
ABSTRACT
Inherited prion diseases are characterized by mutations in the PRNP gene, which account for 5–15% of human prion diseases. Here we reported 3 Chinese genetic Creutzfeldt-Jacob disease cases (gCJD) with a rare mutation in PRNP leading to an exchange of amino acid from glutamic acid (E) to alanine (A) at codon 196 (E196A). All three patients were Han Chinese without any sibship among them. They showed various unspecific symptoms at onset and displayed typical clinical manifestations of sporadic CJD with progress of disease. The same time, 2 cases showed psychotic symptoms during the clinical courses. 14-3-3 proteins were positive in cerebrospinal fluid (CSF) and special abnormality were detected in MRI of all the cases. The polymorphism of codon 129 was methionin homozygote and that of codon 219 was glutamate homozygote in all 3 patients. The disease durations of the 3 cases varied from 10 to 22 months and no disease associated family history was figured out in all the cases.
INTRODUCTION
Human prion diseases consist of Creutzfeldt–Jakob disease (CJD), Kuru, Gerstmann–Sträussler–Scheinker syndrome (GSS) and fatal familial insomnia (FFI).Citation1 About 85% of all CJD cases are sporadic, 10–15% are inherited and less than 1% is acquired.Citation2 Normal cellular prion protein (PrPC) is encoded by PRNP gene that locates at chromosome 20 of humans.Citation3 Till now, more than 55 mutations in the PRNP coding sequence have been described associating with human genetic prion diseases, whose clinical manifestations and neuropathological abnormalities may vary largely depending on the various genotypes.Citation4 More than 60 cases of various genetic prion diseases have been identified by Chinese National CJD Surveillance Network since 2006, FFI, T188K gCJD and E200K gCJD were the most leading genetic prion diseases.Citation5 Here we reported 3 Chinese gCJD cases with a substitution from glutamic acid to alanine at codon 196 (E196A) by PRNP sequencing and all patients were methionine homozygous at codon 129 and glutamic acid homozygous at codon 219.
METHODS
Collection of Clinical Data and Case Identification
The surveillance for human prion disease was conducted by Chinese National Surveillance Network for CJD (CNSNC) under the leadership of China Center for Disease Control and Prevention (CCDC) as described previously.Citation6 The clinical data of the suspected CJD patients were collected by the neurologists in the sentinel hospitals and the epidemiological data were collected by the staff from local provincial CDCs. The final diagnosis was given by the expert team consisting of neurologists, epidemiologists and laboratory staffs based on the diagnostic criteria for CJD recommended by WHO.Citation7 The follow-up surveys for the patients were performed by the staff of CNSNC by telephone.
Laboratory Tests
Blood samples and cerebrospinal fluid (CSF) were collected by the medical staffs in local hospitals and transferred to our center at the condition of low temperature. CSF was split charging for 50 μl and stored under −80°C until usage. CSF 14-3-3 protein was detected by Western blot described briefly as following. A total of 20 μl CSF sample was separated in 12% SDS-PAGE by addition of 5 times of loading buffer. Following electrophoresis, the fractionated proteins were electronically transferred to nitrocellulose (NC) membranes (Whatman, USA) by the semi-90 dry method in transferring buffer. After blocking, the membrane was incubated with 1:1000 diluted 14-3-3 polyclonal antibody (Santa Cruz Biological, Santa Cruz, USA) at room temperature for 2 h. Membranes were subsequently incubated with goat anti-rabbit HRP-conjugated secondary antibody and reactive signals were visualized using an enhanced chemiluminescence kit (Amersham-Pharmacia Biotech, USA). The genomic DNA of the patients was extracted from the peripheral blood samples. Than the PRNP gene was amplified by an established polymerase chain reaction (PCR) with the specific primers described before (forward primer: 5′-GGC AAA CCT TGG ATG CTG G-3′ and reverse primer: 5′-CCC ACT ATC AGG AAG ATG AGG-3′). After amplification with a fixed PCR protocol, the PRNP gene was analyzed by direct sequencing.Citation8
RESULTS
CJD surveillance network has been conducted in China since 2006. Three E196A gCJD cases were identified with definitely genetic confirmation in 2011, 2012 and 2015, respectively. All three patients were Han Chinese without any sibship among them. Any similar symptoms or other neurological diseases were not identified in the patients' parents or in the members of those 3 families. Brain postmortem or brain biopsy was not performed.
Clinical Information
Case 1, a 76 year-old male, who was a retired engineer, complained intermittent confusion and dystrophy for about 20 d upon admission. The detailed clinical features were described previously.Citation9
Case 2, a 54 year-old women, who was a retired client. Four months ago, she started to complain abnormalities such as persistent intelligence decline, swirl and walking unsteadily. The symptoms worsened gradually after admission. Physical examination revealed that she was unconsciousness without response to any question. Orbital pressure reflection disappeared. The tendon reflex of the pair upper limb was strong positive, while that of the pair lower limb was not releasable. Limb muscle tension declined. She did not have any improvement during hospitalization and discharged several weeks later. She maintained unconsciousness when she was back to home. Her family members reported convulsion in her limb and face frequently. She died 17 months after discharged from hospital. Total clinical course was about 22 months.
Case 3, a 57 year-old women, who was retired medical staff, complained speak thickly and left hand weakness. One month before being admitted to hospital, she appeared difficulty in talking and reading. Her left hand felt weak and was unable to pick up anything. Physical examination at hospitalization showed that she had dysarthria and emotional instability. Her angle of mouth deflected to right and her left nasolabial groove was slightly shallow. Then, she was unable to walk straightly. When she was being tested the Romberg's sign, the patient could not stand stably whenever opening the eyes or closing the eyes. The general condition worsened gradually and she appeared myoclone and psychotic symptoms. She discharged from hospital 2 months later. And now, she was still alive but in the state of mutism at the follow-up survey half year later.
Clinical Examination
Electroencephalogram (EEG) and magnetic resonance imaging (MRI) were performed during hospitalization. Typical periodic sharp wave complexes (PSWCs) was detected in Case 1. Bilateral diffuse waves, occasionally with periodic sharp waves in EEG was observed in Case 2 showed, while that of Case 3 revealed moderate abnormality especially at right hemisphere. MRI of Case 1 showed high signals at the surfaces of bilateral frontal parietal lobes and left occipital lobe in diffusion weighted imaging (DWI). High signals in caudate and putamen were observed in Case 2 and Case 3. DWI revealed ribbon-like signals on the surface of cortex of Case 3.
Laboratory Tests
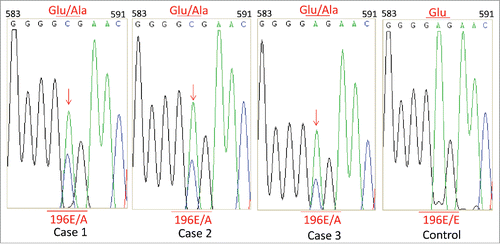
CSF biochemistry assays of 3 patients did not show special abnormality in Case 2 and 3, while high protein amount (2.01 g/L, normal value 150–400 mg/L) in Case 1. Further reviewing of the medical record confirmed this CSF sample was contaminated with blood during lumber puncture. All three cases were positive for CSF 14-3-3 protein in 14-3-3 specific Western blots (), in which the CSF sample of Case 1 was collected again and free of blood. Direct sequencing assays of the PRNP gene PCR products from the 3 patients identified the same missense mutation at codon 196 of (A to C), leading to a substitution from glutamic acid (Glu) to alanine (Ala) (). The polymorphism of codon 129 was methionin homozygote and that of codon 219 was glutamate homozygote in all 3 patients. No additional nucleotide exchanges were found in other regions of the PRNP sequences. As refused by the family members, we did not conduct PRNP sequencing for the lineal consanguinity.
FIGURE 1. Western blot analysis of protein 14-3-3 in CSF samples. Positive Ctrl: 10% goat brain homogenate. Negative Ctrl: a human CSF sample previously confirmed to be 14-3-3 negative. Case sample: the CSF samples of the E196A gCJD cases. M: commercially supplied prestained protein molecular markers whose relative molecular weights are showed on the left.
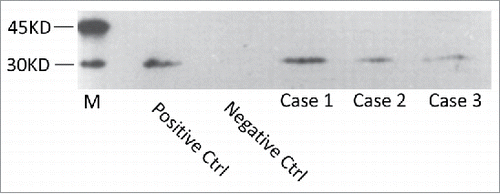
FIGURE 2. Graphic presentation of the sequencing analysis of PRNP. DNA sequence from the 3 patients at codon 196 shows a E to A heterozygous transition at codon 196 in one PRNP allele, leading to and exchange from Glu (E) to Ala (A). The right graph shows a control of DNA sequence of a person containing Glu/Glu homozygote at codon 196 in PRNP alleles.
DISCUSSION
CJD has an incidence of 1 or 2 cases per million populations per year worldwide. Majority of CJD is sporadic form (sCJD) with rapidly progressive dementia, visual or cerebellar problems, myoclonus, pyramidal or extrapyramidal features and akinetic mutism at late stage.Citation10 The features of three E196A gCJD patients are summarized in . Clinically, three E196A gCJD cases displayed the manifestations like sCJD. Myoclonic jerks appeared relatively later and akinetic mutism were noticed at terminal stages. Two cases showed different psychotic symptoms during the clinical courses. CSF 14-3-3 positive and special abnormalities in MRI were noticed in all 3 cases. All three patients were Han Chinese without any special diet habit. Case 3 was retired staff in a hospital, but she was a office client and had no chance to contact the potential prion-associated risk materials and tissue specimen. None of the 3 patients showed disease-associate family history, so that they were suspected as sCJD prior to PRNP sequencing. It emphasizes again the importance of PRNP sequencing in identifying the case of genetic human prion disease without family history.
TABLE 1. Characteristics of 3 Chinese patients with E196A gCJD.
E196A gCJD is rarely reported worldwide. Review the published articles on PubMed identifies only one case (Case 1 in this study) reported by a Chinese group,Citation9 which belongs to China CJD Surveillance System. However, another point-mutation in this codon, E196K, has been described in Caucasian, which is causally linked to human prion disease.Citation11 Majority E196K gCJD cases distributes in Germany,Citation12-14 one in France,Citation15 one in Italy Citation16 and one in China.Citation17 E196K gCJD patients usually present nonspecific symptoms at onset and display the features typical of sCJD during disease progression. All E196K gCJD cases are CSF 14-3-3 positive. Those data propose the similarity in the phenotypes of 2 different point-mutation at the codon 196. However, there are some differences between those 2 mutations. Four German E196K gCJD cases belong to 2 families,Citation12 while none of three E196A gCJD cases has disease-associated family history. The median onset age of E196A gCJD cases are 71 y and disease duration is relatively short (6.5 months).Citation9 Two of E196A cases are in their 50 y of age and disease durations of 3 cases are distinctly longer. Certainly, the exact similarity and difference between E196A and E196K mutations need more information. As we do not have any brain autopsy, the neuropathology of E196A remains unknown.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
Funding
This work was supported by Chinese National Natural Science Foundation Grants (81301429, 81572048), China Mega-Project for Infectious Disease (2011ZX10004-101, 2012ZX10004215) and SKLID Development Grant (2012SKLID102, 2015SKLID503).
REFERENCES
- Imran M, Mahmood S. An overview of human prion diseases. Virol J 2011; 8:559; PMID:22196171; http://dx.doi.org/10.1186/1743-422X-8-559
- Prusiner SB. The prion diseases. Brain Pathol 1998; 8:499-513; PMID:9669700; http://dx.doi.org/10.1111/j.1750-3639.1998.tb00171.x
- Zeng L, Zou W, Wang G. Cellular prion protein (PrP(C)) and its role in stress responses. Int J Clin Exp Med 2015; 8:8042-50; PMID:26221369
- Jeong BH, Kim YS. Genetic studies in human prion diseases. J Korean Med Sci 2014; 29:623-32; PMID:24851016; http://dx.doi.org/10.3346/jkms.2014.29.5.623
- Shi Q, Zhou W, Chen C, Zhang BY, Xiao K, Zhang XC, Shen XJ, Li Q, Deng LQ, Dong JH, et al. The features of genetic prion diseases based on Chinese surveillance program. PLoS One 2015; 10(10):e0139552; PMID:26488179; http://dx.doi.org/10.1371/journal.pone.0139552
- Gao C, Shi Q, Tian C, Chen C, Han J, Zhou W, Zhang BY, Jiang HY, Zhang J, Dong XP. The epidemiological, clinical, and laboratory features of sporadic Creutzfeldt-Jakob disease patients in China: surveillance data from 2006 to 2010. PLoS One 2011; 6(8):e24231; PMID:21904617; http://dx.doi.org/10.1371/journal.pone.0024231
- Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, Heinemann U, Breithaupt M, Varges D, Meissner B, Ladogana A, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain 2009; 132:2659-68; PMID:19773352; http://dx.doi.org/10.1093/brain/awp191
- Shi Q, Shen XJ, Zhou W, Xiao K, Zhang XM, Zhang BY, Dong XP. Rare V180I mutation in PRNP gene of a Chinese patient with Creutzfeldt-Jakob disease. Prion 2014; 8:411-4; PMID:25482600; http://dx.doi.org/10.4161/19336896.2014.967040
- Zhang H, Wang M, Wu L, Zhang H, Jin T, Wu J, Sun L. Novel prion protein gene mutation at codon 196 (E196A) in a septuagenarian with Creutzfeldt-Jakob disease. J Clin Neurosci 2014; 21:175-8; PMID:23787189; http://dx.doi.org/10.1016/j.jocn.2013.03.016
- Kim MO, Geschwind MD. Clinical update of Jakob-Creutzfeldt disease. Curr Opin Neurol 2015; 28:302-10; PMID:25923128; http://dx.doi.org/10.1097/WCO.0000000000000197
- Peoc'h K, Manivet P, Beaudry P, Attane F, Besson G, Hannequin D, Delasnerie-Lauprêtre N, Laplanche JL. Identification of three novel mutations (E196K, V203I, E211Q) in the prion protein gene (PRNP) in inherited prion diseases with Creutzfeldt-Jakob disease phenotype. Hum Mutat 2000; 15:482; PMID:10790216; http://dx.doi.org/10.1002/(SICI)1098-1004(200005)15:5%3c482::AID-HUMU16%3e3.0.CO;2-1
- Krasnianski A, Heinemann U, Ponto C, Kortt J, Kallenberg K, Varges D, Schulz-Schaeffer WJ, Kretzschmar HA, Zerr I. Clinical findings and diagnosis in genetic prion diseases in Germany. Eur J Epidemiol 2016; 31(2):187-96; PMID:26076917; http://dx.doi.org/10.1007/s10654-015-0049-y
- Eigenbrod S, Frick P, Giese A, Schelzke G, Zerr I, Kretzschmar HA. Comprehensive neuropathologic analysis of genetic prion disease associated with the E196K mutation in PRNP reveals phenotypic heterogeneity. J Neuropathol Exp Neurol 2011; 70:192-200; PMID:21293298; http://dx.doi.org/10.1097/NEN.0b013e31820cd8a4
- Schelzke G, Eigenbrod S, Romero C, Varges D, Breithaupt M, Taratuto AL, Kretzschmar HA, Zerr I. Genetic prion disease with codon 196 PRNP mutation: clinical and pathological findings. Neurobiol Aging 2011; 32: 756 e751-9; PMID:NOT_FOUND; http://dx.doi.org/10.1016/j.neurobiolaging.2010.11.023
- Bejot Y, Osseby GV, Caillier M, Moreau T, Laplanche JL, Giroud M. Rare E196K mutation in the PRNP gene of a patient exhibiting behavioral abnormalities. Clin Neurol Neurosurg 2010; 112:244-7; PMID:20005032; http://dx.doi.org/10.1016/j.clineuro.2009.11.002
- Clerici F, Elia A, Girotti F, Contri P, Mariani C, Tagliavini F, Di Fede G. Atypical presentation of Creutzfeldt-Jakob disease: the first Italian case associated with E196K mutation in the PRNP gene. J Neurol Sci 2008; 275:145-7; PMID:18706660; http://dx.doi.org/10.1016/j.jns.2008.06.036
- Shi Q, Chen C, Song XN, Gao C, Tian C, Zhou W, Song XH, Yao LS, Han J, Dong XP. A Chinese Creutzfeldt-Jakob disease patient with E196K mutation in PRNP. Prion 2011; 5:117-20; PMID:21597335; http://dx.doi.org/10.4161/pri.5.2.15846