
Abstract
Prion-like proteins can undergo conformational rearrangements from an intrinsically disordered to a highly ordered amyloid state. This ability to change conformation is encoded in distinctive domains, termed prion domains (PrDs). Previous work suggests that PrDs change conformation to affect protein function and create phenotypic diversity. More recent work shows that PrDs can also undergo many weak interactions when disordered, allowing them to organize the intracellular space into dynamic compartments. However, mutations within PrDs and altered aggregation properties have also been linked to age-related diseases in humans. Thus, the physiological role of prion-like proteins, the mechanisms regulating their conformational promiscuity and the links to disease are still unclear. Here, we summarize recent work with prion-like proteins in Dictyostelium discoideum. This work was motivated by the finding that D. discoideum has the highest content of prion-like proteins of all organisms investigated to date. Surprisingly, we find that endogenous and exogenous prion-like proteins remain soluble in D. discoideum and do not misfold and aggregate. We provide evidence that this is due to specific adaptations in the protein quality control machinery, which may allow D. discoideum to tolerate its highly aggregation-prone proteome. We predict that D. discoideum will be an important model to study the function of prion-like proteins and their mechanistic links to disease.
CHOOSING THE RIGHT MODEL
The ability of prion-like proteins to misfold poses a challenge to cellular and organismal health. It is therefore of utmost importance to understand the mechanisms that control their conformational promiscuity. The majority of studies on misfolding-prone prion-like proteins have been done with classical model organisms, such as yeast, worms or flies. These studies have provided valuable insights. However, the narrow focus on a few models may have biased our perception of prion-like proteins.
Model organisms are tremendously powerful drivers of biomedical research, and the choice of a model organism often plays an important role in the success of a research project. Experimental accessibility has long been the key criterion for choosing a model organism. However, with increasing advances in technology, such as CRISPR/Cas9-mediated genome engineering, this feature is becoming less important. Instead, the challenge today is to find a model with specific biological features. These features may have been amplified or exaggerated by evolution, and could thus provide insight into an important aspect of biology. Prominent examples are organisms with an extraordinary capacity to self-renew, such as planarian flatworms or axolotl.Citation1 Studies carried out in these organisms allowed important insight into molecular mechanisms of regeneration. Thus, a model organism with amplified features can provide a unique window into biology, often with very broad implications.
DICTYOSTELIUM: A MODEL FOR PROTEIN MISFOLDING AND AGGREGATION
Protein aggregates are a hallmark of many age-related neurodegenerative diseases. The aggregated proteins share a common feature: they posses regions that are devoid of protein structure, so-called intrinsically disordered regions. The compositional complexity of these sequences is low and they often contain a high content of polar, uncharged amino acids, such as glutamine (Q) and asparagine (N). Sequences with such amino acid compositions show similarity to regions in yeast prion proteins and are thus often referred to as prion-like domains. Yeast prion proteins can switch from a non-prion to an aggregated prion state, and this switch is associated with a change in protein function.Citation2,3 Once formed, these prion states are highly stable and can spread as epigenetic elements in a population of yeast cells. They have been proposed to be benign drivers of phenotypic diversity in clonal populations of single-celled organisms such as budding yeast.Citation4 However, in humans, proteins with prion-like domains have been implicated in neurodegenerative diseases,Citation5,6 and mutations in PrDs often accelerate disease progression.Citation7-9
Research on yeast prion proteins has provided important insight into the structural basis and the functional role of amyloids and prions.Citation10,11 In an effort to gain further insight into this class of proteins, we recently expanded the repertoire of model organisms for the study of prion-like proteins. This was driven by the observation that proteomes, or, more specifically, the amino acid composition of proteomes, are vastly different from organism to organism. Previous comparative studies revealed that the highest content of Q/N-rich low complexity sequences is found in the social amoebae Dictyostelium discoideum.Citation12 Thus, we hypothesized that this natural enrichment of Q/N-rich domains might have enabled the evolution of sophisticated mechanisms to control and maintain them.
To investigate this idea, we first reevaluated the Q/N richness of the D. discoideum proteome. In agreement with previous results, we observed that the number of proteins containing homopolymeric runs of Qs or Ns exceeds by far the numbers observed in other organisms such as S. cerevisiae or humans.Citation13 Moreover, the polyQ or polyN runs often extend beyond the aggregation threshold of 35–40 residues (see ). In addition, we identified 1,733 proteins in the proteome of D. discoideum with prion-like sequence features. These findings indicate that the proteome of D. discoideum might have a much higher aggregation propensity compared to other organisms. Given this observation, we hypothesized that D. discoideum could provide a unique window into the universal problem of protein misfolding and aggregation.
TABLE 1. Proteins with homopolymeric N or Q stretches above the aggregation threshold of 35 residues
PRION-LIKE DOMAINS ARE CONSERVED AND MAY PLAY AN IMPORTANT ROLE DURING DEVELOPMENT
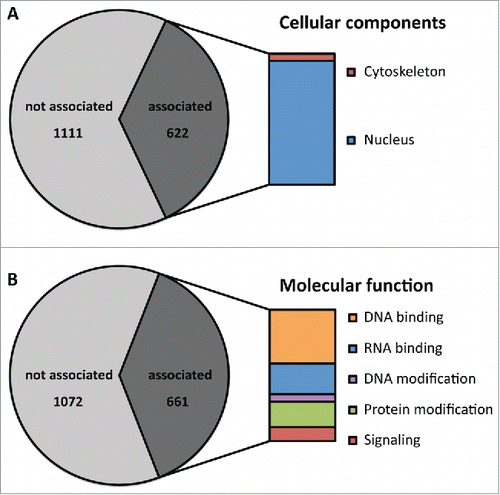
The accumulation of prion-like domains within the proteome of D. discoideum seems puzzling, because the high aggregation propensity of these domains can be an extraordinary liability to a cell. Therefore, we hypothesized that there may be a strong selective pressure against the continued presence of PrDs. Interestingly, however, we found ∼900 prion-like proteins in the proteome of the close relative D. purpureum.Citation13 Of these, 505 proteins are shared between both species. We also found a comparable overlap within the associated protein super-domain families. This contradicts the notion of a random distribution of prion-like domains and argues that the domains have functions that are under positive selection. This notion is further substantiated by the observation that the identified prion-like domains in D. discoideum are significantly associated with RNA binding domains (RRM), an observation that was also made in S. cerevisiae, humans and other organisms.Citation14,15 Furthermore, a fraction of the identified prion-like proteins cluster into specific gene ontology (GO) groups in D. discoideum () as well as in other organisms.Citation13 Interestingly, the identified groups in all organisms represent similar cellular components and molecular functions. These findings point toward a shared biological function of prion-like domains in different organisms.
FIGURE 1. Prion-like proteins cluster into specific gene ontology groups. Within the group of prion-like domains, specific gene ontology (GO) groups are enriched. (A) Proteins associated with the cytoskeleton and the nucleus are specifically enriched among prion-like proteins. 622 proteins of the identified 1733 correspond to these groups. (B) Prion-like proteins are enriched for GO terms associated with DNA binding, RNA binding, DNA modification, protein modification and signaling. 661 proteins of the identified 1733 correspond to these groups.
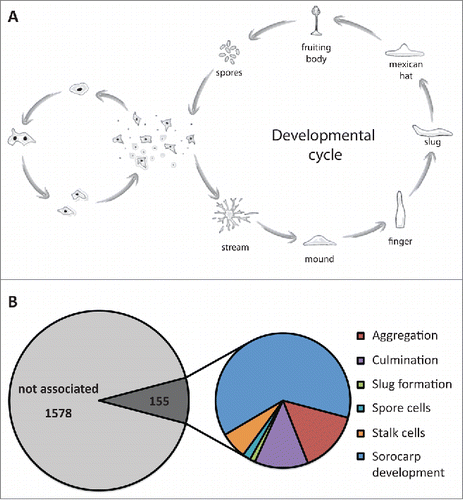
The content of intrinsically disordered proteins within a proteome differs between species. The amount of disorder seems to be related to the habitat of a species, with increasing levels of disordered proteins found in organisms, which live in very unstable environments.Citation16 D. discoideum is a soil living amoebae and therefore faces constant environmental fluctuations. Moreover, it has a fascinating live cycle, where, upon nutrient depletion, the organism changes from a unicellular state to a multi-cellular state (). During this transition, differentiation takes place, and cells are primed to either differentiate into surviving spores or into dying stalk cells. Surprisingly, we found a significant functional enrichment of prion-like proteins in developmental processes (6.7% in prion-like proteins compared to 3,7% in the whole proteome, p < 0.005) (). This indicates that prion-like proteins may be involved in regulating the developmental cycle of D. discoideum. This hypothesis agrees with previous suggestions, which link an increase in morphological complexity with increased levels of intrinsic disorder in a proteome.Citation16 However, the question remains how prion-like proteins function on the molecular level. Recent findings by the Verstreppen lab suggest that repeat variations in Q/N-rich domains can tune the solubility of proteins and thus their functional state.Citation17 Because the frequency of mutations in repeat regions is much higher than anywhere else in a protein sequence, one possible function of these repeats could be that they accelerate adaptive mutations in highly fluctuating environments.
FIGURE 2. Prion-like proteins may be involved in the development of D. discoideum. (A) Live cycle of D. discoideum. Upon nutrient depletion, cells enter the developmental cycle and proceed through several morphogenic states during which the cells differentiate into spore cells or stalk cells. Spore cells remain dormant until the conditions become more favorable. (B) Prion-like proteins are specifically enriched for proteins associated with developmental processes. 155 proteins are involved in general or specific processes during sorocarp development.
D. DISCOIDEUM EFFICIENTLY CONTROLS THE AGGREGATION OF PRION-LIKE PROTEINS
The accumulation of aggregation-prone prion-like proteins in the proteome of D. discoideum raises the question whether this organism has evolved mechanisms to control the aggregation of these proteins. To approach this question, we studied the aggregation behavior of several well-characterized prion-like proteins. These proteins have been studied extensively in other organisms such as yeast, C. elegans, or mammalian cells and found to form cytosolic aggregates.Citation18-20
First, we studied proteins containing homopolymeric polyQ or polyN runs. We used a polyQ-containing version of the human Huntingtin exon 1 with 103 glutamines (Q103) and a synthetic variant in which the glutamines were replaced by 47 asparagines (N47). Both proteins exhibit similar aggregation properties in S. cerevisiae.Citation21 In contrast to observations made in other organisms, when expressed in D. discoideum cells, these proteins remained diffusely distributed in almost all cells.Citation13 However, we detected small amounts of aggregated material in biochemical assays, which suggests that the proteins may aggregate in a small fraction of the cells. We think that these few cells are stressed, mostly likely though damage or aging. The amount of aggregated material was slightly higher in cells expressing N47 than in cells expressing Q103, suggesting that N47 is more aggregation-prone than Q103. We attribute this to the different effects of asparagine and glutamine on amyloid formation, as reported previously.Citation21,22 Interestingly, in a small number of N47-expressing cells, N47 also accumulated in the nucleus.Citation13 Thus, we conclude that N47 can accumulate in the nucleus, but that N47 and Q103 remain mostly soluble in D. discoideum.
Next, we analyzed the aggregation behavior of prion-like proteins, first focusing on the PrD (NM) of the yeast prion proteins Sup35. Consistent with observations made for Q103 and N47, we observed a diffuse distribution of NM throughout the cytosol and barely any accumulation of aggregated material.Citation13 When we expressed a modified version of NM, which was engineered to have a higher aggregation propensity in yeast, we observed a slight increase in the aggregated material and a higher incidence of nuclear accumulations. In addition, we analyzed the behavior of endogenous prion-like domains. These proteins remained likewise soluble in D. discoideum, while they formed cytosolic aggregates when expressed in S. cerevisiae.Citation13 These observations indicate that proteins, which have a high tendency to aggregate in other organisms, remain soluble when expressed in D. discoideum. This supports the hypothesis that D. discoideum possesses sophisticated pathways to control the aggregation of prion-like proteins.
We next performed a detailed analysis of the nuclear signal. We noticed that 75% of the cells showed a diffuse nuclear distribution of prion-like proteins with occasional small puncta. In the remaining 25%, the proteins localized to 2 or 3 distinct nuclear foci.Citation13 Subsequent analysis by correlative light and electron microscopy (CLEM) and immunostainings of histones and DNA suggest that these structures represent nucleoli. To test whether the nuclear accumulations contain aggregated proteins, we measured their mobility using fluorescence recovery after photobleaching (FRAP). Surprisingly, the nuclear foci showed a high mobility, thus excluding the possibility that the nuclear accumulations consist of amyloid. Rather, this suggests that prion-like proteins are enriched in nuclear compartments like the nucleolus.
Why do prion-like proteins accumulate in the nucleus of D. discoideum? Several previous studies showed that cytosolic aggregation-prone proteins accumulate in the nucleus and are subsequently degraded.Citation23,24 The nuclear localization of N47 and modified NM indicates that similar processes might occur in D. discoideum. Indeed, we found that proteins accumulating in the nucleus are ubiquitinated and their degradation is reduced when proteasomal inhibitors are added.Citation13 These observations substantiate the notion that the nucleus of D. discoideum serves as a compartment for protein quality control, wherein aggregation-prone proteins accumulate and are degraded by the ubiquitin proteasome system (UPS). Interestingly, previous studies showed that, in other organisms, the UPS is impaired through accumulating polyQ proteins, most likely because they are difficult to degrade.Citation25 This suggests that D. discoideum may have undergone specific adaptations, which decrease the vulnerability of the ubiquitin proteasome system to these proteins.
AGGREGATION OF PRION-LIKE PROTEINS IS CONTROLLED BY MOLECULAR CHAPERONES
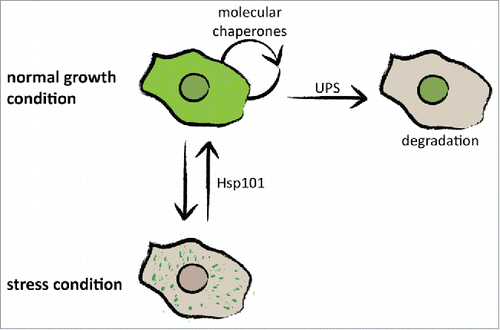
The surprising observation that prion-like proteins, both exogenous and endogenous, remain soluble in D. discoideum suggests that cytosolic pathways control protein aggregation. Indeed, we found that impairment of the molecular chaperone system, either by specific chaperone inhibitors or heat stress, causes massive formation of cytosolic aggregates in D. discoideum.Citation13 Moreover, we observed that the formed cytosolic aggregates could be dissolved once the stress was removed. We attribute this function to the disaggregase Hsp101, a chaperone of the Hsp100 family and ortholog of the yeast Hsp104. Overexpression of the disaggregase prevented heat-induced formation of aggregates, and inhibition reduced the dissolution of aggregates in the recovery phase.Citation13 This suggests that D. discoideum has evolved mechanisms to prevent aggregation under normal growth conditions and to reverse aggregation after stress. During both molecular processes chaperones play a key role. Taken together, these findings show that D. discoideum has evolved several molecular mechanisms to control aggregation-prone proteins. During normal growth, proteins can be efficiently degraded by the proteasome and can be kept soluble by molecular chaperones (). In a second layer of defense, D. discoideum utilizes the disaggregase Hsp101 and probably also other chaperones to ensure survival during stress conditions ().
FIGURE 3. Molecular mechanisms controlling protein aggregation in D. discoideum. D. discoideum cells have evolved at least 2 mechanisms to control protein aggregation. During normal growth conditions, proteins are kept soluble by molecular chaperones and can be degraded in the nucleus. The disaggregase Hsp101, probably in combination with other chaperones, can rescue stress-induced protein aggregation and restore cellular integrity.
In addition to the mechanisms described above, cells can also use mitosis to generate aggregate-free progeny. This has been shown for asymmetrically dividing cells such as S. cerevisiae,Citation26 but also in higher eukaryotes.Citation27 Although D. discoideum divides predominantly symmetrically, it undergoes asymmetric cell sorting during development. Thus, by sorting damage-containing cells to prestalk regions during development (). discoideum may be able to clean the population of unfit cells. The mechanisms underlying the fate decision in D. discoideum have been controversially discussed for many years. Interestingly, however, stressful conditions increase the fraction of cells that commit to the stalk cell fate.Citation28 This suggests that D. discoideum might use its developmental program to purge the population of cells that have accumulated protein damage. Investigations are ongoing to study whether cell-sorting mechanisms have a role in purging D. discoideum of protein aggregates.
CAN D. DISCOIDEUM HELP US DETERMINE THE FUNCTIONAL ROLE OF PRION-LIKE DOMAINS?
Although we have gained important insight into the mechanisms regulating the conformational promiscuity of prion-like proteins, we still have a very limited understanding of their function in living cells. It is unlikely that all prion-like proteins in a given proteome form amyloid-like assemblies. Such an overwhelming presence of amyloid structures would be difficult to control. Instead, a newly emerging concept suggests that prion-like domains may primarily act through transiently forming protein-protein contacts, which aid in the formation of dynamic and disordered assemblies in living cells.Citation9,29 According to this view, the functional state of many prion-like domains is not the amyloid-like state but the disordered state. Thus, we think that D. discoideum will not only help us understand the mechanisms controlling prion-like proteins, but it will also aid in solving the controversy of the functional role of prion-like domains.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
Acknowledgments
We thank members of the Alberti lab for critical comments on the manuscript.
Funding
We acknowledge funding by the Max-Planck Society.
Related Research Data
REFERENCES
- Tanaka EM, Reddien PW. The cellular basis for animal regeneration. Dev Cell 2011; 21:172-85; PMID:21763617; http://dx.doi.org/10.1016/j.devcel.2011.06.016
- Shorter J, Lindquist S. Prions as adaptive conduits of memory and inheritance. Nat Rev Genet 2005; 6:435-50; PMID:15931169; http://dx.doi.org/10.1038/nrg1616
- Sugiyama S, Tanaka M. Self-propagating amyloid as a critical regulator for diverse cellular functions. J Biochem 2014; 155:345-51; PMID:24711463; http://dx.doi.org/10.1093/jb/mvu026
- Halfmann R, Jarosz DF, Jones SK, Chang A, Lancaster AK, Lindquist S. Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature 2012; 482:363-8; PMID:22337056; http://dx.doi.org/10.1038/nature10875
- Gitler AD, Shorter J. RNA-binding proteins with prion-like domains in ALS and FTLD-U. Prion 2011; 5:179-87; PMID:21847013; http://dx.doi.org/10.4161/pri.5.3.17230
- Maniecka Z, Polymenidou M. From nucleation to widespread propagation: A prion-like concept for ALS. Virus Res 2015; 207:94-105; PMID:25656065; http://dx.doi.org/10.1016/j.virusres.2014.12.032
- Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013; 495:467-73; PMID:23455423; http://dx.doi.org/10.1038/nature11922
- Nomura T, Watanabe S, Kaneko K, Yamanaka K, Nukina N, Furukawa Y. Intranuclear Aggregation of Mutant FUS/TLS as a molecular pathomechanism of amyotrophic lateral sclerosis. J Biol Chem 2014; 289:1192-202; PMID:24280224; http://dx.doi.org/10.1074/jbc.M113.516492
- Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, et al. A Liquid-to-Solid Phase transition of the ALS protein FUS accelerated by disease mutation. Cell 2015; 162:1066-1077; PMID:26317470; http://dx.doi.org/10.1016/j.cell.2015.07.047
- Liebman SW, Chernoff YO. Prions in yeast. Genetics 2012; 191:1041-72; PMID:22879407; http://dx.doi.org/10.1534/genetics.111.137760
- Wickner RB, Shewmaker FP, Bateman DA, Edskes HK, Gorkovskiy A, Dayani Y, Bezsonov EE. Yeast prions: structure, biology and prion-handling systems. Microbiol Mol Biol Rev 2015; 79:1-17; PMID:25631286; http://dx.doi.org/10.1128/MMBR.00041-14
- Michelitsch MDM, Weissman JSJ. A census of glutamine/asparagine-rich regions: implications for their conserved function and the prediction of novel prions. Proc Natl Acad Sci USA 2000; 97:11910-5; PMID:11050225; http://dx.doi.org/10.1073/pnas.97.22.11910
- Malinovska L, Palm S, Gibson K, Verbavatz JM, Alberti S. Dictyostelium discoideumhas a highly Q/N-rich proteome and shows an unusual resilience to protein aggregation. Proc Natl Acad Sci USA 2015; 112(20):E2620-9, 201504459-10
- King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res 2012; 1462:61-80; PMID:22445064; http://dx.doi.org/10.1016/j.brainres.2012.01.016
- Malinovska L, Kroschwald S, Alberti S. Protein disorder, prion propensities, and self-organizing macromolecular collectives. Biochim Biophys Acta 2013; 1834:1-14; PMID:23451353; http://dx.doi.org/10.1016/j.bbapap.2012.08.018
- Uversky VN. A decade and a half of protein intrinsic disorder: Biology still waits for physics. Protein Sci 2013; 22:693-724; PMID:23553817; http://dx.doi.org/10.1002/pro.2261
- Gemayel R, Chavali S, Pougach K, Legendre M, Zhu B, Boeynaems S, van der Zande E, Gevaert K, Rousseau F, Schymkowitz J, et al. Variable Glutamine-Rich repeats modulate transcription factor activity. Mol Cell 2015; 59:615-27; PMID:26257283; http://dx.doi.org/10.1016/j.molcel.2015.07.003
- Krobitsch S, Lindquist S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc Natl Acad Sci USA 2000; 97:1589-94; PMID:10677504; http://dx.doi.org/10.1073/pnas.97.4.1589
- Brignull HR, Morley JF, Garcia SM, Morimoto RI. Modeling polyglutamine pathogenesis in C elegans Methods in Enzymology. Elsevier 2006; 412:256-82.
- Jana NR, Tanaka M, Wang GH, Nukina N. Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum Mol Genet 2000; 9:2009-18; PMID:10942430; http://dx.doi.org/10.1093/hmg/9.13.2009
- Halfmann R, Alberti S, Krishnan R, Lyle N, Lyle N, O'Donnell CW, O'Donnell CW, King OD, Berger B, Berger B, et al. Opposing effects of glutamine and asparagine govern prion formation by intrinsically disordered proteins. Mol Cell 2011; 43:72-84; PMID:21726811; http://dx.doi.org/10.1016/j.molcel.2011.05.013
- Lu X, Murphy RM. Asparagine repeat peptides: aggregation kinetics and comparison with glutamine repeats. Biochemistry 2015; 54:4784-94; PMID:26204228; http://dx.doi.org/10.1021/acs.biochem.5b00644
- Latonen L. Nucleolar aggresomes as counterparts of cytoplasmic aggresomes in proteotoxic stress. Bioessays 2011; 33:386-95; PMID:21425306; http://dx.doi.org/10.1002/bies.201100008
- Park SH, Kukushkin Y, Gupta R, Chen T, Konagai A, Hipp MS, et al. PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the SIS1P chaperone. Cell 2013; 154:134-45; PMID:23791384; http://dx.doi.org/10.1016/j.cell.2013.06.003
- Ortega Z. Ubiquitin-proteasome system involvement in Huntington's disease. Frontiers Mol Neuroscience 2014; 7:77; PMID:25324717; http://dx.doi.org/10.3389/fnmol.2014.00077
- Aguilaniu H, Gustafsson L, Rigoulet M, Nystrom T. Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science 2003; 299:1751-3; PMID:12610228; http://dx.doi.org/10.1126/science.1080418
- Rujano MA, Bosveld F, Salomons FA, Dijk F, van Waarde MAWH, et al. Polarised asymmetric inheritance of accumulated protein damage in higher eukaryotes. PLoS Biol 2006; 4:e417; PMID:17147470; http://dx.doi.org/10.1371/journal.pbio.0040417
- Castillo DI, Queller DC, Strassmann JE. Cell condition, competition, and chimerism in the social amoeba Dictyostelium discoideum. Ethology Ecol Evolution 2011; 23:262-73; http://dx.doi.org/10.1080/03949370.2011.568526
- Kroschwald S, Maharana S, Mateju D, Malinovska L, Nüske E, Poser I, Richter D, Alberti S. Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. Elife 2015; 4:e06807; PMID:26238190; http://dx.doi.org/10.7554/eLife.06807