
ABSTRACT
Prion diseases, or transmissible spongiform encephalopathies (TSEs) are progressive, fatal neurodegenerative diseases with no effective treatment. The pathology of these diseases involves the conversion of a protease sensitive form of the cellular prion protein (PrPC) into a protease resistant infectious form (PrPres). The efficiency of this conversion is predicated upon a number of factors, most notably a strong homology between cellular PrPC and PrPres. In our recently published study, we infected mice with the RML-Chandler strain of scrapie and treated them with heterologous hamster prion proteins. This treatment was seen to reduce clinical signs of prion disease, to delay the onset of clinical symptoms and to prolong survival. In this current article we discuss potential mechanisms of action of treatment with heterologous prion proteins. We also discuss potential extensions of these studies using a heterologous rabbit PrP-based treatment strategy or a peptide based strategy, and improvement of treatment delivery including a lentiviral-based system.
The prion diseases, or transmissible spongiform encephalopathies (TSEs) are progressive, uniformly-fatal neurodegenerative diseases of infectious, genetic, or sporadic origin. TSEs include diseases that affect both humans (e.g. Creutzfeldt-Jakob disease [CJD], Gerstmann-Straussler-Scheinker syndrome [GSS], and fatal familial insomia [FFI]) and animals (e.g., bovine spongiform encephalopathy [BSE], chronic wasting disease [CWD] of cervids, and scrapie in sheep. Following a typically long incubation period, those affected by a TSE experience progressive, and ultimately fatal, neurocognitive decline. No effective therapy for prion diseases has been identified and nearly all patients with the most common form of prion disease, CJD, die within 14 months of their diagnosis.Citation1 While unique in their own right, the molecular and biochemical mechanisms, cellular infectivity, the progressive accumulation of misfolded protein mechanistically aligns the prion diseases with a larger group of protein misfolding neurodegenerative disease, including Alzheimer's, Parkinson's, and Huntington's diseases.
The hallmark event in the prion disorders is the misfolding of the normal cellular prion protein (denoted PrPC) into a misfolded isoform (commonly denoted as PrPres or PrPSc). It is widely accepted that the PrPres formation is the key initiating event in prion disease pathogenesis and that its presence correlates with infectivity. As such, PrPres is also the mostly widely used diagnostic surrogate marker for affected individuals and animals. Although the accumulation of PrPres is the hallmark feature of prion diseases and the microscopic neuropathology is stereotypic (coincident gliosis, synaptic dysfunction, spongiosis, and neuronal loss), the mechanism(s) by which the misfolded prion protein exerts its toxic effects is unknown. However, it is likely that the consequences of prion infection result from a toxic gain of function of misfolded PrP, as depletion of PrPC does not appear to be detrimental.Citation2
The conversion of PrPC to PrPres is a highly specific process of templated conversion requiring direct interaction between the normal and abnormal forms of the protein.Citation3 The efficiency of this conversion is predicated upon a number of specific reaction conditions, including the secondary structure of PrPres, homology of the primary and secondary structures between PrPC and PrPres, and the architecture of the PrPC-PrPres complex.Citation4,5 Increased contact between PrPC-PrPres at residue 129 and the relative rigidity of the β2-α2 loops in PrPC are 2 important factors in mediating the efficiency of PrPres formation and TSE susceptibility.Citation5-7 In addition to steric factors, the formation of PrPres is favored by destabilization of PrPC as a number of destabilizing pathogenic mutations in PrP are linked with increasing misfolding rates.Citation8-10
In light of this work demonstrating the importance of a precise interaction in the PrPC-PrPres complex, it seems reasonable to probe the possibility that interference with this interface might be a potential therapeutic avenue. That is, to test the hypothesis proposed by Singh and Udgaonkar in their comprehensive review on PrP misfolding, namely that “…any ligand, whether small or large, that binds to the native conformation of the [PrPC] protein would stabilize that state and can therefore be expected to decrease the native-state dynamics that drive misfolding.”Citation11 Support for such an approach has been validated by antibody-based studies, which have stabilized the α1 region of PrPC and prevented prion disease in animals.Citation12,13
The concept of heterologous prion proteins (HetPrP) as potential therapeutics is not new. Horiuchi et al demonstrate that inclusion of a heterologous species PrPc in a cell-free conversion system was capable of interfering with the formation of PrPres between 2 homologous species.Citation14 When they divide the process of PrPres formation into 2 steps, namely initial binding between PrPC and PrPres followed by acquisition of protease resistance, the interfering effect of HetPrP appears to occur during the latter.Citation14 Further in vitro studies demonstrate that expression of hamster prion protein (HaPrP) in persistently-infected mouse cells nearly completely eliminates the accumulation of PrPres,Citation15 suggesting that heterologous HaPrP may either inhibit PrPres production or enhance its clearance. Moreover, scrapie-infected mouse cells that are induced to express a rabbit prion gene produce substantially less PrPres as compared to mouse cells that do not express rabbit prion proteins, suggesting that rabbit prion proteins may interfere with mouse PrPres formation.Citation16
In our work, we extended these in vitro observations into the mouse using the RML-Chandler strain of scrapie and HetPrP therapy using bacterially expressed and purified recombinant HaPrP amino acids 23-231.Citation17 In brief, mice were intracerebrally inoculated with a 0.01% RML-Chandler strain brain homogenate combined with 1 of 2 dosages of either HaPrP or vehicle control. Mice were then treated with HaPrP orally the following day. To assess the effect of the HaPrP dosage, 2 treatment groups were used, including a high dose of recombinant protein (0.7 mg/ml, high dose) and a low dose (0.35 mg/ml). Lastly, 2 control groups were included, namely a mock treatment group comprised of infected treated with vehicle only and an untreated / uninfected group. To assess the impact of treatment on clinical disease, mice were evaluated daily following infection, weekly during the first months and then daily in later months for signs of scrapie-related symptoms including ataxic gait, hind limb paresis, decreased motility, dull eyes, flattened stature, weight loss and kyphosis.
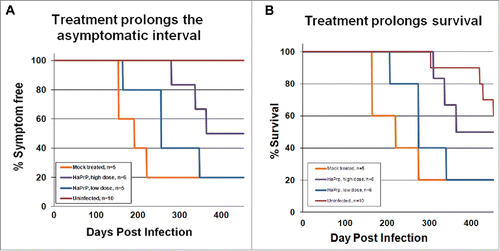
Clinically, treatment with high dose HaPrP effectively and significantly delayed the onset of clinical symptoms, including motor and non-motor deficits, and prolonged survival as compared to vehicle-treated animals ().Citation17 Moreover, when the study was terminated at 452 d post-infection, half of the high dose treated animals were still free of scrapie symptoms. In addition to abrogating the clinical signs of prion disease, mice receiving the high-dose of HaPrP accumulated significantly less PrPres in both brain and spleen as compared to mice treated with a low dose of HaPrP or with vehicle only. Additionally, HaPrP partially mitigated the neuropathologic consequences of prion infection as high dose treated animals showed a trend toward fewer activated astrocytes as revealed by immunohistochemistry for glial fibrillary acidic protein and less severe neuropil spongiosis.
FIGURE 1. Treatment with heterologous recombinant HaPrP delayed the onset of symptoms and prolonged survival. Kaplan-Meier plots showing the time at which mock-treated (orange, n = 5), low-dose-treated (blue, n = 5), high-dose-treated mice (purple, n = 6) and uninfected (red, n = 10) developed (A) detectable symptoms associated with scrapie infection, including ataxic gait, weight loss, and kyphosis, and (B) time of survival. We tested for differences between groups using a modified version of the Gehan-Wilcoxon test and found a statistically significant difference between the mock infected group and the high dose group (p = 0.0348). The low-dose group was not significantly different than the mock-treated control group. The uninfected control mice showed significantly longer survival times than the 3 groups of infected mice; uninfected versus mock (p = 0.008), uninfected vs. low dose (p = 0.006) and uninfected versus high dose (p = 0.0201).This figure is reprinted from our original article, Skinner et al., 2015, published in PloS ONE.
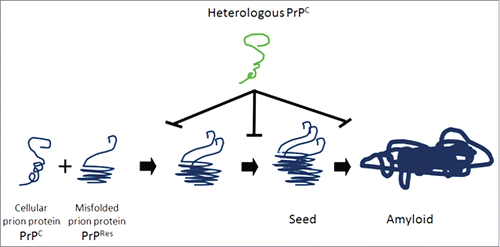
Taken in concert, treatment with HetPrPC inhibits both the formation of PrPres and the clinical consequences of prion infection. However, the mechanism underlying this phenomenon is unknown. We believe that HetPrP binds to both PrPres and PrPC and blocks the production and elongation of PrPres chains and amyloid formation (). The work of Horiuchi et al., offers 2 possible mechanistic models for this interference, based upon number and type of binding sites for PrPC on PrPres.Citation14 In a “one binding system,” they posit that the binding of HetPrP to a growing PrPres oligomer creates a functionally impotent aggregate that is then incapable of generating the steric interactions necessary for the continued production of PrPres. Alternately, in a “2 binding system” they propose that the growing PrPres oligomer contains 2 binding sites, namely a conversion-inducing site and a non-converting site. In such a 2 site system, HetPrP would interfere with the formation of PrPres by binding and blockade of the conversion site without blocking the non-converting site. In addition to biochemical mechanisms, it is possible that the protective effect of HetPrP in our study resulted from an evoked immune response that impacted PrPres formations. However, Western blot analysis of serum from study mice did not reveal the presence of anti-hamster PrP antibodies at 1:500 or 1:1000 dilutions. Lastly, it is important to note that because mice were intracerebrally inoculated simultaneously with both scrapie prions and HaPrP, it is quite likely that the HaPrP in the inoculum served to inactivate the scrapie prion by binding to PrPres and forming an inactive complex due to sequence incongruence.
FIGURE 2. Model of heterologous prion protein inhibition of protein misfolding, nucleation and formation of amyloid. Normal cellular prion proteins, PrPc, bind with and take on the conformation of misfolded, protease resistant prion proteins PrPres. This process continues with leads to seeds of oligomers of misfolded proteins, which elongate and form amyloid deposits. Heterologous prion proteins inhibit this process. We hypothesize that the mechanism mediating this inhibition is that heterologous prion proteins bind directly to PrPC and PrPres and do not conform to the shape of PrPres, and block the progression of seed and amyloid formation.
Based upon in vitro and in vivo studies, it is increasingly apparent that HetPrP treatment safely inhibits the PrPC to PrPres conversion process. These results render feasible the prospect of treating prion diseases with HetPrP. While demonstrating efficacy, the treatment regime used in our study (intracerebral instillation of HetPrP at the time of infection followed by oral ingestion of heterologous PrPC) is not ideal for treating patients with existing prion disease, and delivery via intracerebral injection is certainly not anticipated to allow HetPrP to make contact with and inactivate all PrPres in the system. As such, future studies should emphasize the development of more practical HetPrP delivery modalities and the evaluation of more effective HetPrP sequences.
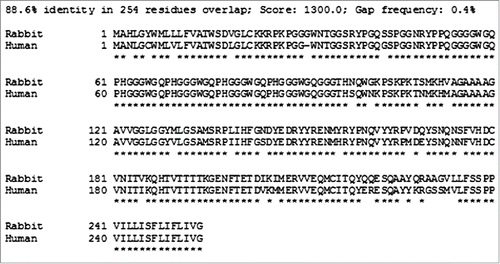
While a wide range of mammal species are susceptible to prion infection, the efficiency of interspecies prion infection is varied and governed by a so-called "species barrier,” the integrity of which is inversely proportional to the strength of the interaction between host PrPC and incoming PrPres. Among tested species, rabbits have been shown to be unusually resistant to prion disease inoculation, as attempts to transmit Kuru, CJD, sheep scrapie, TME, and mouse-adapted scrapie have failed.Citation18,19 While subsequent groups have confirmed that the rabbit is not absolutely resistant or prion infection, there is general agreement that they are only minimally susceptible.Citation20,21 The degree of primary sequence homology is important in determining the robustness of the species barrier and the rabbit prion protein shows relatively low sequence homology to other species prion proteins. To illustrate this, shows an amino acid sequence alignmentCitation22 of human and rabbit prion proteins. Based upon this work, we propose that a rabbit PrP-based HetPrP treatment strategy may be more effective than HaPrP at inhibiting prion disease.
FIGURE 3. Comparison of human and rabbit prion protein amino acid sequences. The SIM - Local similarity program (Xiaoquin and Miller, 1991) was used to make the alignments.
While IC injection of HetPrP was used in our study, the clinical evolution of this approach necessitates a more effective and simpler means of delivery. One such approach would be delivery via the blood stream to deliver HetPrP to all areas of the brain as previous studies have demonstrated the ability of PrP to cross the blood-brain-barrier.Citation23 In addition, it may be possible to use peptides derived from HetPrP rather than whole proteins. Indeed Chabry et al., showed in vitro inhibition of PrP conversation with synthetic peptides derived from mouse and hamster PrP.Citation24,25 Another such possibility for HetPrP treatment is the adoption of a gene therapy-based approach using lentiviral vectors. There is increasing precedence for gene therapy based approaches to prion diseases. Mallucci et al., generated an adult-onset PrP knockout mouse model with delayed, neuron-specific deletion of PrPC which mitigated the clinical and neuropathologic consequences of prion disease.Citation26 In an alternate gene therapy study, the same group used RNAi-driven gene silencing to reduce PrPC expression. Using lenti-shRNA directed against PrPC, treated mice experienced a significant down-regulation of PrPC expression and a delay in prion disease progression.Citation27 Moreover, Toupet et al demonstrate that chronic injection of a recombinant lentiviral vector expressing a dominant negative prion protein directly into the brains of prion disease infected mice at 80 and 90 d post-infection was shown to reduce astrocytic gliosis and extend survival.Citation28
These initial studies demonstrate proof of principal that HetPrP may serve as useful therapeutic agents for prion diseases. However, further studies are warranted to optimize both the form of HetPrP as well as its mode of delivery. The increasing awareness of the role of protein misfolding in many neurodegenerative processes makes the development of an effective treatment strategy for prion diseases a high priority. Strategies that work for treating prion diseases may also be effective when applied to other neurodegerative diseases.
ABBREVIATIONS
| TSEs | = | transmissible spongiform encephalopathies |
| CJD | = | Creutzfeldt-Jakob disease |
| GSS | = | Gerstmann-Straussler-Scheinker syndrome |
| FFI | = | fatal familial insomia |
| BSE | = | bovine spongiform encephalopathy |
| CWD | = | chronic wasting disease |
| denoted PrPC | = | cellular prion protein |
| commonly denoted as PrPres or PrPSc | = | misfolded isoform |
| HetPrP | = | heterologous prion proteins |
| HaPrP | = | hamster prion protein |
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
REFERENCES
- Valleron AJ, Boelle PY, Will R, Cesbron JY. Estimation of epidemic size and incubation time based on age characteristics of vCJD in the United Kingdom. Science 2001; 294:1726-8; PMID:11721058; http://dx.doi.org/10.1126/science.1066838
- Hetz C, Maundrell K, Soto C. Is loss of function of the prion protein the cause of prion disorders? Trends Mol Med 2003; 9:237-43; PMID:12829011; http://dx.doi.org/10.1016/S1471-4914(03)00069-8
- Caughey B, Chesebro B. Transmissible spongiform encephalopathies and prion protein interconversions. Adv Virus Res 2001; 56:277-311; PMID:11450303; http://dx.doi.org/10.1016/S0065-3527(01)56031-5
- Prusiner SB, Scott M, Foster D, Pan KM, Groth D, Mirenda C, Torchia M, Yang SL, Serban D, Carlson GA, et al. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 1990; 63:673-86; PMID:1977523; http://dx.doi.org/10.1016/0092-8674(90)90134-Z
- Mallik S, Yang W, Norstrom EM, Mastrianni JA. Live cell fluorescence resonance energy transfer predicts an altered molecular association of heterologous PrPSc with PrPC. J Biol Chem 285:8967-75; PMID:20086009; http://dx.doi.org/10.1074/jbc.M109.058107
- Bett C, Fernandez-Borges N, Kurt TD, Lucero M, Nilsson KP, Castilla J, Sigurdson CJ. Structure of the beta2-alpha2 loop and interspecies prion transmission. FASEB J 26:2868-76; PMID:22490928; http://dx.doi.org/10.1096/fj.11-200923
- Giachin G, Biljan I, Ilc G, Plavec J, Legname G. Probing early misfolding events in prion protein mutants by NMR spectroscopy. Molecules 18:9451-76; PMID:23966072; http://dx.doi.org/10.3390/molecules18089451
- Singh J, Udgaonkar JB. Structural effects of multiple pathogenic mutations suggest a model for the initiation of misfolding of the prion protein. Angew Chem Int Ed Engl 54:7529-33; PMID:25959220; http://dx.doi.org/10.1002/anie.201501011
- Liemann S, Glockshuber R. Influence of amino acid substitutions related to inherited human prion diseases on the thermodynamic stability of the cellular prion protein. Biochemistry 1999 38:3258-67; PMID:10079068; http://dx.doi.org/10.1021/bi982714g
- Apetri AC, Surewicz K, Surewicz WK. The effect of disease-associated mutations on the folding pathway of human prion protein. J Biol Chem 2004; 279:18008-14; PMID:14761942; http://dx.doi.org/10.1074/jbc.M313581200
- Singh J, Udgaonkar JB. Molecular Mechanism of the Misfolding and Oligomerization of the Prion Protein: Current Understanding and Its Implications. Biochemistry 54:4431-42; PMID:26171558; http://dx.doi.org/10.1021/acs.biochem.5b00605
- Heppner FL, Musahl C, Arrighi I, Klein MA, Rulicke T, Oesch B, Zinkernagel RM, Kalinke U, Aguzzi A. Prevention of scrapie pathogenesis by transgenic expression of anti-prion protein antibodies. Science 2001; 294:178-82; PMID:11546838; http://dx.doi.org/10.1126/science.1063093
- White AR, Enever P, Tayebi M, Mushens R, Linehan J, Brandner S, Anstee D, Collinge J, Hawke S. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature 2003; 422:80-3; PMID:12621436; http://dx.doi.org/10.1038/nature01457
- Horiuchi M, Priola SA, Chabry J, Caughey B. Interactions between heterologous forms of prion protein: binding, inhibition of conversion, and species barriers. Proc Natl Acad Sci U S A 2000; 97:5836-41; PMID: 10811921; http://dx.doi.org/10.1073/pnas.110523897
- Priola SA, Caughey B, Race RE, Chesebro B. Heterologous PrP molecules interfere with accumulation of protease-resistant PrP in scrapie-infected murine neuroblastoma cells. J Virol 1994; 68:4873-8; PMID:7913509
- Vorberg I, Groschup MH, Pfaff E, Priola SA. Multiple amino acid residues within the rabbit prion protein inhibit formation of its abnormal isoform. J Virol 2003; 77:2003-9; PMID:12525634; http://dx.doi.org/10.1128/JVI.77.3.2003-2009.2003
- Skinner PJ, Kim HO, Bryant D, Kinzel NJ, Reilly C, Priola SA, Ward AE, Goodman PA, Olson K, Seelig DM. Treatment of Prion Disease with Heterologous Prion Proteins. PLoS One 10:e013; 1993.
- Gibbs CJ, Jr., Gajdusek DC. Experimental subacute spongiform virus encephalopathies in primates and other laboratory animals. Science 1973; 182:67-8; PMID:4199733; http://dx.doi.org/10.1126/science.182.4107.67
- Barlow RM, Rennie JC. The fate of ME7 scrapie infection in rats, guinea-pigs and rabbits. Res Vet Sci 1976; 21:110-1; PMID:821118
- Chianini F, Fernandez-Borges N, Vidal E, Gibbard L, Pintado B, de Castro J, Priola SA, Hamilton S, Eaton SL, Finlayson J, et al. Rabbits are not resistant to prion infection. Proc Natl Acad Sci U S A 109:5080-5; PMID:22416127; http://dx.doi.org/10.1073/pnas.1120076109
- Vidal E, Fernandez-Borges N, Pintado B, Ordonez M, Marquez M, Fondevila D, Torres JM, Pumarola M, Castilla J. Bovine spongiform encephalopathy induces misfolding of alleged prion-resistant species cellular prion protein without altering its pathobiological features. J Neurosci 33:7778-86; PMID:23637170; http://dx.doi.org/10.1523/JNEUROSCI.0244-13.2013
- Huang XQ, Hardison RC, Miller W. A space-efficient algorithm for local similarities. Comput Appl Biosci 1990; 6:373-81; PMID:2257499
- Begley DJ. Transport of prion proteins across the blood-brain barrier. Exp Neurol 2009; 220:217-8; PMID:19682986; http://dx.doi.org/10.1016/j.expneurol.2009.08.006
- Chabry J, Caughey B, Chesebro B. Specific inhibition of in vitro formation of protease-resistant prion protein by synthetic peptides. J Biol Chem 1998; 273:13203-7; PMID:9582363; http://dx.doi.org/10.1074/jbc.273.21.13203
- Chabry J, Priola SA, Wehrly K, Nishio J, Hope J, Chesebro B. Species-independent inhibition of abnormal prion protein (PrP) formation by a peptide containing a conserved PrP sequence. J Virol 1999; 73:6245-50; PMID:10400714
- Mallucci GR, Ratte S, Asante EA, Linehan J, Gowland I, Jefferys JG, Collinge J. Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. EMBO J 2002; 21:202-10; PMID:11823413; http://dx.doi.org/10.1093/emboj/21.3.202
- White MD, Farmer M, Mirabile I, Brandner S, Collinge J, Mallucci GR. Single treatment with RNAi against prion protein rescues early neuronal dysfunction and prolongs survival in mice with prion disease. Proc Natl Acad Sci U S A 2008; 105:10238-43; PMID:18632556; http://dx.doi.org/10.1073/pnas.0802759105
- Toupet K, Compan V, Crozet C, Mourton-Gilles C, Mestre-Frances N, Ibos F, Corbeau P, Verdier JM, Perrier V. Effective gene therapy in a mouse model of prion diseases. PLoS One 2008; 3:e2773; http://dx.doi.org/10.1371/journal.pone.0002773