
ABSTRACT
Mutations in the profilin 1 (PFN1) gene have been identified as a cause of familial amyotrophic lateral sclerosis (ALS), and neuropathological studies indicate that TDP-43 is accumulated in brains of patients with PFN1 mutation. Here, we investigated the role of PFN1 mutations in the formation of prion-like abnormal TDP-43. Expression of PFN1 with pathogenic mutations resulted in the formation of cytoplasmic aggregates positive for p62 and ubiquitin, and these aggregates sequestered endogenous TDP-43. TDP-43 accumulation was facilitated in the presence of proteasome or lysosome inhibitor. Co-expression of mutant PFN1 and TDP-43 increased the levels of detergent-insoluble and phosphorylated TDP-43, and this increase required the C-terminal region of TDP-43. Moreover, detergent-insoluble fractions prepared from cells expressing ALS-linked mutant PFN1 induced seed-dependent accumulation of TDP-43. These findings indicate that expression of PFN1 mutants induces accumulation of TDP-43, and promotes conversion of normal TDP-43 into an abnormal form. These results provide new insight into the mechanisms of TDP-43 proteinopathies and other diseases associated with amyloid-like protein deposition.
Transactivation response DNA-binding protein 43 (TDP-43), encoded by the TARDBP gene, is a highly conserved, ubiquitously expressed heterogenous nuclear ribonucleoprotein (hnRNP) involved in exon splicing, gene transcription, regulation of mRNA stability and biosynthesis, and formation of nuclear bodies.Citation1-5 Structurally, TDP-43 is characterized by 2 RNA-recognition motifs (RRM1 and RRM2), and the C-terminal region includes a glycine-rich domain and a glutamine/asparagine (Q/N)-rich domain that are implicated in interactions with other proteins.Citation6-9 In pathological conditions, such as frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS), TDP-43 is accumulated mainly as ubiquitin-positive tau-negative inclusions in the cytoplasmic compartment of neuronal and glial cells in the brain and spinal cord of patients.Citation10,11 Biochemical and histological studies have demonstrated that TDP-43 is deposited in a filamentous form and most of the filaments are abnormally phosphorylated.Citation12 Therefore, both loss of normal function of nuclear TDP-43 due to cytoplasmic mislocalization and gain of toxic function due to TDP-43 aggregation in the cytoplasm are thought to be involved in the pathogenesis of ALS and FTLD accompanied with TDP-43 inclusions (FTLD-TDP).Citation13
The discovery of mutations in the TARDBP gene in familial and sporadic ALS indicated a pivotal role of TDP-43 in the pathogenesis of ALS and FTLD.Citation14-16 These mutations are predominantly located in the C-terminal region, suggesting that conformational change of this region is closely related to the pathogenesis.Citation17 Actually, the glycine-rich and Q/N-rich domains in this region contribute to amyloid-like fibril formation as well as aggregation propensity of TDP-43.Citation18-21 The RRM2 domain also plays a key role in TDP-43 aggregation.Citation22 On the other hand, we previously demonstrated that insoluble TDP-43 aggregates in brains of ALS and FTLD-TDP patients have prion-like properties.Citation20 When insoluble TDP-43 from ALS or FTLD-TDP brains was introduced as seeds into cells expressing TDP-43, seed-dependent TDP-43 accumulation was induced. Interestingly, the C-terminal fragment-banding patterns of converted host proteins resemble those of insoluble TDP-43 used as seeds, suggesting that the conversion is template-dependent. Moreover, mass spectrometric analysis of insoluble TDP-43 from brains of ALS patients suggested that RRM2, the glycine-rich domain, and a part of the Q/N-rich domain form the core region of TDP-43 aggregates.Citation23 These results suggest that seed-dependent accumulation of prion-like, conformationally changed TDP-43 via interaction at the C-terminal region has a pivotal role in the pathogenesis of ALS and FTLD-TDP. However, it is largely unknown how and when such abnormal TDP-43 is formed in cells.
Recently, mutations in the profilin 1 (PFN1) gene have been identified as a cause of familial ALS.Citation24 PFN1 is involved in various cellular functions by binding to actin monomer, phosphoinositides and proline-rich proteins.Citation25-27 Previous studies have found that loss-of-function of PFN1 resulting from mutations linked to familial ALS causes cytoskeletal disruption and altered stress granule dynamics.Citation24,28 On the other hand, classical TDP-43 pathology was present in the brains of patients with autosomal dominant mutations in the PFN1 geneCitation29 and co-aggregation of PFN1 and TDP-43 was observed in cells expressing ALS-linked mutant PFN1.Citation24 These findings imply that gain-of-toxic function PFN1 mutations cause TDP-43 aggregation.
To elucidate how mutant PFN1 induces TDP-43 pathology, we transiently expressed wild-type (WT) or ALS-linked PFN1 mutants (C71G, M114T, E117G, or G118V) in SH-SY5Y neuroblastoma cells. As shown previously, PFN1 mutants that cause ALS (C71G, M114T, and G118V) are destabilized and are prone to aggregate in cells. These aggregates were also positive for ubiquitin and p62 (characteristic of inclusions detected in FTLD or ALS) and were predominantly localized in cytoplasm.Citation30 On the other hand, the E117G PFN1 mutant, a moderate risk factor of familial ALS, was diffusely localized in cells, like WT PFN1, suggesting that the propensity of PFN1 mutants to aggregate is implicated in the pathogenesis of ALS. In support of this hypothesis, another pathogenic mutant A20T PFN1 displays a high aggregation propensity.Citation29 Interestingly, in some cells expressing mutant PFN1, co-aggregates of PFN1 and endogenous TDP-43 were formed in the cytoplasm, the downstream functions of TDP-43 were disturbed, and TDP-43 disappeared from the nucleus. Further, immunoprecipitation analysis demonstrated that PFN1 interacts with TDP-43, but not with fused in sarcoma (FUS), a hnRNP implicated in FTLD and ALS.Citation31 These findings indicate that TDP-43 is sequestered into PFN1 aggregates due to its affinity for PFN1. This mechanism might at least partly explain why nuclear protein TDP-43 forms inclusions in the cytoplasm, and why inclusions mainly composed of TDP-43 are formed. That is, TDP-43 localized in the cytoplasm (either produced or transported there) might be sequestered into structures with affinity for TDP-43, and start forming inclusions in the cytoplasm.
Deficits in the protein degradation system, including the ubiquitin-proteasome and autophagy-lysosome systems, with aging are implicated in the pathogenesis of various neurodegenerative diseases.Citation32 In the case of PFN1 mutations, inhibition of proteasome by MG132 increased PFN1 and TDP-43 aggregation in cells transfected with mutant PFN1, primarily because the total amount of mutant PFN1 and TDP-43 was increased. On the other hand, PFN1 aggregates suppressed the formation of aggresome-like structures when proteasome was inhibited. Although it is not clearly known to what extent this suppression is involved in the increased TDP-43 aggregation, formation of aggresomes is recognized as a cytoprotective response serving to sequester potentially toxic misfolded proteins and facilitate their clearance by autophagy.Citation33 Therefore, increased misfolded TDP-43 and decreased function of ubiquitin-proteasome systems might coordinately increase TDP-43 aggregation in cells harboring PFN1 aggregates in the presence of a proteasome inhibitor. PFN1 aggregates also suppressed the conversion of LC3-I to LC3-II, an essential event for autophagy, possibly because of sequestration of LC3 into PFN1 aggregates. It is not clear whether LC3 is directly sequestered into PFN1 aggregates or whether the process is mediated by the binding of other proteins such as p62 sequestered into PFN1 aggregates. Importantly, TDP-43 aggregates increased in the presence of the PFN1 aggregates when lysosomal function was inhibited by bafilomycin A1, a specific inhibitor of vacuolar ATPase. Bafilomycin A1 did not markedly increase PFN1 aggregates, different from MG132. Therefore, this finding may imply that prion-like conformationally changed TDP-43 is formed in the presence of PFN1 aggregates, and seed-dependent TDP-43 accumulation is facilitated by decreased lysosomal degradation of TDP-43 fibrils that function as a template for seed-dependent accumulation of TDP-43. These effects of PFN1 aggregates on the degradation system, and the increased endogenous TDP-43 accumulation when the degradation system is inhibited, might partly explain why ALS pathogenesis develops with aging. Further studies are needed in order to elucidate in detail the mechanism through which PFN1 aggregates suppress the degradation system.
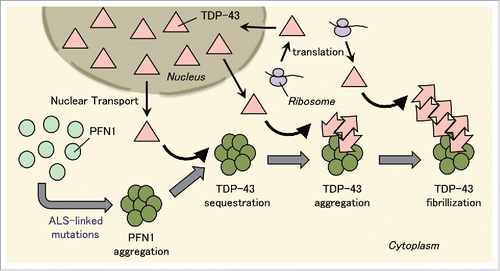
The process of seed-dependent TDP-43 accumulation progresses through the conversion of normal TDP-43 into conformationally changed (abnormal) TDP-43 that serves as a template of TDP-43 fibrils, leading to TDP-43 filament formation.Citation20,34 To investigate the effect of PFN1 aggregates on seed-dependent TDP-43 accumulation, TDP-43 monomer was supplied by means of overexpression. Insoluble TDP-43 accumulation markedly increased in cells co-transfected with mutant PFN1 and TDP-43, indicating that seed-dependent TDP-43 accumulation progressed in the presence of PFN1 aggregates. Moreover, insoluble phosphorylated TDP-43, a hallmark of pathological TDP-43, was detected dependently on the amount of insoluble TDP-43 accumulation. Thus, TDP-43 might be phosphorylated after seed-dependent TDP-43 accumulation. We propose that TDP-43 sequestered into PFN1 aggregates in the cytoplasm has a prion-like property and induces seed-dependent TDP-43 accumulation. To test the idea that TDP-43 is sequestered into PFN1 aggregates in the cytoplasm, we co-expressed mutant PFN1 and TDP-43 lacking the nuclear localization signal (ΔNLS) in cells. This resulted in higher accumulation of insoluble TDP-43 and phosphorylated TDP-43 than did co-expression of mutant PFN1 and TDP-43. The results of co-expression of C71G PFN1 with GFP-tagged full-length, N-terminal, or C-terminal TDP-43 also revealed that increased TDP-43 aggregation in the presence of PFN1 aggregates requires the C-terminal region of TDP-43, which includes the prion-like domain.Citation18,21 Furthermore, seed-dependent TDP-43 accumulation and insoluble TDP-43 phosphorylation were increased in cells after introduction of detergent-insoluble fraction from cells transfected with ALS-linked mutant PFN1, indicating that conformationally changed, prion-like TDP-43 was formed in the presence of mutant PFN1. Interestingly, detergent-insoluble fraction from cells expressing soluble PFN1 E117G mutant also induced seed-dependent TDP-43 accumulation. This finding indicates that conformational change of TDP-43 induced by mutant PFN1 does not necessarily require PFN1 aggregation. Possibly, TDP-43 might not be easy to aggregate under physiological conditions due to the presence of multiple interacting partners.Citation35 Although the precise mechanism remains unknown, PFN1 mutations linked to ALS might increase the affinity of PFN1 for TDP-43, and this in turn might disrupt interactions with other partners and induce conformational change of TDP-43. We propose that structures, such as PFN1 aggregates, that sequester TDP-43 might be a trigger to induce TDP-43 proteinopathy ().
FIGURE 1. Conversion of normal TDP-43 into prion-like species by ALS-linked PFN1 mutants. PFN1 with mutations that cause ALS forms aggregates in the cytoplasm. TDP-43 localized in the cytoplasm for its production or transportation from nucleus is sequestered into PFN1 aggregates, where it is converted into prion-like species, and forms TDP-43 aggregates. Furthermore, these TDP-43 aggregates serve as a template for conversion of normal TDP-43 into prion-like TDP-43 aggregates. As a result, TDP-43 filaments are formed. Thus, TDP-43 aggregation is facilitated firstly by sequestration of TDP-43 into PFN1 aggregates and secondly by self-template-type aggregation of prion-like TDP-43.
Interestingly, as seed-dependent TDP-43 accumulation increased over time, endogenous PFN1 was increased in the detergent-insoluble fraction, indicating that TDP-43 aggregates also sequestered endogenous PFN1. In support of this idea, PFN1 is accumulated in inclusions positive for phosphorylated TDP-43 in the brain of sporadic ALS or progranulin-deficient FTLD patients. Therefore, endogenous PFN1 would be locally lost in cells harboring TDP-43 aggregates. It has been reported that phosphorylated TDP-43 aggregates sequester RNA polymerase II, and induce cell death via a non-apoptotic mechanism in SH-SY5Y cells and FTLD-TDP patients.Citation36 Considering that PFN1 is involved in various cellular functions, localized loss of endogenous PFN1 might also mediate the toxicity of TDP-43 inclusions. Further, it is possible that WT PFN1 aggregates sequester TDP-43. In the brain of ALS or FTLD patients without PFN1 mutations, PFN1 accumulation might be simply a result of sequestration into TDP-43 aggregates. On the other hand, PFN1 accumulation might precede TDP-43 accumulation. Post-translational modifications of PFN1, such as phosphorylation or acetylation, which accumulate with aging, might induce conformational change of PFN1 and lead to its aggregation.Citation37-39 Further studies are needed to examine whether accumulated PFN1 in ALS or FTLD patients is post-translationally modified or whether post-translational modifications of PFN1 induce conformational change of PFN1. We propose that co-aggregation of PFN1 and TDP-43 is a potential therapeutic target for TDP-43 proteinopathy.
In summary, PFN1 mutations associated with ALS induce formation of PFN1 aggregates, which sequester TDP-43 and form co-aggregates of PFN1 and TDP-43 that are positive for ubiquitin and p62, which is characteristic of inclusions observed in ALS and FTLD-TDP brains. Furthermore, TDP-43 sequestered into PFN1 aggregates functions as a template for seed-dependent TDP-43 accumulation, suggesting that PFN1 aggregates provide a scaffold for conformational change of TDP-43. Our findings provide a clue as to how prion-like TDP-43 may be generated, and highlight the importance of the formation of conformationally changed, prion-like TDP-43 in the pathogenesis of ALS and FTLD-TDP.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
The authors report no potential conflicts of interest.
ACKNOWLEDGMENTS
We thank Genjiro Suzuki for his comments on the manuscript.
Funding
Funding was provided by JSPS KAKENHI Grant Number 15K18370, 14J03307 (to YT), 23228004 (to MH), MEXT KAKENHI Grant Numbers 26117005, 23240050 (to MH), and MHLW Grant ID Number 12946221 (to MH).
REFERENCES
- Ayala YM, De Conti L, Avendano-Vazquez SE, Dhir A, Romano M, D'Ambrogio A, Tollervey J, Ule J, Baralle M, Buratti E, et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J 2011; 30:277-88; PMID:21131904; http://dx.doi.org/10.1038/emboj.2010.310
- Ayala YM, Zago P, D'Ambrogio A, Xu YF, Petrucelli L, Buratti E, Baralle FE. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci 2008; 121:3778-85; PMID:18957508; http://dx.doi.org/10.1242/jcs.038950
- Buratti E, Baralle FE. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci 2008; 13:867-78; PMID:17981595; http://dx.doi.org/10.2741/2727
- Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol 1995; 69:3584-96; PMID:7745706
- Wang IF, Reddy NM, Shen CK. Higher order arrangement of the eukaryotic nuclear bodies. Proc Natl Acad Sci U S A 2002; 99:13583-8; PMID:12361981; http://dx.doi.org/10.1073/pnas.212483099
- Ayala YM, Pantano S, D'Ambrogio A, Buratti E, Brindisi A, Marchetti C, Romano M, Baralle FE. Human, Drosophila, and C.elegans TDP43: nucleic acid binding properties and splicing regulatory function. J Mol Biol 2005; 348:575-88; PMID:15826655; http://dx.doi.org/10.1016/j.jmb.2005.02.038
- Buratti E, Baralle FE. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem 2001; 276:36337-43; PMID:11470789; http://dx.doi.org/10.1074/jbc.M104236200
- Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, Baralle FE. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem 2005; 280:37572-84; PMID:16157593; http://dx.doi.org/10.1074/jbc.M505557200
- Wang IF, Wu LS, Shen CK. TDP-43: an emerging new player in neurodegenerative diseases. Trends Mol Med 2008; 14:479-85; PMID:18929508; http://dx.doi.org/10.1016/j.molmed.2008.09.001
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 2006; 351:602-11; PMID:17084815; http://dx.doi.org/10.1016/j.bbrc.2006.10.093
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006; 314:130-3; PMID:17023659; http://dx.doi.org/10.1126/science.1134108
- Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, Beach TG, Buratti E, Baralle F, Morita M, et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol 2008; 64:60-70; PMID:18546284; http://dx.doi.org/10.1002/ana.21425
- Lee EB, Lee VM, Trojanowski JQ. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci 2012; 13:38-50
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 2008; 40:572-4; PMID:18372902; http://dx.doi.org/10.1038/ng.132
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008; 319:1668-72; PMID:18309045; http://dx.doi.org/10.1126/science.1154584
- Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 2010; 9:995-1007; PMID:20864052; http://dx.doi.org/10.1016/S1474-4422(10)70195-2
- Pesiridis GS, Lee VM, Trojanowski JQ. Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum Mol Genet 2009; 18:R156-62; PMID:19808791; http://dx.doi.org/10.1093/hmg/ddp303
- Guo W, Chen Y, Zhou X, Kar A, Ray P, Chen X, Rao EJ, Yang M, Ye H, Zhu L, et al. An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat Struct Mol Biol 2011; 18:822-30; PMID:21666678; http://dx.doi.org/10.1038/nsmb.2053
- Wang IF, Chang HY, Hou SC, Liou GG, Way TD, James Shen CK. The self-interaction of native TDP-43 C terminus inhibits its degradation and contributes to early proteinopathies. Nat Commun 2012; 3:766; PMID:22473010; http://dx.doi.org/10.1038/ncomms1766
- Nonaka T, Masuda-Suzukake M, Arai T, Hasegawa Y, Akatsu H, Obi T, Yoshida M, Murayama S, Mann DM, Akiyama H, et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep 2013; 4:124-34; PMID:23831027; http://dx.doi.org/10.1016/j.celrep.2013.06.007
- Shimonaka S, Nonaka T, Suzuki G, Hisanaga S, Hasegawa M. Templated aggregation of TAR DNA-binding protein of 43 kDa (TDP-43) by seeding with TDP-43 peptide fibrils. J Biol Chem 2016; 291:8896-907; PMID:26887947; http://dx.doi.org/10.1074/jbc.M115.713552
- Wang YT, Kuo PH, Chiang CH, Liang JR, Chen YR, Wang S, Shen JC, Yuan HS. The truncated C-terminal RNA recognition motif of TDP-43 protein plays a key role in forming proteinaceous aggregates. J Biol Chem 2013; 288:9049-57; PMID:23372158; http://dx.doi.org/10.1074/jbc.M112.438564
- Kametani F, Obi T, Shishido T, Akatsu H, Murayama S, Saito Y, Yoshida M, Hasegawa M. Mass spectrometric analysis of accumulated TDP-43 in amyotrophic lateral sclerosis brains. Sci Rep 2016; 6:23281; PMID:26980269; http://dx.doi.org/10.1038/srep23281
- Wu CH, Fallini C, Ticozzi N, Keagle PJ, Sapp PC, Piotrowska K, Lowe P, Koppers M, McKenna-Yasek D, Baron DM, et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 2012; 488:499-503; PMID:22801503; http://dx.doi.org/10.1038/nature11280
- Carlsson L, Nystrom LE, Sundkvist I, Markey F, Lindberg U. Actin polymerizability is influenced by profilin, a low molecular weight protein in non-muscle cells. J Mol Biol 1977; 115:465-83; PMID:563468; http://dx.doi.org/10.1016/0022-2836(77)90166-8
- Staiger CJ, Yuan M, Valenta R, Shaw PJ, Warn RM, Lloyd CW. Microinjected profilin affects cytoplasmic streaming in plant cells by rapidly depolymerizing actin microfilaments. Curr Biol 1994; 4:215-9; PMID:7922326; http://dx.doi.org/10.1016/S0960-9822(00)00050-6
- Witke W. The role of profilin complexes in cell motility and other cellular processes. Trends Cell Biol 2004; 14:461-9; PMID:15308213; http://dx.doi.org/10.1016/j.tcb.2004.07.003
- Figley MD, Bieri G, Kolaitis RM, Taylor JP, Gitler AD. Profilin 1 associates with stress granules and ALS-linked mutations alter stress granule dynamics. J Neurosci 2014; 34:8083-97; PMID:24920614; http://dx.doi.org/10.1523/JNEUROSCI.0543-14.2014
- Smith BN, Vance C, Scotter EL, Troakes C, Wong CH, Topp S, Maekawa S, King A, Mitchell JC, Lund K, et al. Novel mutations support a role for profilin 1 in the pathogenesis of ALS. Neurobiol Aging 2015; 36:1602.e17-27; PMID:25499087; http://dx.doi.org/10.1016/j.neurobiolaging.2014.12.037
- Tanaka Y, Nonaka T, Suzuki G, Kametani F, Hasegawa M. Gain-of-function profilin 1 mutations linked to familial amyotrophic lateral sclerosis cause seed-dependent intracellular TDP-43 aggregation. Hum Mol Genet 2016; 25:1420-33; PMID:26908597; http://dx.doi.org/10.1093/hmg/ddw024
- Ratti A, Buratti E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J Neurochem 2016; Epub ahead of print; PMID:27015757
- Martinez-Vicente M, Sovak G, Cuervo AM. Protein degradation and aging. Exp Gerontol 2005; 40:622-33; PMID:16125351; http://dx.doi.org/10.1016/j.exger.2005.07.005
- Olzmann JA, Li L, Chin LS. Aggresome formation and neurodegenerative diseases: therapeutic implications. Curr Med Chem 2008; 15:47-60; PMID:18220762; http://dx.doi.org/10.2174/092986708783330692
- Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem 2009; 284:20329-39; PMID:19465477; http://dx.doi.org/10.1074/jbc.M109.010264
- Freibaum BD, Chitta RK, High AA, Taylor JP. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res 2010; 9:1104-20; PMID:20020773 http://dx.doi.org/10.1021/pr901076y
- Yamashita M, Nonaka T, Hirai S, Miwa A, Okado H, Arai T, Hosokawa M, Akiyama H, Hasegawa M. Distinct pathways leading to TDP-43-induced cellular dysfunctions. Hum Mol Genet 2014; 23:4345-56; PMID:24698978; http://dx.doi.org/10.1093/hmg/ddu152
- Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 2006; 23:607-18; PMID:16916647; http://dx.doi.org/10.1016/j.molcel.2006.06.026
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009; 325:834-40; PMID:19608861; http://dx.doi.org/10.1126/science.1175371
- Ingre C, Landers JE, Rizik N, Volk AE, Akimoto C, Birve A, Hubers A, Keagle PJ, Piotrowska K, Press R, et al. A novel phosphorylation site mutation in profilin 1 revealed in a large screen of US, Nordic, and German amyotrophic lateral sclerosis/frontotemporal dementia cohorts. Neurobiol Aging 2013; 34:1708.e1-6; PMID:23141414; http://dx.doi.org/10.1016/j.neurobiolaging.2012.10.009