowal AS, Singer MA, Lindquist S. Protein disaggregation mediated by heat-shock protein Hsp104. Nature 1994; 372:475-8; PMID:7984243; http://dx.doi.org/10.1038/372475a0
- DeSantis ME, Shorter J. The elusive middle domain of Hsp104 and ClpB: location and function. Biochim Biophys Acta 2012; 1823:29-39; PMID:21843558; http://dx.doi.org/10.1016/j.bbamcr.2011.07.014
- Parsell DA, Kowal AS, Lindquist S. Saccharomyces cerevisiae Hsp104 protein. Purification and characterization of ATP-induced structural changes. J Biol Chem 1994; 269:4480-7; PMID:8308017
- Tessarz P, Mogk A, Bukau B. Substrate threading through the central pore of the Hsp104 chaperone as a common mechanism for protein disaggregation and prion propagation. Mol Microbiol 2008; 68:87-97; PMID:18312264; http://dx.doi.org/10.1111/j.1365-2958.2008.06135.x
- Haslberger T, Zdanowicz A, Brand I, Kirstein J, Turgay K, Mogk A, Bukau B. Protein disaggregation by the AAA+ chaperone ClpB involves partial threading of looped polypeptide segments. Nat Struct Mol Biol 2008; 15:641-50; PMID:18488042; http://dx.doi.org/10.1038/nsmb.1425
- Lum R, Niggemann M, Glover JR. Peptide and protein binding in the axial channel of Hsp104. Insights into the mechanism of protein unfolding. J Biol Chem 2008; 283:30139-50; PMID:18755692; http://dx.doi.org/10.1074/jbc.M804849200
- Lum R, Tkach JM, Vierling E, Glover JR. Evidence for an unfolding/threading mechanism for protein disaggregation by Saccharomyces cerevisiae Hsp104. J Biol Chem 2004; 279:29139-46; PMID:15128736; http://dx.doi.org/10.1074/jbc.M403777200
- Shorter J. The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS One 2011; 6:e26319; PMID:22022600; http://dx.doi.org/10.1371/journal.pone.0026319
- Rampelt H, Kirstein-Miles J, Nillegoda NB, Chi K, Scholz SR, Morimoto RI, Bukau B. Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J 2012; 31:4221-35; PMID:22990239; http://dx.doi.org/10.1038/emboj.2012.264
- Sweeny EA, Shorter J. Prion proteostasis: Hsp104 meets its supporting cast. Prion 2008; 2:135-40; PMID:19242125; http://dx.doi.org/10.4161/pri.2.4.7952
- Torrente MP, Shorter J. The metazoan protein disaggregase and amyloid depolymerase system: Hsp110, Hsp70, Hsp40, and small heat shock proteins. Prion 2013; 7:457-63; PMID:24401655; http://dx.doi.org/10.4161/pri.27531
- Doyle SM, Shorter J, Zolkiewski M, Hoskins JR, Lindquist S, Wickner S. Asymmetric deceleration of ClpB or Hsp104 ATPase activity unleashes protein-remodeling activity. Nat Struct Mol Biol 2007; 14:114-22; PMID:17259993; http://dx.doi.org/10.1038/nsmb1198
- Lo Bianco C, Shorter J, Regulier E, Lashuel H, Iwatsubo T, Lindquist S, Aebischer P. Hsp104 antagonizes alpha-synuclein aggregation and reduces dopaminergic degeneration in a rat model of Parkinson disease. J Clin Invest 2008; 118:3087-97; PMID:18704197; http://dx.doi.org/10.1172/JCI35781
- Shorter J, Lindquist S. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science 2004; 304:1793-7; PMID:15155912; http://dx.doi.org/10.1126/science.1098007
- Shorter J, Lindquist S. Destruction or potentiation of different prions catalyzed by similar Hsp104 remodeling activities. Mol Cell 2006; 23:425-38; PMID:16885031; http://dx.doi.org/10.1016/j.molcel.2006.05.042
- Shorter J, Lindquist S. Hsp104, Hsp70 and Hsp40 interplay regulates formation, growth and elimination of Sup35 prions. Embo J 2008; 27:2712-24; PMID:18833196; http://dx.doi.org/10.1038/emboj.2008.194
- Wendler P, Shorter J, Plisson C, Cashikar AG, Lindquist S, Saibil HR. Atypical AAA+ subunit packing creates an expanded cavity for disaggregation by the protein-remodeling factor Hsp104. Cell 2007; 131:1366-77; PMID:18160044; http://dx.doi.org/10.1016/j.cell.2007.10.047
- Wendler P, Shorter J, Snead D, Plisson C, Clare DK, Lindquist S, Saibil HR. Motor mechanism for protein threading through Hsp104. Mol Cell 2009; 34:81-92; PMID:19362537; http://dx.doi.org/10.1016/j.molcel.2009.02.026
- Duennwald ML, Echeverria AL, Shorter J. Small Heat Shock Proteins Potentiate Amyloid Dissolution by Protein Disaggregases from Yeast and Humans. PLoS Biol 2012; 9 e1001346; ; http://dx.doi.org/10.1371/journal.pbio.1001346
- DiSalvo S, Derdowski A, Pezza JA, Serio TR. Dominant prion mutants induce curing through pathways that promote chaperone-mediated disaggregation. Nat Struct Mol Biol 2011; 18:486-92; PMID:21423195; http://dx.doi.org/10.1038/nsmb.2031
- Park YN, Zhao X, Yim YI, Todor H, Ellerbrock R, Reidy M, Eisenberg E, Masison DC, Greene LE. Hsp104 overexpression cures Saccharomyces cerevisiae [PSI+] by causing dissolution of the prion seeds. Eukaryot Cell 2014;; 13:635-47
- Klaips CL, Hochstrasser ML, Langlois CR, Serio TR. Spatial quality control bypasses cell-based limitations on proteostasis to promote prion curing. Elife 2014; 3; PMID:25490068; http://dx.doi.org/10.7554/eLife.04288
- Liu YH, Han YL, Song J, Wang Y, Jing YY, Shi Q, Tian C, Wang ZY, Li CP, Han J, et al. Heat shock protein 104 inhibited the fibrillization of prion peptide 106-126 and disassembled prion peptide 106-126 fibrils in vitro. Int J Biochem Cell Biol 2011; 43:768-74; PMID:21296677; http://dx.doi.org/10.1016/j.biocel.2011.01.022
- Shorter J. Emergence and natural selection of drug-resistant prions. Mol Biosyst 2010; 6:1115-30; PMID:20422111; http://dx.doi.org/10.1039/c004550k
- Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 1995; 268:880-4; PMID:7754373; http://dx.doi.org/10.1126/science.7754373
- DeSantis ME, Shorter J. Hsp104 drives "protein-only" positive selection of Sup35 prion strains encoding strong [PSI(+)]. Chem Biol 2012; 19:1400-10; PMID:23177195; http://dx.doi.org/10.1016/j.chembiol.2012.09.013
- Cushman-Nick M, Bonini NM, Shorter J. Hsp104 suppresses polyglutamine-induced degeneration post onset in a drosophila MJD/SCA3 model. PLoS Genet 2013; 9:e1003781; PMID:24039611; http://dx.doi.org/10.1371/journal.pgen.1003781
- Satyal SH, Schmidt E, Kitagawa K, Sondheimer N, Lindquist S, Kramer JM, Morimoto RI. Polyglutamine aggregates alter protein folding homeostasis in Caenorhabditis elegans. Proc Natl Acad Sci U S A 2000; 97:5750-5; PMID:10811890; http://dx.doi.org/10.1073/pnas.100107297
- Vacher C, Garcia-Oroz L, Rubinsztein DC. Overexpression of yeast hsp104 reduces polyglutamine aggregation and prolongs survival of a transgenic mouse model of Huntington's disease. Hum Mol Genet 2005; 14:3425-33; PMID:16204350; http://dx.doi.org/10.1093/hmg/ddi372
- Dandoy-Dron F, Bogdanova A, Beringue V, Bailly Y, Tovey MG, Laude H, Dron M. Infection by ME7 prion is not modified in transgenic mice expressing the yeast chaperone Hsp104 in neurons. Neurosci Lett 2006; 405:181-5; PMID:16884849; http://dx.doi.org/10.1016/j.neulet.2006.05.066
- Murray AN, Kelly JW. Hsp104 gives clients the individual attention they need. Cell 2012; 151:695-7; PMID:23141530; http://dx.doi.org/10.1016/j.cell.2012.10.033
- Escusa-Toret S, Vonk WI, Frydman J. Spatial sequestration of misfolded proteins by a dynamic chaperone pathway enhances cellular fitness during stress. Nat Cell Biol 2013; 15:1231-43; PMID:24036477; http://dx.doi.org/10.1038/ncb2838
- Shorter J. Amyloid remodeling by Hsp104. In: Witt SN, ed. Protein Chaperones and Protection from Neurodegeneration. Hoboken, NJ: Taylor & Francis., 2011; 235-59
- Hallett M, Albanese A, Dressler D, Segal KR, Simpson DM, Truong D, Jankovic J. Evidence-based review and assessment of botulinum neurotoxin for the treatment of movement disorders. Toxicon 2013; 67:94-114; PMID:23380701; http://dx.doi.org/10.1016/j.toxicon.2012.12.004
- Naumann M, Dressler D, Hallett M, Jankovic J, Schiavo G, Segal KR, Truong D. Evidence-based review and assessment of botulinum neurotoxin for the treatment of secretory disorders. Toxicon 2013; 67:141-52; PMID:23178324; http://dx.doi.org/10.1016/j.toxicon.2012.10.020
- Chancellor MB, Elovic E, Esquenazi A, Naumann M, Segal KR, Schiavo G, Smith CP, Ward AB. Evidence-based review and assessment of botulinum neurotoxin for the treatment of urologic conditions. Toxicon 2013; 67:129-40; PMID:23415704; http://dx.doi.org/10.1016/j.toxicon.2013.01.020
- Osborne SL, Latham CF, Wen PJ, Cavaignac S, Fanning J, Foran PG, Meunier FA. The Janus faces of botulinum neurotoxin: sensational medicine and deadly biological weapon. J Neurosci Res 2007; 85:1149-58; PMID:17387703; http://dx.doi.org/10.1002/jnr.21171
- Tighe AP, Schiavo G. Botulinum neurotoxins: mechanism of action. Toxicon 2013; 67:87-93; PMID:23201505; http://dx.doi.org/10.1016/j.toxicon.2012.11.011
- DeSantis ME, Leung EH, Sweeny EA, Jackrel ME, Cushman-Nick M, Neuhaus-Follini A, Vashist S, Sochor MA, Knight MN, Shorter J. Operational plasticity enables hsp104 to disaggregate diverse amyloid and nonamyloid clients. Cell 2012; 151:778-93; PMID:23141537; http://dx.doi.org/10.1016/j.cell.2012.09.038
- Carroni M, Kummer E, Oguchi Y, Wendler P, Clare DK, Sinning I, Kopp J, Mogk A, Bukau B, Saibil HR. Head-to-tail interactions of the coiled-coil domains regulate ClpB activity and cooperation with Hsp70 in protein disaggregation. Elife (Cambridge) 2014; 3:e02481
- Lee S, Sielaff B, Lee J, Tsai FT. CryoEM structure of Hsp104 and its mechanistic implication for protein disaggregation. Proc Natl Acad Sci U S A 2010; 107:8135-40; PMID:20404203; http://dx.doi.org/10.1073/pnas.1003572107
- DeSantis ME, Sweeny EA, Snead D, Leung EH, Go MS, Gupta K, Wendler P, Shorter J. Conserved distal loop residues in the Hsp104 and ClpB middle domain contact nucleotide-binding domain 2 and enable Hsp70-dependent protein disaggregation. J Biol Chem 2014; 289:848-67; PMID:24280225; http://dx.doi.org/10.1074/jbc.M113.520759
- Saibil H. Chaperone machines for protein folding, unfolding and disaggregation. Nat Rev Mol Cell Biol 2013; 14:630-42; PMID:24026055; http://dx.doi.org/10.1038/nrm3658
- Chen-Plotkin AS, Lee VM, Trojanowski JQ. TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol 2010; 6:211-20; PMID:20234357; http://dx.doi.org/10.1038/nrneurol.2010.18
- Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 2010; 9:995-1007; PMID:20864052; http://dx.doi.org/10.1016/S1474-4422(10)70195-2
- McCann H, Stevens CH, Cartwright H, Halliday GM. alpha-Synucleinopathy phenotypes. Parkinsonism Relat Disord 2014; 20 Suppl 1:S62-7; ; http://dx.doi.org/10.1016/S1353-8020(13)70017-8
- Auluck PK, Caraveo G, Lindquist S. alpha-Synuclein: membrane interactions and toxicity in Parkinson's disease. Annu Rev Cell Dev Biol 2010; 26:211-33; PMID:20500090; http://dx.doi.org/10.1146/annurev.cellbio.042308.113313
- Johnson BS, McCaffery JM, Lindquist S, Gitler AD. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc Natl Acad Sci U S A 2008; 105:6439-44; PMID:18434538; http://dx.doi.org/10.1073/pnas.0802082105
- Ju S, Tardiff DF, Han H, Divya K, Zhong Q, Maquat LE, Bosco DA, Hayward LJ, Brown RH, Jr., Lindquist S, et al. A yeast model of FUS/TLS-dependent cytotoxicity. PLoS Biol 2011; 9:e1001052; PMID:21541368; http://dx.doi.org/10.1371/journal.pbio.1001052
- Outeiro TF, Lindquist S. Yeast cells provide insight into α-synuclein biology and pathobiology. Science 2003; 302:1772-5; PMID:14657500; http://dx.doi.org/10.1126/science.1090439
- Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science 2006; 313:324-8; PMID:16794039; http://dx.doi.org/10.1126/science.1129462
- Gitler AD. Beer and bread to brains and beyond: can yeast cells teach us about neurodegenerative disease? Neurosignals 2008; 16:52-62; PMID:18097160; http://dx.doi.org/10.1159/000109759
- Tenreiro S, Munder MC, Alberti S, Outeiro TF. Harnessing the power of yeast to unravel the molecular basis of neurodegeneration. J Neurochem 2013; 127:438-52; PMID:23600759; http://dx.doi.org/10.1111/jnc.12271
- Gitcho MA, Bigio EH, Mishra M, Johnson N, Weintraub S, Mesulam M, Rademakers R, Chakraverty S, Cruchaga C, Morris JC, et al. TARDBP 3'-UTR variant in autopsy-confirmed frontotemporal lobar degeneration with TDP-43 proteinopathy. Acta Neuropathol 2009; 118:633-45; PMID:19618195; http://dx.doi.org/10.1007/s00401-009-0571-7
- Dini Modigliani S, Morlando M, Errichelli L, Sabatelli M, Bozzoni I. An ALS-associated mutation in the FUS 3'-UTR disrupts a microRNA-FUS regulatory circuitry. Nat Commun 2014; 5:4335; PMID:25004804
- Sabatelli M, Moncada A, Conte A, Lattante S, Marangi G, Luigetti M, Lucchini M, Mirabella M, Romano A, Del Grande A, et al. Mutations in the 3' untranslated region of FUS causing FUS overexpression are associated with amyotrophic lateral sclerosis. Hum Mol Genet 2013; 22:4748-55; PMID:23847048; http://dx.doi.org/10.1093/hmg/ddt328
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science 2003; 302:841; PMID:14593171; http://dx.doi.org/10.1126/science.1090278
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet 2004; 364:1167-9; PMID:15451224; http://dx.doi.org/10.1016/S0140-6736(04)17103-1
- Shin CW, Kim HJ, Park SS, Kim SY, Kim JY, Jeon BS. Two Parkinson's disease patients with alpha-synuclein gene duplication and rapid cognitive decline. Mov Disord 2010; 25:957-9; PMID:20222138; http://dx.doi.org/10.1002/mds.23043
- Ahn TB, Kim SY, Kim JY, Park SS, Lee DS, Min HJ, Kim YK, Kim SE, Kim JM, Kim HJ, et al. alpha-Synuclein gene duplication is present in sporadic Parkinson disease. Neurology 2008; 70:43-9; PMID:17625105; http://dx.doi.org/10.1212/01.wnl.0000271080.53272.c7
- Jackson KL, Dayton RD, Orchard EA, Ju S, Ringe D, Petsko GA, Maquat LE, Klein RL. Preservation of forelimb function by UPF1 gene therapy in a rat model of TDP-43-induced motor paralysis. Gene Ther 2015; 22(1):20–28. doi:10.1038/gt.2014.101
- Armakola M, Higgins MJ, Figley MD, Barmada SJ, Scarborough EA, Diaz Z, Fang X, Shorter J, Krogan NJ, Finkbeiner S, et al. Inhibition of RNA lariat debranching enzyme suppresses TDP-43 toxicity in ALS disease models. Nat Genet 2012; 44:1302-9; PMID:23104007; http://dx.doi.org/10.1038/ng.2434
- Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010; 466:1069-75; PMID:20740007; http://dx.doi.org/10.1038/nature09320
- Kim HJ, Raphael AR, LaDow ES, McGurk L, Weber RA, Trojanowski JQ, Lee VM, Finkbeiner S, Gitler AD, Bonini NM. Therapeutic modulation of eIF2alpha phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet 2014; 46:152-60; PMID:24336168; http://dx.doi.org/10.1038/ng.2853
- Narayan P, Ehsani S, Lindquist S. Combating neurodegenerative disease with chemical probes and model systems. Nat Chem Biol 2014; 10:911-20; PMID:25325702; http://dx.doi.org/10.1038/nchembio.1663
- Gitler AD, Chesi A, Geddie ML, Strathearn KE, Hamamichi S, Hill KJ, Caldwell KA, Caldwell GA, Cooper AA, Rochet JC, et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet 2009; 41:308-15; PMID:19182805; http://dx.doi.org/10.1038/ng.300
- Yeger-Lotem E, Riva L, Su LJ, Gitler AD, Cashikar AG, King OD, Auluck PK, Geddie ML, Valastyan JS, Karger DR, et al. Bridging high-throughput genetic and transcriptional data reveals cellular responses to alpha-synuclein toxicity. Nat Genet 2009; 41:316-23; PMID:19234470; http://dx.doi.org/10.1038/ng.337
- Caraveo G, Auluck PK, Whitesell L, Chung CY, Baru V, Mosharov EV, Yan X, Ben-Johny M, Soste M, Picotti P, et al. Calcineurin determines toxic versus beneficial responses to alpha-synuclein. Proc Natl Acad Sci U S A 2014; 111:E3544-52; PMID:25122673; http://dx.doi.org/10.1073/pnas.1413201111
- Chung CY, Khurana V, Auluck PK, Tardiff DF, Mazzulli JR, Soldner F, Baru V, Lou Y, Freyzon Y, Cho S, et al. Identification and rescue of alpha-synuclein toxicity in Parkinson patient-derived neurons. Science 2013; 342:983-7; PMID:24158904; http://dx.doi.org/10.1126/science.1245296
- Su LJ, Auluck PK, Outeiro TF, Yeger-Lotem E, Kritzer JA, Tardiff DF, Strathearn KE, Liu F, Cao S, Hamamichi S, et al. Compounds from an unbiased chemical screen reverse both ER-to-Golgi trafficking defects and mitochondrial dysfunction in Parkinson's disease models. Dis Model Mech 2010; 3:194-208; PMID:20038714; http://dx.doi.org/10.1242/dmm.004267
- Tardiff DF, Jui NT, Khurana V, Tambe MA, Thompson ML, Chung CY, Kamadurai HB, Kim HT, Lancaster AK, Caldwell KA, et al. Yeast reveal a "druggable" Rsp5/Nedd4 network that ameliorates alpha-synuclein toxicity in neurons. Science 2013; 342:979-83; PMID:24158909; http://dx.doi.org/10.1126/science.1245321
- Tardiff DF, Khurana V, Chung CY, Lindquist S. From yeast to patient neurons and back again: powerful new discovery platform. Mov Disord 2014; 29:1231-40; PMID:25131316; http://dx.doi.org/10.1002/mds.25989
- Tardiff DF, Tucci ML, Caldwell KA, Caldwell GA, Lindquist S. Different 8-hydroxyquinolines protect models of TDP-43 protein, alpha-synuclein, and polyglutamine proteotoxicity through distinct mechanisms. J Biol Chem 2012; 287:4107-20; PMID:22147697; http://dx.doi.org/10.1074/jbc.M111.308668
- Bonini NM, Gitler AD. Model organisms reveal insight into human neurodegenerative disease: ataxin-2 intermediate-length polyglutamine expansions are a risk factor for ALS. J Mol Neurosci 2011; 45:676-83; PMID:21660502; http://dx.doi.org/10.1007/s12031-011-9548-9
- Gispert S, Kurz A, Waibel S, Bauer P, Liepelt I, Geisen C, Gitler AD, Becker T, Weber M, Berg D, et al. The modulation of Amyotrophic Lateral Sclerosis risk by ataxin-2 intermediate polyglutamine expansions is a specific effect. Neurobiol Dis 2012; 45:356-61; PMID:21889984; http://dx.doi.org/10.1016/j.nbd.2011.08.021
- Hart MP, Brettschneider J, Lee VM, Trojanowski JQ, Gitler AD. Distinct TDP-43 pathology in ALS patients with ataxin 2 intermediate-length polyQ expansions. Acta Neuropathol 2012; 124:221-30; PMID:22526021; http://dx.doi.org/10.1007/s00401-012-0985-5
- Hart MP, Gitler AD. ALS-associated ataxin 2 polyQ expansions enhance stress-induced caspase 3 activation and increase TDP-43 pathological modifications. J Neurosci 2012; 32:9133-42; PMID:22764223; http://dx.doi.org/10.1523/JNEUROSCI.0996-12.2012
- Lee T, Li YR, Chesi A, Hart MP, Ramos D, Jethava N, Hosangadi D, Epstein J, Hodges B, Bonini NM, et al. Evaluating the prevalence of polyglutamine repeat expansions in amyotrophic lateral sclerosis. Neurology 2011; 76:2062-5; PMID:21562248; http://dx.doi.org/10.1212/WNL.0b013e31821f4447
- Lee T, Li YR, Ingre C, Weber M, Grehl T, Gredal O, de Carvalho M, Meyer T, Tysnes OB, Auburger G, et al. Ataxin-2 intermediate-length polyglutamine expansions in European ALS patients. Hum Mol Genet 2011; 20:1697-700; PMID:21292779; http://dx.doi.org/10.1093/hmg/ddr045
- Yu Z, Zhu Y, Chen-Plotkin AS, Clay-Falcone D, McCluskey L, Elman L, Kalb RG, Trojanowski JQ, Lee VM, Van Deerlin VM, et al. PolyQ repeat expansions in ATXN2 associated with ALS are CAA interrupted repeats. PLoS One 2011; 6:e17951; PMID:21479228; http://dx.doi.org/10.1371/journal.pone.0017951
- Couthouis J, Hart MP, Erion R, King OD, Diaz Z, Nakaya T, Ibrahim F, Kim HJ, Mojsilovic-Petrovic J, Panossian S, et al. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet 2012; 21:2899-911; PMID:22454397; http://dx.doi.org/10.1093/hmg/dds116
- Couthouis J, Hart MP, Shorter J, DeJesus-Hernandez M, Erion R, Oristano R, Liu AX, Ramos D, Jethava N, Hosangadi D, et al. A yeast functional screen predicts new candidate ALS disease genes. Proc Natl Acad Sci U S A 2011; 108:20881-90; PMID:22065782; http://dx.doi.org/10.1073/pnas.1109434108
- Meriin AB, Zhang X, He X, Newnam GP, Chernoff YO, Sherman MY. Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J Cell Biol 2002; 157:997-1004; PMID:12058016; http://dx.doi.org/10.1083/jcb.200112104
- Duennwald ML, Jagadish S, Giorgini F, Muchowski PJ, Lindquist S. A network of protein interactions determines polyglutamine toxicity. Proc Natl Acad Sci U S A 2006; 103:11051-6; PMID:16832049; http://dx.doi.org/10.1073/pnas.0604548103
- Gokhale KC, Newnam GP, Sherman MY, Chernoff YO. Modulation of prion-dependent polyglutamine aggregation and toxicity by chaperone proteins in the yeast model. J Biol Chem 2005; 280:22809-18; PMID:15824100; http://dx.doi.org/10.1074/jbc.M500390200
- Cashikar AG, Schirmer EC, Hattendorf DA, Glover JR, Ramakrishnan MS, Ware DM, Lindquist SL. Defining a pathway of communication from the C-terminal peptide binding domain to the N-terminal ATPase domain in a AAA protein. Mol Cell 2002; 9:751-60; PMID:11983167; http://dx.doi.org/10.1016/S1097-2765(02)00499-9
- Sielaff B, Tsai FT. The M-domain controls Hsp104 protein remodeling activity in an Hsp70/Hsp40-dependent manner. J Mol Biol 2010; 402:30-7; PMID:20654624; http://dx.doi.org/10.1016/j.jmb.2010.07.030
- Miot M, Reidy M, Doyle SM, Hoskins JR, Johnston DM, Genest O, Vitery MC, Masison DC, Wickner S. Species-specific collaboration of heat shock proteins (Hsp) 70 and 100 in thermotolerance and protein disaggregation. Proc Natl Acad Sci U S A 2011; 108:6915-20; PMID:21474779; http://dx.doi.org/10.1073/pnas.1102828108
- Lee J, Kim JH, Biter AB, Sielaff B, Lee S, Tsai FT. Heat shock protein (Hsp) 70 is an activator of the Hsp104 motor. Proc Natl Acad Sci U S A 2013; 110:8513-8; PMID:23650362; http://dx.doi.org/10.1073/pnas.1217988110
- Schirmer EC, Homann OR, Kowal AS, Lindquist S. Dominant gain-of-function mutations in Hsp104p reveal crucial roles for the middle region. Mol Biol Cell 2004; 15:2061-72; PMID:14978213; http://dx.doi.org/10.1091/mbc.E02-08-0502
- Sweeny EA, Jackrel ME, Go MS, Sochor MA, Razzo BM, DeSantis ME, Gupta K, Shorter J. The Hsp104 N-Terminal Domain Enables Disaggregase Plasticity and Potentiation. Mol Cell 2015; 57(5):836–849. doi:10.1016/j.molcel.2014.12.021; PMID:25620563
- Qiu H, Lee S, Shang Y, Wang WY, Au KF, Kamiya S, Barmada SJ, Finkbeiner S, Lui H, Carlton CE, et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J Clin Invest 2014; 124:981-99; PMID:24509083; http://dx.doi.org/10.1172/JCI72723
- Swarup V, Phaneuf D, Bareil C, Robertson J, Rouleau GA, Kriz J, Julien JP. Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain 2011; 134:2610-26; PMID:21752789; http://dx.doi.org/10.1093/brain/awr159
- Pesiridis GS, Lee VM, Trojanowski JQ. Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum Mol Genet 2009; 18:R156-62; PMID:19808791; http://dx.doi.org/10.1093/hmg/ddp303
- Da Cruz S, Cleveland DW. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr Opin Neurobiol 2011; 21:904-19; PMID:21813273; http://dx.doi.org/10.1016/j.conb.2011.05.029
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al. Mutation in the α-Synuclein Gene Identified in Families with Parkinson's Disease. Science 1997; 276:2045-7; PMID:9197268; http://dx.doi.org/10.1126/science.276.5321.2045
- Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, et al. The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 2004; 55:164-73; PMID:14755719; http://dx.doi.org/10.1002/ana.10795
- Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT. Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson's disease: Implications for pathogenesis and therapy. Proc Natl Acad Sci USA 2000; 97:571-6; PMID:10639120; http://dx.doi.org/10.1073/pnas.97.2.571
- Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009; 323:1208-11; PMID:19251628; http://dx.doi.org/10.1126/science.1165942
- Huang EJ, Zhang J, Geser F, Trojanowski JQ, Strober JB, Dickson DW, Brown RH, Jr., Shapiro BE, Lomen-Hoerth C. Extensive FUS-immunoreactive pathology in juvenile amyotrophic lateral sclerosis with basophilic inclusions. Brain Pathol 2010; 20:1069-76; PMID:20579074; http://dx.doi.org/10.1111/j.1750-3639.2010.00413.x
- Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B, et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J 2010; 29:2841-57; PMID:20606625; http://dx.doi.org/10.1038/emboj.2010.143
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 2008; 319:1668-72; PMID:18309045; http://dx.doi.org/10.1126/science.1154584
- Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, Hatanpaa KJ, White CL 3rd, Bigio EH, Caselli R, et al. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol 2008; 63:535-8; PMID:18288693; http://dx.doi.org/10.1002/ana.21344
- Mackenzie IRA, Neumann M. FET proteins in frontotemporal dementia and amyotrophic lateral sclerosis. Brain Res 2012; 1462:40-3; PMID:22261247; http://dx.doi.org/10.1016/j.brainres.2011.12.010
- Tan AY, Manley JL. The TET family of proteins: functions and roles in disease. J Mol Cell Biol 2009; 1:82-92; PMID:19783543; http://dx.doi.org/10.1093/jmcb/mjp025
- Oguchi Y, Kummer E, Seyffer F, Berynskyy M, Anstett B, Zahn R, Wade RC, Mogk A, Bukau B. A tightly regulated molecular toggle controls AAA+ disaggregase. Nat Struct Mol Biol 2012; 19:1338-46; PMID:23160353; http://dx.doi.org/10.1038/nsmb.2441
- Sanchez Y, Lindquist SL. HSP104 required for induced thermotolerance. Science 1990; 248:1112-5; PMID:2188365; http://dx.doi.org/10.1126/science.2188365
- Sanchez Y, Taulien J, Borkovich KA, Lindquist S. Hsp104 is required for tolerance to many forms of stress. EMBO J 1992; 11:2357-64; PMID:1600951
- Takahashi A, Hara H, Kurahashi H, Nakamura Y. A systematic evaluation of the function of the protein-remodeling factor Hsp104 in [PSI+] prion propagation in S. cerevisiae by comprehensive chromosomal mutations. Prion 2007; 1:69-77; PMID:19164920; http://dx.doi.org/10.4161/pri.1.1.4060
- Wang JD, Herman C, Tipton KA, Gross CA, Weissman JS. Directed evolution of substrate-optimized GroEL/S chaperonins. Cell 2002; 111:1027-39; PMID:12507429; http://dx.doi.org/10.1016/S0092-8674(02)01198-4
- Quan S, Wang L, Petrotchenko EV, Makepeace KA, Horowitz S, Yang J, Zhang Y, Borchers CH, Bardwell JC. Super Spy variants implicate flexibility in chaperone action. Elife 2014; 3:e01584; PMID:24497545; http://dx.doi.org/10.7554/eLife.01584
- Martin A, Baker TA, Sauer RT. Diverse pore loops of the AAA+ ClpX machine mediate unassisted and adaptor-dependent recognition of ssrA-tagged substrates. Mol Cell 2008; 29:441-50; PMID:18313382; http://dx.doi.org/10.1016/j.molcel.2008.02.002
- Aponte RA, Zimmermann S, Reinstein J. Directed evolution of the DnaK chaperone: mutations in the lid domain result in enhanced chaperone activity. J Mol Biol 2010; 399:154-67; PMID:20381501; http://dx.doi.org/10.1016/j.jmb.2010.03.060
- Schweizer RS, Aponte RA, Zimmermann S, Weber A, Reinstein J. Fine tuning of a biological machine: DnaK gains improved chaperone activity by altered allosteric communication and substrate binding. Chembiochem 2011; 12:1559-73; PMID:21656889; http://dx.doi.org/10.1002/cbic.201000786
- Sharma D, Masison DC. Single methyl group determines prion propagation and protein degradation activities of yeast heat shock protein (Hsp)-70 chaperones Ssa1p and Ssa2p. Proc Natl Acad Sci U S A 2011; 108:13665-70; PMID:21808014; http://dx.doi.org/10.1073/pnas.1107421108
- Pratt WB, Gestwicki JE, Osawa Y, Lieberman AP. Targeting Hsp90/Hsp70-based protein quality control for treatment of adult onset neurodegenerative diseases. Annu Rev Pharmacol Toxicol 2015; 55:353–371; doi:10.1146/annurev-pharmtox-010814-124332
- Wang AM, Miyata Y, Klinedinst S, Peng HM, Chua JP, Komiyama T, Li X, Morishima Y, Merry DE, Pratt WB, et al. Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat Chem Biol 2013; 9:112-8; PMID:23222885; http://dx.doi.org/10.1038/nchembio.1140