
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Protein sequences are evolved to encode generally one folded structure, out of a nearly infinite array of possible folds. Underlying this code is a funneled free energy landscape that guides folding to the native conformation. Protein misfolding and aggregation are also a manifestation of free-energy landscapes. The detailed mechanisms of these processes are poorly understood, but often involve rare, transient species and a variety of different pathways. The inherent complexity of misfolding has hampered efforts to measure aggregation pathways and the underlying energy landscape, especially using traditional methods where ensemble averaging obscures important rare and transient events. We recently studied the misfolding and aggregation of prion protein by examining 2 monomers tethered in close proximity as a dimer, showing how the steps leading to the formation of a stable aggregated state can be resolved in the single-molecule limit and the underlying energy landscape thereby reconstructed. This approach allows a more quantitative comparison of native folding versus misfolding, including fundamental differences in the dynamics for misfolding. By identifying key steps and interactions leading to misfolding, it should help to identify potential drug targets. Here we describe the importance of characterizing free-energy landscapes for aggregation and the challenges involved in doing so, and we discuss how single-molecule studies can help test proposed structural models for PrP aggregates.
INTRODUCTION
Many neurodegenerative diseases are believed to be caused at least in part by the misfolding and aggregation of specific proteins, including Alzheimer's (Aβ peptide), Parkinson's (α-synuclein), ALS (SOD1) and the spongiform encephalopathies (prion protein PrP).Citation1,2 In each case, the protein misfolds and aggregates into a form that is rich in β-sheets, ultimately forming insoluble amyloid fibrils. Given the importance of protein misfolding and aggregation to disease, it is extremely important to understand the molecular mechanisms behind this structural conversion. However, detailed examination of misfolding and aggregation mechanisms is challenging owing to the heterogeneous and complex pathways involved. Early stages of these pathways likely involve rare, transient intermediates, ultimately leading to larger, insoluble oligomers—both of which present challenges for detailed structural and biophysical characterization. In the case of PrP, neither the structure of the infectious scrapie isoform (PrPSc) nor the mechanism for conversion of native PrP (PrPC) is known, although various models have been proposed for the structure of PrPScCitation3-6 and the conversion mechanism.Citation2,7
Underpinning the molecular events that lead to protein aggregation is the energy landscape (), which represents the energy of the protein as a function of all possible conformations. The landscape encodes the relative stabilities of different states (e.g. native, partially folded intermediate, unfolded, misfolded, soluble oligomer, or insoluble aggregate) and the energy barriers that separate them. The current view of energy landscapes for protein misfolding and aggregation is generally qualitative rather than quantitative, due to a paucity of appropriate experimental data. In a funnelled energy landscape, high-energy, high-entropy unfolded states at the top of the funnel fold along any variety of paths down to the low-energy, low-entropy native conformation. Native folding is relatively efficient because the native state is comprised of a network of mutually supportive stabilizing contacts – they are said to be minimally frustrated.Citation8 In contrast, misfolding and aggregation are characterized by multiple competing conformations separated by substantial kinetic barriers; the landscape for inter-molecular aggregation, which is generally believed to be linked to the folding landscape via non-native structures, is thus imagined as being much rougher than for native folding.Citation9
FIGURE 1. Energy landscape cartoons depicting native folding (left) and aggregation (right). Non-native species often connect the 2 regimes. The landscape is expected to be more rugged for aggregation, having deeper kinetic traps. Single-molecule force spectroscopy can measure the critical landscape properties like the energetic stabilities of the different states (including intermediates), the heights of the energy barriers between states, the position of the barriers along the reaction coordinate, and the diffusion coefficient that connects the landscape properties to the observed kinetics of structure formation.
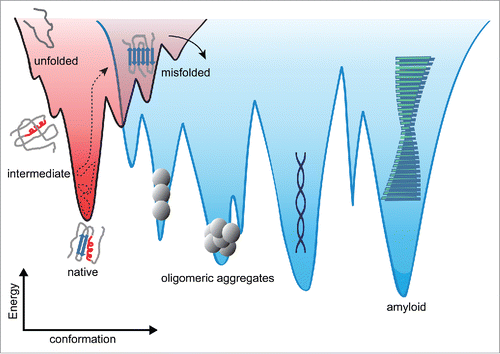
This view is supported experimentally by the observation that misfolding and aggregation are complex, apparently involving many different intermediates and competing pathways.Citation10-13 Energy landscapes for misfolding and aggregation have been calculated from simulationsCitation14-16 and partial free-energy surfaces—for example describing the native and near-native conformations—have been examined both by experiment and simulation.Citation17-20 Thermodynamic stabilities and activation energies for amyloid formation were also examined experimentally.Citation21,22 However, energy landscapes for aggregation have not been reconstructed experimentally, limiting the understanding of the fundamental determinants of aggregation.
ENERGY LANDSCAPES AND KINETICS IN FOLDING
Misfolding and aggregation are dynamic, with timescales ranging from µs–ms (for the earliest events that may initiate aggregation) to minutes or hours (oligomerization) and hours to weeks (fibrillization). The underlying energy landscapes can be used to understand this progression from a natively folded protein into a large aggregate. In particular, the barriers and ‘bumps’ along the energy landscapes determine the timescale for misfolding and aggregation. Kinetics are very important in protein aggregationCitation23-25 and thus to the development of therapeutics.Citation26 If the aggregated form is more thermodynamically stable than the native fold,Citation21 then it is only the kinetic barriers that prevent spontaneous misfolding and aggregation. Modulating kinetic barriers may offer a therapeutic strategy to treat protein misfolding disease, for example by designing ligands that kinetically stabilize the native conformationCitation27 so as to delay unfolding and subsequent aggregation. For PrP, previous work suggested that the native structure is kinetically trapped by a ∼20 kcal/mol barrier separating it from a more thermodynamically stable, β-rich oligomeric state, and mutations or ‘seed’ aggregates (PrPSc) were postulated to enhance aggregation by stabilizing the misfolding transition state.Citation28 Such a scenario suggests possible approaches to inhibiting aggregation by blocking the binding to PrPSc via transition-state analogsCitation29 or kinetically trapping PrPSc intermediates to prevent progress down the pathological aggregation pathway.Citation30
Kinetic barriers in folding are typically viewed as arising from the competing effects of enthalpy and entropy during structural changes: structured states are generally enthalpically favored but entropically disfavoured, and mismatches in the free-energy changes caused by enthalpy and entropy reduction result in barriers.Citation31 Although they play a critical role in folding because they dominate the dynamics, barriers are difficult to characterize quantitatively in a reliable manner. The barrier height, ΔG‡, is usually inferred from the rate constant, k, which is easily determined experimentally:[1]
[1] where T is temperature, kB is Boltzmann's constant, and k0 is the rate prefactor or ‘attempt frequency’. The challenge for relating rates to ΔG‡ is determining k0. One approach uses the Eyring equation from transition-state theory: k0 = κ(kBT/h), where h is Planck's constant and κ the transmission coefficient (relating to the probability that the molecule crosses the barrier, often assumed to be 1).Citation32 However, this approach overestimates k0 for protein folding, because it neglects diffusive barrier recrossing and thus also overestimates ΔG‡.
Kramers' theory for diffusive barrier crossingCitation32 provides a better framework for determining k0:[2]
[2] where D is the intrachain diffusion coefficient and κw and κb relate to the local curvature of the landscape in the potential well and barrier top, respectively. Here, D reports on the configurational dynamics of the protein chain, rather than on translational diffusion. Note that in Kramers' theory, the prefactor can vary from one protein to the next, or even for different transitions in the same protein (for multi-state folding), because of changes in κ and D. Hence imputing changes in rates at a given temperature (e.g., upon mutation of the protein or comparing native and non-native folding) solely to changes in ΔG‡, as often done,Citation33 is not generally valid.Citation34 Kramers' theory has been shown to describe protein folding well both in simulationsCitation35,36 and experiments.Citation37,38
Although applying Kramers' theory to ensemble measurements of protein folding can be challenging, Citation39 single-molecule measurements provide fertile ground. This is especially true for single-molecule force spectroscopy (SMFS), in which the extension of a protein is measured as its structure changes in response to a denaturing force applied to its ends.Citation40 SMFS has been used extensively to study the folding energy landscapes of both proteins and nucleic acids.Citation41 Because SMFS data capture the statistical mechanics of the structural fluctuations, they can be used to measure energy landscapes in greater detail than possible at the ensemble level. Multiple methods can be used to quantify the landscape, from reconstructions of the full energy profileCitation42-46 to descriptions of the most critical features like barrier heights and positionsCitation47,48 and diffusion coefficients.Citation38,44,49,50
OBSERVING PrP AT THE SINGLE-MOLECULE LEVEL: FOLDING AND MISFOLDING ENERGY LANDSCAPES
The dynamics of PrP relevant to misfolding and aggregation have been studied extensively at the ensemble level by experimentCitation10,28,51-53 and at the molecular scale by computation.Citation17-19,54-56 Single-molecule studies have been less common,Citation57 however, even though they are ideally suited for probing aggregation because of their ability to detect and characterize sub-populations and transient or rare states.Citation58 We previously used SMFS to show that isolated monomers of hamster PrP(90-231) frequently sampled a variety of misfolded conformations off the pathway for native folding,Citation38,59 but did not form partially-folded on-pathway intermediates postulated to mediate misfolding.Citation60-62 None of the misfolded conformations was thermodynamically stable, consistent with the view that misfolded PrP is stable only within aggregatesCitation7 but contradicting the report of a stable monomeric misfolded form.Citation63 These measurements led to a reconstruction of the full energy profile for native folding of PrP,Citation38 but they could not speak to the landscape for aggregation because they did not study the interactions between monomers that stabilize misfolding.
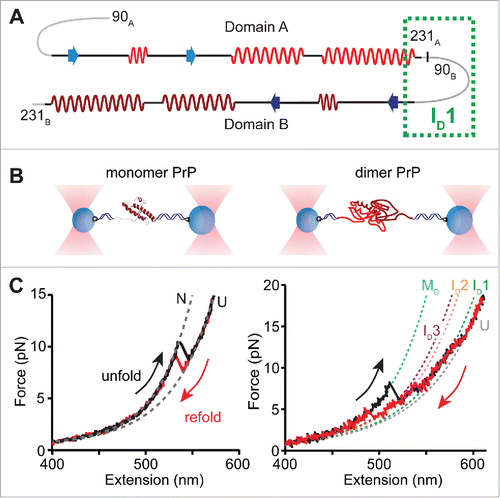
To observe directly the formation of stable misfolded structures, we investigated the folding of individual dimers of PrP, as the smallest form of oligomer, using SMFS.Citation64 Monomers of hamster PrP(90-231) were covalently connected in tandem to generate dimers (). Covalent linking greatly increases the effective local concentration to promote aggregation, providing a platform to focus on early events in misfolding and aggregation.Citation15,65 PrP dimers were held in dual-beam optical tweezers (), and the force ramped up and down to unfold and refold the protein, generating force-extension curves. Abrupt changes in the force and extension creating ‘rips’ in the force-extension curves reflected structural transitions in the protein and were characterized by the force at which they occurred and the change in contour length (ΔLc) observed. In contrast to the 2-state behavior of monomers, dimers passed through at least 3 intermediates, as determined by the minimum number of transitions needed to fit the dimer curves (). Furthermore, ΔLc upon unfolding the dimer was more than twice that for monomers, indicating a structure involving ∼240 amino acids (compared to the 104 in the monomer structureCitation66). The lengths, forces, and patterns of intermediate states implied that neither domain within the tandem dimer formed either the native fold or any of the monomeric misfolded structures seen previously.Citation59 Instead, a new set of misfolded structures not seen in monomers was observed, stabilized by interdomain contacts, i.e. the dimer formed a non-native aggregate.
FIGURE 2. Summary of single-molecule force spectroscopy experiments. (A) Design of the PrP tandem dimer, indicating the native structural elements and the proposed region for ID1 formation. (B) In the experimental set-up of optical tweezers, PrP molecules are tethered between 2 polystyrene beads via DNA handles, and the beads are trapped using high-intensity laser beams. Moving the beads apart and back together again ramps the force on the protein up and down to cause unfolding and refolding. (C) Examples of pulling curves showing unfolding and refolding of a PrP monomer (left) and a PrP dimer (right), where each transition gives rise to a ‘rip’ in the force-extension curve. Dashed lines indicate fits using an analytical model that describes stretching of the unstructured parts of the construct (handles and unfolded protein), used to determine the size of the structure that unfolded/refolded and the number of intermediates observed. Neither domain of the dimer folded natively; instead the dimer misfolded via several intermediates to an aggregated state (cartoon in panel B). Adapted from Ref. 64.
These measurements allowed the energy landscape for PrP aggregation to be measured. By combining fits to Kramers' theory of the rates implied by the observed unfolding forces to obtain barrier heights and positionsCitation48 with complementary measurements of the free-energy changes between states, the energy profile for this multi-state process was reconstructed (). Here the coordinate representing the degree of folding is the length change (ΔLc) between each state, which is related to the number of residues that are folded/unfolded. Comparison of the energy landscapes for dimer misfoldingCitation64 and native foldingCitation38 reveals striking differences (). Dimer misfolding involves several intermediates, with the last one (ID3) being close in energy to the fully misfolded state (MD). Conversion between ID3 and MD is kinetically restricted, with a rate of only 0.5 s−1. In this sense, the measured dimer misfolding landscape matches the cartoon notion of aggregation landscapes (), in that it is more rugged (contains more intermediates) than the native landscape and includes a kinetic trap (ID3). However, contrary to the notion of aggregation involving a heterogeneous mix of metastable states and competing pathways, only one pathway was ever observed during dimer misfolding.
FIGURE 3. Comparison of the experimentally measured energy landscapes for native folding, misfolding and aggregation of PrP. Native folding is 2-state with no observed intermediates between the native and unfolded states. In the context of a tandem dimer, the near-barrierless access to the misfolded ID1 leads the dimer down the misfolding pathway. Several misfolded intermediates are observed, leading to the final misfolded/aggregated state MD. Inset indicates the additional 3kBT of roughness over the misfolding transition barriers that slows diffusion along the misfolding pathway 1000 fold compared to the native pathway. Adapted from Refs. 38 and 64.
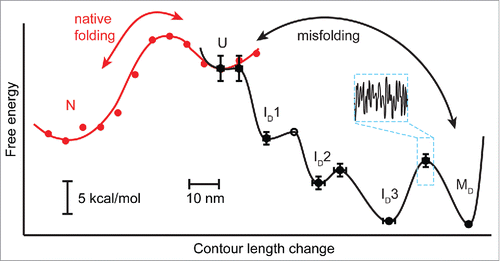
Comparing the native and misfolded landscapes revealed that the misfolded dimer is 2 kcal/mol more stable than 2 native monomers. The dimer thus forms the smallest thermodynamically stable misfolded state of PrP, given that misfolded forms of monomeric PrP are not stable.Citation38 Interestingly, this differential stability (compared to native PrP) is the same as found from an earlier study of β-oligomers.Citation28 Although it is very difficult to deduce secondary structure from SMFS data, CD spectroscopy of the tandem dimers showed that they indeed formed extensive β-sheet structure, raising suggestive parallels to the earlier work.
DIFFUSION AND ROUGHNESS IN THE LANDSCAPE
The homogeneous dimer misfolding pathway appears on the surface quite similar to a generic, well-funnelled, multistate pathway leading to a native structure, but the folding kinetics hint at something different. The folding rate is 5-fold slower for crossing the first barrier in the misfolding landscape than for crossing the native barrier, even though the initial misfolding barrier is marginal (). One may wonder, why does misfolding occur so slowly given that there is essentially no kinetic barrier? This effect does not reflect the need first to unfold from a native conformation before misfolding, since the initial misfolding transition starts from the unfolded state. Rather, it reflects the often-underappreciated fact that the dynamics depend not only on the barrier height, but also on the intrachain diffusion coefficient, D (Eqn 2). Whereas ΔG‡ influences rates by determining the time required for a thermal fluctuation of sufficient magnitude for barrier crossing to occur, D characterizes the microscopic dynamics of the protein and thus sets the timescale for how quickly the protein moves along the landscape. D is thus a crucial parameter for characterizing folding, misfolding and aggregation mechanisms. Previous studies have measured D for peptides and disordered or denatured proteins, typically finding D ∼107–108 nm2/s,Citation67-69 but it has never been possible to compare D for native folding and misfolding directly and thus probe the microscopic differences in the dynamics.
The force-dependent kinetics in the misfolding were used to determine D from Kramers' theory, using the reconstructed landscape (). Strikingly, D (103 nm2/s) was 1000-fold slower than for native folding, implying that misfolding transitions occur over a much longer timescale. This hypothesis was tested by holding the dimer at a force where it fluctuated in equilibrium between the unfolded state (U) and the first misfolded intermediate (ID1), allowing the motions across the barrier to be observed directly. The time required for each passage over the barrier, the transit time, was indeed found to be on average ∼300 times slower than for native folding of PrP, consistent with a much-reduced D for misfolding.
One way to understand the reduction in D during misfolding is in terms of additional roughness in the energy landscape surface, visualised as ‘micro-barriers’ layered over the large-scale barriers separating the well-defined states.Citation70 The micro-barriers then create short-lived local traps that slow down the motion over the landscape, reducing the effective diffusion coefficient observed over larger length scales. Such roughness can be attributed to ‘internal friction’ in the protein,Citation71 consisting of processes like non-native contact formation and dynamics orthogonal to the reaction path that slow down the progress to the folded state.Citation72,73 Enhanced internal friction was previously identified as the cause of 3000-fold differences in folding rates between homologous spectrin proteins.Citation34 Analogously, enhanced internal friction during misfolding likely explains the reduction in D observed here. This result also indicates that PrP dimer misfolding involves a much greater level of frustration—that is, competition among isoenergetic non-native contacts—and in this sense also matches the cartoon vision of aggregation landscapes.
STRUCTURE OF THE MISFOLDED DIMER
SMFS yields a wealth of information about the steps during misfolding, but it does not provide high-resolution structures. Nevertheless, it does provide constraints for building and testing structural models. For example, the key intermediate that initiated misfolding, ID1, was not observed in monomeric PrP,Citation59 indicating that it must involve interactions between residues in both domains of the dimer. From the length change upon unfolding, we estimated that it consists of ∼50 amino acids. It thus almost certainly encompasses the region spanning the link between the 2 domains, i.e., the C-terminal residues of the first domain and the N-terminal residues of the second domain (). Residues 109-122 of PrP are predicted to have a strong propensity to form α-helical structure,Citation74 and a structurally-ambivalent “chameleon sequence” capable of forming different secondary structures depending on the context provided by neighboring sequences was identified in residues 114-125,Citation75 suggesting that this region could indeed form the nucleus for a stable structure in the context of interactions with the C-terminal residues of a neighboring domain, despite the fact that it is unstructured in monomeric PrP. Molecular dynamics simulations have also shown that N-terminal residues 90-100 and 110-127 transiently sample β-strand and α-strand conformations in the monomer,Citation54 supporting the notion that this region may initiate misfolding. The unusual α-strand structure has been implicated as being amyloidogenic,Citation76 and peptides spanning PrP residues 106-135 were neurotoxic in cells.Citation77
Various models for misfolded PrP are available for comparison with our data on the PrP dimer. The crystal structure of human PrP(90-231)Citation78 shows a domain-swapped dimer in which each monomeric domain is structured as in monomeric PrPC but helix 3 swaps positions in the 2 domains. This domain-swapped structure could, in principle, form in the tandem dimer, but it is incompatible with the force spectroscopy results: the stable intermediate ID1 is formed in part from the residues in the N-terminus that remain disordered in the crystal structure. A dimeric version of a structural model proposed for PrPSc based on simulationsCitation3 would yield ΔLc upon unfolding of ∼90 nm, somewhat larger than our observed value (81 ± 1nm), whereas a parallel β-sheet model of PrP amyloidCitation79 would imply ΔLc ∼70 nm for the case of a dimer, somewhat smaller than observed. In contrast, the observed ΔLc is much lower than what would be predicted by models in which each monomeric domain is structured from residues 90 to 230, ΔLc ∼100 nm.Citation5,80 The high stability of our dimers also contrasts with the suggestion of a partially-denatured dimeric amyloid precursor existing in an equilibrium with monomeric PrP.Citation81
Investigating the secondary structure content of the misfolded dimer, CD spectroscopy revealed a β-rich structure (11% α-helical, 35% β-sheet) at both pH 4 and pH 7. The low helical content is consistent with work showing that the helical C terminus of PrPC likely converts fully to β-strands in PrPSc,Citation5 in contrast to earlier models positing the retention of significant C-terminal helix content.Citation3,4 This interpretation is also consistent with the results of previous SM fluorescence studies of PrP aggregation, which found evidence for the rapid formation of β-rich dimers as the first step in aggregation.Citation82
RELEVANCE OF THE MISFOLDED DIMER TO PRION DISEASE
Dimerization has long been suspected to play an important role in pathogenic conversion of PrPC,Citation83 although larger oligomers seem to be more infectious.Citation84 Recombinant PrP forms dimers both at low pHCitation85 and upon dilution from 0.2 to 0.05% SDS,Citation86 while brain-derived PrP dimerizes in vitro.Citation87 A model for conversion of PrPC to amyloid based on dimeric domain-swapping has been proposed,Citation88 inspired by the domain-swapped dimer found in the crystal structure of human PrP.Citation78 Most interestingly, synthetic PrP dimers were toxic to neurons both in cultureCitation89,90 and in mouse models of prion disease,Citation90 and antibodies raised against tandem dimers of PrP showed anti-prion activity in vivo.Citation91 More recently, it was found that recombinant PrP could be converted into a toxic dimer using PMCA.Citation92
Despite the evidence for the relevance of dimeric states, it is very difficult to ascertain whether any of the species observed at the single-molecule level are in fact related to pathogenesis in vivo. Nevertheless, although any connection to disease is only speculative, our results show that PrP seems to be uniquely pre-disposed to conversion into misfolded structures through intermolecular interactions. Indeed, even the smallest oligomer, a dimer, can rapidly and reliably convert to an apparently β-rich form that is more stable than PrPC. This misfolded structure may well act as a first step along the aggregation pathway, as reflected in the fact that the dimer aggregates much more rapidly than monomeric PrPC.
FUTURE STUDIES
By providing a platform for making direct comparisons between native folding and aggregation at the single-molecule level, studies like this oneCitation64 represent an exciting new approach for understanding the microscopic mechanisms of structural conversion in disease-related proteins. The aggregation of PrP into the misfolded dimeric state offers a controlled environment for testing the effects of aggregation-inhibiting ligands with known anti-prion activity,Citation93 to investigate in greater detail their mechanism of action and thereby gain insight into how to design more effective anti-prion agents, or to study the action of molecular chaperones in the context of misfolding diseases.Citation94 Of course to date only dimers linked between the C and N termini, which may limit the accessible conformations, have been studied so far. Studying other link topologies (C-C, N-N) in dimers, to relax the topological constraints, and exploring larger oligomers, to see how aggregates change with size,Citation95 should allow the quantitative picture of the aggregation landscape to be extended via comparisons of well-defined systems. Combining experiments with computational simulations may also provide insight into the structures of the misfolded states.Citation96
As a last point, we note that during folding measurements, proteins spend most of their time in the low-energy wells of the landscape, and only a very brief time crossing the barriers between these wells. Yet critical details of folding, misfolding and aggregation occur during these brief transitions. Information about the barrier crossing events themselves, known as transition paths, is a key target of current efforts, because they contain all the information about the transition states dominating the dynamics, but transition paths remain very difficult to study directly.Citation97 Because of the very slow diffusion during dimer misfolding, we were able to observe transition paths directly for the first time, in any molecule.Citation64 Future transition-path studies hold great promise for elucidating the mechanistic details of misfolding, both directly from experiment and by comparing measured transition path properties to atomistic simulations.Citation73
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
Funding
This work was supported by the Alberta Prion Research Institute, Alberta Innovates Health Solutions, and Alberta Innovates Technology Futures.
REFERENCES
- Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 2006; 75:333-66; PMID:16756495; http://dx.doi.org/10.1146/annurev.biochem.75.101304.123901
- Colby DW, Prusiner SB. Prions. Cold Spring Harb Perspect Biol 2011; 3:a006833; PMID:21421910; http://dx.doi.org/10.1101/cshperspect.a006833
- DeMarco ML, Daggett V. From conversion to aggregation: Protofibril formation of the prion protein. Proc Natl Acad Sci U S A 2004; 101:2293-8; PMID:14983003; http://dx.doi.org/10.1073/pnas.0307178101
- Govaerts C, Wille H, Prusiner SB, Cohen FE. Evidence for assembly of prions with left-handed ß-helices into trimers. Proc Natl Acad Sci U S A 2004; 101:8342-7; PMID:15155909; http://dx.doi.org/10.1073/pnas.0402254101
- Smirnovas V, Baron GS, Offerdahl DK, Raymond GJ, Caughey B, Surewicz WK. Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat Struct Mol Biol 2011; 18:504-6; PMID:21441913; http://dx.doi.org/10.1038/nsmb.2035
- Diaz-Espinoza R, Soto C. High-resolution structure of infectious prion protein: The final frontier. Nat Struct Mol Biol 2012; 19:370-7; PMID:22472622; http://dx.doi.org/10.1038/nsmb.2266
- Cobb NJ, Surewicz WK. Prion diseases and their biochemical mechanisms. Biochemistry 2009; 48:2574-85; PMID:19239250; http://dx.doi.org/10.1021/bi900108v
- Ferreiro DU, Komives EA, Wolynes PG. Frustration in biomolecules. Q Rev Biophys 2014; 47:285-363; PMID:25225856; http://dx.doi.org/10.1017/S0033583514000092
- Jahn TR, Radford SE. Folding vs. aggregation: Polypeptide conformations on competing pathways. Arch Biochem Biophys 2008; 469:100-17; PMID:17588526; http://dx.doi.org/10.1016/j.abb.2007.05.015
- Larda ST, Simonetti K, Al-Abdul-Wahid MS, Sharpe S, Prosser RS. Dynamic equilibria between monomeric and oligomeric misfolded states of the mammalian prion protein measured by 19F NMR. J Am Chem Soc 2013; 135:10533-41; PMID:23781904; http://dx.doi.org/10.1021/ja404584s
- Eichner T, Kalverda AP, Thompson GS, Homans SW, Radford SE. Conformational conversion during amyloid formation at atomic resolution. Mol Cell 2011; 41:161-72; PMID:21255727; http://dx.doi.org/10.1016/j.molcel.2010.11.028
- Neudecker P, Robustelli P, Cavalli A, Walsh P, Lundström P, Zarrine-Afsar A, Sharpe S, Vendruscolo M, Kay LE. Structure of an intermediate state in protein folding and aggregation. Science 2012; 336:362-6; PMID:22517863; http://dx.doi.org/10.1126/science.1214203
- Cremades N, Cohen SA, Deas E, Abramov A, Chen A, Orte A, Sandal M, Clarke R, Dunne P, Aprile F, et al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 2012; 149:1048-59; PMID:22632969; http://dx.doi.org/10.1016/j.cell.2012.03.037
- Zheng W, Schafer NP, Wolynes PG. Frustration in the energy landscapes of multidomain protein misfolding. Proc Natl Acad Sci U S A 2013; 110:1680-5; PMID:23319605; http://dx.doi.org/10.1073/pnas.1222130110
- Zheng W, Schafer NP, Wolynes PG. Free energy landscapes for initiation and branching of protein aggregation. Proc Natl Acad Sci U S A 2013; 110:20515-2020; PMID:24284165; http://dx.doi.org/10.1073/pnas.1320483110
- Baftizadeh F, Biarnes X, Pietrucci F, Affinito F, Laio A. Multidimensional view of amyloid fibril nucleation in atomistic detail. J Am Chem Soc 2012; 134:3886-94; PMID:22276669; http://dx.doi.org/10.1021/ja210826a
- Benetti F, Biarnés X, Attanasio F, Giachin G, Rizzarelli E, Legname G. Structural determinants in prion protein folding and stability. J Mol Biol 2014; 426:3796-810; PMID:25280897; http://dx.doi.org/10.1016/j.jmb.2014.09.017
- Chen J, Thirumalai D. Helices 2 and 3 are the initiation sites in the PrP(C) → PrP(SC) transition. Biochemistry 2013; 52:310-9; PMID:23256626; http://dx.doi.org/10.1021/bi3005472
- Barducci A, Chelli R, Procacci P, Schettino V, Gervasio FL, Parrinello M. Metadynamics simulation of prion protein: Beta-structure stability and the early stages of misfolding. J Am Chem Soc 2006; 128:2705-10; PMID:16492057; http://dx.doi.org/10.1021/ja057076l
- De Simone A, Dhulesia A, Soldi G, Vendruscolo M, Hsu ST, Chiti F, Dobson CM. Experimental free energy surfaces reveal the mechanisms of maintenance of protein solubility. Proc Natl Acad Sci U S A 2011; 108:21057-62; PMID:22160682; http://dx.doi.org/10.1073/pnas.1112197108
- Baldwin AJ, Knowles TP, Tartaglia GG, Fitzpatrick AW, Devlin GL, Shammas SL, Waudby CA, Mossuto MF, Meehan S, Gras SL, et al. Metastability of native proteins and the phenomenon of amyloid formation. J Am Chem Soc 2011; 133:14160-3; PMID:21650202; http://dx.doi.org/10.1021/ja2017703
- Buell AK, Dhulesia A, White DA, Knowles TP, Dobson CM, Welland ME. Detailed analysis of the energy barriers for amyloid fibril growth. Angew Chem Int Ed 2012; 51:5247-51; http://dx.doi.org/10.1002/anie.201108040
- Wetzel R. Kinetics and thermodynamics of amyloid fibril assembly. Acc Chem Res 2006; 39:671-9; PMID:16981684; http://dx.doi.org/10.1021/ar050069h
- Ricchiuto P, Brukhno AV, Auer S. Protein aggregation: Kinetics versus thermodynamics. J Phys Chem B 2012; 116:5384-90; PMID:22512540; http://dx.doi.org/10.1021/jp302797c
- Pellarin R, Schuetz P, Guarnera E, Caflisch A. Amyloid fibril polymorphism is under kinetic control. J Am Chem Soc 2010; 132:14960-70; PMID:20923147; http://dx.doi.org/10.1021/ja106044u
- Arosio P, Vendruscolo M, Dobson CM, Knowles TP. Chemical kinetics for drug discovery to combat protein aggregation diseases. Trends Pharmacol Sci 2014; 35:127-35; PMID:24560688; http://dx.doi.org/10.1016/j.tips.2013.12.005
- Johnson SM, Wiseman RL, Sekijima Y, Green NS, Adamski-Werner S, Kelly JW. Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: A focus on the transthyretin amyloidoses. Acc Chem Res 2005; 38:911-21; PMID:16359163; http://dx.doi.org/10.1021/ar020073i
- Baskakov IV, Legname G, Prusiner SB, Cohen FE. Folding of prion protein to its native α-helical conformation is under kinetic control. J Biol Chem 2001; 276:19687-90; PMID:11306559; http://dx.doi.org/10.1074/jbc.C100180200
- Schramm VL. Transition states, analogues and drug development. ACS Chem Biol 2013; 8:71-81; PMID:23259601; http://dx.doi.org/10.1021/cb300631k
- Ayrolles-Torro A, Imberdis T, Torrent J, Toupet K, Baskakov IV, Poncet-Montange G, Grégoire C, Roquet-Baneres F, Lehmann S, Rognan D, et al. Oligomeric-induced activity by thienyl pyrimidine compounds traps prion infectivity. J Neurosci 2011; 31:14882-92; PMID:22016521; http://dx.doi.org/10.1523/JNEUROSCI.0547-11.2011
- Oliveberg M, Wolynes PG. The experimental survey of protein-folding energy landscapes. Q Rev Biophys 2005; 38:245-88; PMID:16780604; http://dx.doi.org/10.1017/S0033583506004185
- Hänggi P, Talkner P, Borkovec M. Reaction-rate theory: Fifty years after kramers. Rev Mod Phys 1990; 62:251-341; http://dx.doi.org/10.1103/RevModPhys.62.251
- Fersht A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. New York: Freeman, 1998
- Wensley BG, Batey S, Bone FAC, Chan ZM, Tumelty NR, Steward A, Kwa LG, Borgia A, Clarke J. Experimental evidence for a frustrated energy landscape in a three-helix-bundle protein family. Nature 2010; 463:685-8; PMID:20130652; http://dx.doi.org/10.1038/nature08743
- Socci ND, Onuchic JN, Wolynes PG. Diffusive dynamics of the reaction coordinate for protein folding funnels. J Chem Phys 1996; 104:5860-8; http://dx.doi.org/10.1063/1.471317
- Klimov DK, Thirumalai D. Viscosity dependence of the folding rates of proteins. Phys Rev Lett 1997; 79:317-20; http://dx.doi.org/10.1103/PhysRevLett.79.317
- Cellmer T, Henry ER, Hofrichter J, Eaton WA. Measuring internal friction of an ultrafast-folding protein. Proc Natl Acad Sci U S A 2008; 105:18320-5; PMID:19020085; http://dx.doi.org/10.1073/pnas.0806154105
- Yu H, Gupta AN, Liu X, Neupane K, Brigley AM, Sosova I, Woodside MT. Energy landscape analysis of native folding of the prion protein yields the diffusion constant, transition path time, and rates. Proc Natl Acad Sci U S A 2012; 109:14452-7; PMID:22908253; http://dx.doi.org/10.1073/pnas.1206190109
- Plotkin SS. Determination of barrier heights and prefactors from protein folding rate data. Biophys J 2005; 88:3762-9; PMID:15764665; http://dx.doi.org/10.1529/biophysj.104.052548
- Ritchie DB, Woodside MT. Probing the structural dynamics of proteins and nucleic acids with optical tweezers. Curr Opin Struct Biol 2015; 34:43-51; PMID:26189090; http://dx.doi.org/10.1016/j.sbi.2015.06.006
- Woodside MT, Block SM. Reconstructing the folding energy landscape by single-molecule force spectroscopy. Annu Rev Biophys 2014; 43:19-39; PMID:24895850; http://dx.doi.org/10.1146/annurev-biophys-051013-022754
- Woodside MT, Anthony PC, Behnke-Parks WM, Larizadeh K, Herschlag D, Block SM. Direct measurement of the full, sequence-dependent folding landscape of a nucleic acid. Science 2006; 314:1001-4; PMID:17095702; http://dx.doi.org/10.1126/science.1133601
- Gupta AN, Vincent A, Neupane K, Yu H, Woodside MT. Experimental validation of free energy landscape reconstruction from non-equilibrium single-molecule force spectroscopy experiments. Nature Phys 2011; 7:631-4; http://dx.doi.org/10.1038/nphys2022
- Lannon H, Haghpanah JS, Montclare JK, Vanden-Eijnden E, Brujic J. Force-clamp experiments reveal the free-energy profile and diffusion coefficient of the collapse of protein molecules. Phys Rev Lett 2013; 110:128301; PMID:25166851; http://dx.doi.org/10.1103/PhysRevLett.110.128301
- Engel MC, Ritchie DB, Foster DA, Beach KS, Woodside MT. Reconstructing folding energy landscape profiles from nonequilibrium pulling curves with an inverse weierstrass integral transform. Phys Rev Lett 2014; 113:238104; PMID:25526163; http://dx.doi.org/10.1103/PhysRevLett.113.238104
- Manuel AP, Lambert J, Woodside MT. Reconstructing folding energy landscapes from splitting probability analysis of single-molecule trajectories. Proc Natl Acad Sci U S A 2015; 112:7183-8; PMID:26039984; http://dx.doi.org/10.1073/pnas.1419490112
- Dudko OK, Hummer G, Szabo A. Intrinsic rates and activation free energies from single-molecule pulling experiments. Phys Rev Lett 2006; 96:108101; PMID:16605793; http://dx.doi.org/10.1103/PhysRevLett.96.108101
- Dudko OK, Hummer G, Szabo A. Theory, analysis, and interpretation of single-molecule force spectroscopy experiments. Proc Natl Acad Sci U S A 2008; 105:15755-60; PMID:18852468; http://dx.doi.org/10.1073/pnas.0806085105
- Neupane K, Ritchie DB, Yu H, Foster DAN, Wang F, Woodside MT. Transition path times for nucleic acid folding determined from energy-landscape analysis of single-molecule trajectories. Phys Rev Lett 2012; 109:068102; PMID:23006308; http://dx.doi.org/10.1103/PhysRevLett.109.068102
- Woodside MT, Lambert J, Beach KSD. Determining intra-chain diffusion coefficients for biopolymer dynamics from single-molecule force spectroscopy measurements. Biophys J 2014; 107:1647-53; PMID:25296317; http://dx.doi.org/10.1016/j.bpj.2014.08.007
- Wildegger G, Liemann S, Glockshuber R. Extremely rapid folding of the C-terminal domain of the prion protein without kinetic intermediates. Nat Struct Biol 1999; 6:550-3; PMID:10360358; http://dx.doi.org/10.1038/9323
- Hart T, Hosszu LL, Trevitt CR, Jackson GS, Waltho JP, Collinge J, Clarke AR. Folding kinetics of the human prion protein probed by temperature jump. Proc Natl Acad Sci U S A 2009; 106:5651-6; PMID:19321423; http://dx.doi.org/10.1073/pnas.0811457106
- Khan MQ, Sweeting B, Mulligan VK, Arslan PE, Cashman NR, Pai EF, Chakrabartty A. Prion disease susceptibility is affected by β-structure folding propensity and local side-chain interactions in PrP. Proc Natl Acad Sci U S A 2010; 107:19808-13; PMID:21041683; http://dx.doi.org/10.1073/pnas.1005267107
- Chen W, van der Kamp MW, Daggett V. Structural and dynamic properties of the human prion protein. Biophys J 2014; 106:1152-63; PMID:24606939; http://dx.doi.org/10.1016/j.bpj.2013.12.053
- van der Kamp MW, Daggett V. Influence of pH on the human prion protein: Insights into the early steps of misfolding. Biophys J 2010; 99:2289-98; PMID:20923664; http://dx.doi.org/10.1016/j.bpj.2010.07.063
- Guest WC, Cashman NR, Plotkin SS. Electrostatics in the stability and misfolding of the prion protein: Salt bridges, self energy, and solvation. Biochem Cell Biol 2010; 88:371-81; PMID:20453937; http://dx.doi.org/10.1139/O09-180
- Yu H, Dee DR, Woodside MT. Single-molecule approaches to prion protein misfolding. Prion 2013; 7:140-6; PMID:23357831; http://dx.doi.org/10.4161/pri.23303
- Hoffmann A, Neupane K, Woodside MT. Single-molecule assays for investigating protein misfolding and aggregation. Phys Chem Chem Phys 2013; 15:7934-48; PMID:23612887; http://dx.doi.org/10.1039/c3cp44564j
- Yu H, Liu X, Neupane K, Gupta AN, Brigley AM, Solanki A, Sosova I, Woodside MT. Direct observation of multiple misfolding pathways in a single prion protein molecule. Proc Natl Acad Sci U S A 2012; 109:5283-8; PMID:22421432; http://dx.doi.org/10.1073/pnas.1107736109
- Apetri AC, Maki K, Roder H, Surewicz WK. Early intermediate in human prion protein folding as evidenced by ultrarapid mixing experiments. J Am Chem Soc 2006; 128:11673-8; PMID:16939293; http://dx.doi.org/10.1021/ja063880b
- Cohen FE, Pan KM, Huang Z, Baldwin M, Fletterick RJ, Prusiner SB. Structural clues to prion replication. Science 1994; 264:530-1; PMID:7909169; http://dx.doi.org/10.1126/science.7909169
- Jenkins DC, Sylvester ID, Pinheiro TJ. The elusive intermediate on the folding pathway of the prion protein. FEBS J 2008; 275:1323-35; PMID:18279390; http://dx.doi.org/10.1111/j.1742-4658.2008.06293.x
- Zhou M, Ottenberg G, Sferrazza GF, Lasmezas CI. Highly neurotoxic monomeric α-helical prion protein. Proc Natl Acad Sci U S A 2012; 109:3113-8; PMID:22323583; http://dx.doi.org/10.1073/pnas.1118090109
- Yu H, Dee DR, Liu X, Brigley AM, Sosova I, Woodside MT. Protein misfolding occurs by slow diffusion across multiple barriers in a rough energy landscape. Proc Natl Acad Sci U S A 2015; 112:8308-13; PMID:26109573; http://dx.doi.org/10.1073/pnas.1419197112
- Liu F, Gruebele M. Mapping an aggregation nucleus one protein at a time. J Phys Chem Lett 2010; 1:16-9; http://dx.doi.org/10.1021/jz9000856
- James TL, Liu H, Ulyanov NB, Farr-Jones S, Zhang H, Donne DG, Kaneko K, Groth D, Mehlhorn I, Prusiner SB, et al. Solution structure of a 142-residue recombinant prion protein corresponding to the infectious fragment of the scrapie isoform. Proc Natl Acad Sci U S A 1997; 94:10086-91; PMID:9294167; http://dx.doi.org/10.1073/pnas.94.19.10086
- Hagen SJ, Hofrichter J, Szabo A, Eaton WA. Diffusion-limited contact formation in unfolded cytochrome c: Estimating the maximum rate of protein folding. Proc Natl Acad Sci U S A 1996; 93:11615-7; PMID:8876184; http://dx.doi.org/10.1073/pnas.93.21.11615
- Nettels D, Gopich IV, Hoffmann A, Schuler B. Ultrafast dynamics of protein collapse from single-molecule photon statistics. Proc Natl Acad Sci U S A 2007; 104:2655-60; PMID:17301233; http://dx.doi.org/10.1073/pnas.0611093104
- Ahmad B, Chen Y, Lapidus LJ. Aggregation of a-synuclein is kinetically controlled by intramolecular diffusion. Proc Natl Acad Sci U S A 2012; 109:2336-41; PMID:22308332; http://dx.doi.org/10.1073/pnas.1109526109
- Bryngelson JD, Wolynes PG. Intermediates and barrier crossing in a random energy-model (with applications to protein folding). J Phys Chem 1989; 93:6902-15; http://dx.doi.org/10.1021/j100356a007
- Ansari A, Jones C, Henry E, Hofrichter J, Eaton W. The role of solvent viscosity in the dynamics of protein conformational changes. Science 1992; 256:1796-8; PMID:1615323; http://dx.doi.org/10.1126/science.1615323
- Borgia A, Wensley BG, Soranno A, Nettels D, Borgia MB, Hoffmann A, Pfeil SH, Lipman EA, Clarke J, Schuler B. Localizing internal friction along the reaction coordinate of protein folding by combining ensemble and single-molecule fluorescence spectroscopy. Nat Commun 2012; 3:1195; PMID:23149740; http://dx.doi.org/10.1038/ncomms2204
- Chung HS, Piana-Agostinetti S, Shaw DE, Eaton WA. Structural origin of slow diffusion in protein folding. Science 2015; 349:1504-10; PMID:26404828; http://dx.doi.org/10.1126/science.aab1369
- Zhang H, Kaneko K, Nguyen JT, Livshits TL, Baldwin MA, Cohen FE, James TL, Prusiner SB. Conformational transformations in peptides containing two putative α-helices of the prion protein. J Mol Biol 1995; 250:514-26; PMID:7542350; http://dx.doi.org/10.1006/jmbi.1995.0395
- Kuznetsov IB, Rackovsky S. Comparative computational analysis of prion proteins reveals two fragments with unusual structural properties and a pattern of increase in hydrophobicity associated with disease-promoting mutations. Protein Sci 2004; 13:3230-44; PMID:15557265; http://dx.doi.org/10.1110/ps.04833404
- Daggett V. Alpha-sheet: The toxic conformer in amyloid diseases? Acc Chem Res 2006; 39:594-602; PMID:16981675; http://dx.doi.org/10.1021/ar0500719
- Haik S, Peyrin JM, Lins L, Rosseneu MY, Brasseur R, Langeveld JP, Tagliavini F, Deslys JP, Lasmezas C, Dormont D. Neurotoxicity of the putative transmembrane domain of the prion protein. Neurobiol Dis 2000; 7:644-56; PMID:11114262; http://dx.doi.org/10.1006/nbdi.2000.0316
- Knaus KJ, Morillas M, Swietnicki W, Malone M, Surewicz WK, Yee VC. Crystal structure of the human prion protein reveals a mechanism for oligomerization. Nat Struct Biol 2001; 8:770-4; PMID:11524679; http://dx.doi.org/10.1038/nsb0901-770
- Cobb NJ, Sönnichsen FD, Mchaourab H, Surewicz WK. Molecular architecture of human prion protein amyloid: A parallel, in-register β-structure. Proc Natl Acad Sci 2007; 104:18946-51; http://dx.doi.org/10.1073/pnas.0706522104
- Yang S, Levine H, Onuchic JN, Cox DL. Structure of infectious prions: Stabilization by domain swapping. FASEB J 2005; 19:1778-82; PMID:16260647; http://dx.doi.org/10.1096/fj.05-4067hyp
- Stohr J, Weinmann N, Wille H, Kaimann T, Nagel-Steger L, Birkmann E, Panza G, Prusiner SB, Eigen M, Riesner D. Mechanisms of prion protein assembly into amyloid. Proc Natl Acad Sci U S A 2008; 105:2409-14; PMID:18268326; http://dx.doi.org/10.1073/pnas.0712036105
- Post K, Pitschke M, Schäfer O, Wille H, Appel TR, Kirsch D, Mehlhorn I, Serban H, Prusiner SB, Riesner D. Rapid acquisition of β-sheet structure in the prion protein prior to multimer formation. Biol Chem 1998; 379:1307-17; PMID:9865603; http://dx.doi.org/10.1515/bchm.1998.379.11.1307
- Tompa P, Tusnády GE, Friedrich P, Simon I. The role of dimerization in prion replication. Biophys J 2002; 82:1711-8; PMID:11916832; http://dx.doi.org/10.1016/S0006-3495(02)75523-9
- Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, Caughey B. The most infectious prion protein particles. Nature 2005; 437:257-61; PMID:16148934; http://dx.doi.org/10.1038/nature03989
- O'Sullivan DBD, Jones CE, Abdelraheim SR, Thompsett AR, Brazier MW, Toms H, Brown DR, Viles JH. NMR characterization of the pH 4 β-intermediate of the prion protein: The N-terminal half of the protein remains unstructured and retains a high degree of flexibility. Biochem J 2007; 401:533-40; PMID:16958619; http://dx.doi.org/10.1042/BJ20060668
- Kaimann T, Metzger S, Kuhlmann K, Brandt B, Birkmann E, Holtje HD, Riesner D. Molecular model of an α-helical prion protein dimer and its monomeric subunits as derived from chemical cross-linking and molecular modeling calculations. J Mol Biol 2008; 376:582-96; PMID:18158160; http://dx.doi.org/10.1016/j.jmb.2007.11.035
- Meyer RK, Lustig A, Oesch B, Fatzer R, Zurbriggen A, Vandevelde M. A monomer-dimer equilibrium of a cellular prion protein (PrPC) not observed with recombinant PrP. J Biol Chem 2000; 275:38081-7; PMID:10967124; http://dx.doi.org/10.1074/jbc.M007114200
- Lee S, Eisenberg D. Seeded conversion of recombinant prion protein to a disulfide-bonded oligomer by a reduction-oxidation process. Nat Struct Biol 2003; 10:725-30; PMID:12897768; http://dx.doi.org/10.1038/nsb961
- Roostaee A, Côté S, Roucou X. Aggregation and amyloid fibril formation induced by chemical dimerization of recombinant prion protein in physiological-like conditions. J Biol Chem 2009; 284:30907-16; PMID:19710507; http://dx.doi.org/10.1074/jbc.M109.057950
- Simoneau S, Rezaei H, Salès N, Kaiser-Schulz G, Lefebvre-Roque M, Vidal C, Fournier J, Comte J, Wopfner F, Grosclaude J, et al. In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog 2007; 3:e125; PMID:17784787; http://dx.doi.org/10.1371/journal.ppat.0030125
- Gilch S, Wopfner F, Renner-Müller I, Kremmer E, Bauer C, Wolf E, Brem G, Groschup MH, Schätzl HM. Polyclonal anti-PrP auto-antibodies induced with dimeric PrP interfere efficiently with PrPSc propagation in prion-infected cells. J Biol Chem 2003; 278:18524-31; PMID:12637572; http://dx.doi.org/10.1074/jbc.M210723200
- Yang X, Yang L, Zhou X, Khan SH, Wang H, Yin X, Yuan Z, Song Z, Wu W, Zhao D. Using protein misfolding cyclic amplification generates a highly neurotoxic PrP dimer causing neurodegeneration. J Mol Neurosci 2013; 51:655-62; PMID:23771785; http://dx.doi.org/10.1007/s12031-013-0039-z
- Sim VL, Caughey B. Recent advances in prion chemotherapeutics. Infect Disord Drug Targets 2009; 9:81-91; PMID:19200018; http://dx.doi.org/10.2174/1871526510909010081
- Mashaghi A, Kramer G, Lamb DC, Mayer MP, Tans SJ. Chaperone action at the single-molecule level. Chem Rev 2014; 114:660-76; PMID:24001118; http://dx.doi.org/10.1021/cr400326k
- Neupane K, Solanki A, Sosova I, Belov M, Woodside MT. Diverse metastable structures formed by small oligomers of α-synuclein probed by force spectroscopy. PLoS ONE 2014; 9:e86495; PMID:24475132; http://dx.doi.org/10.1371/journal.pone.0086495
- Zhang Y, Lyubchenko YL. The structure of misfolded amyloidogenic dimers: computational analysis of force spectroscopy data. Biophys J 2014; 107:2903-10; PMID:25517155; http://dx.doi.org/10.1016/j.bpj.2014.10.053
- Chung HS, McHale K, Louis JM, Eaton WA. Single-molecule fluorescence experiments determine protein folding transition path times. Science 2012; 335:981-4; PMID:22363011; http://dx.doi.org/10.1126/science.1215768