
ABSTRACT
Prion diseases, or transmissible spongiform encephalopathies, have revealed the bewildering phenomenon of transmissibility in neurodegenerative diseases. Hence, the experimental transmissibility of prion-like neurodegenerative diseases via template directed misfolding has become the focus of intense research. Gerstmann-Sträussler-Scheinker disease (GSS) is an inherited prion disease associated with mutations in the prion protein gene. However, with the exception of a few GSS cases with P102L mutation characterized by co-accumulation of protease-resistant PrP core (PrPres) of ∼21 kDa, attempts to transmit to rodents GSS associated to atypical misfolded prion protein with ∼8 kDa PrPres have been unsuccessful. As a result, these GSS subtypes have often been considered as non-transmissible proteinopathies rather than true prion diseases. In a recent study we inoculated bank voles with GSS cases associated with P102L, A117V and F198S mutations and found that they transmitted efficiently and produced distinct pathological phenotypes, irrespective of the presence of 21 kDa PrPres in the inoculum. This study demonstrates that GSS is a genuine prion disease characterized by both transmissibility and strain variation. We discuss the implications of these findings for the understanding of the heterogeneous clinic-pathological phenotypes of GSS and of the molecular underpinnings of prion infectivity.
Transmissible Spongiform Encephalopathies (TSEs) or prion diseases are fatal neurodegenerative diseases affecting humans and animals which have been historically defined on the basis of their experimental transmissibility. TSEs encompass a range of neurological diseases with disparate origin and epidemiology, and are unique in medicine in that they can occur as sporadic, familial or acquired forms. This group of neurodegenerative diseases is caused by spontaneous or induced misfolding of cellular prion protein monomers (PrPC) into insoluble and autocatalytically self-replicating prion protein aggregates (PrPSc). PrPSc is usually resistant to proteinase K (PK), and the protease-resistant core is often referred to as PrPres. PrPSc is thought to be the main component of prions, the proteinaceous infectious agents which are responsible for TSEs. Prions exist as strains, which are thought to be encoded in conformational variants of PrPSc. Different PrPSc conformers can be identified by biochemical fingerprinting, which is of help in the molecular discrimination and classification of TSEs. The neuropathological hallmark of TSEs is the spongiform degeneration, which appears with distinctive morphology and brain distribution in different prion strains. Spongiosis coexists with various degree of neuronal loss, gliosis and white matter degeneration, and with the accumulation of various forms of misfolded PrP aggregates in the brain parenchyma.
Scrapie in small ruminants and Chronic Wasting Disease in cervids are emblematic examples of infectious TSEs, which efficiently spread by natural transmission under field conditions. Conversely, most human TSEs are though to originate from spontaneous misfolding of PrP and occur as sporadic cases or are associated with pathogenic mutations of PrP. Still, brain tissues from affected individuals usually contain prions which are transmissible, as shown by bioassay in animal models. The experimental transmissibility is far from being a mere scientific issue, as it underlies the risk of transmitting TSEs under particular circumstances with dramatic public health consequences. This was the case for the iatrogenic epidemics of Creutzfeld-Jacob disease (CJD) caused by human growth hormone treatments and dura mater grafts, the vCJD epidemic in UK linked to the zoonotic transmission of Bovine Spongiform Encephalopathy through food, and the human-to-human iatrogenic transmission of vCJD by blood transfusion.Citation1 These acquired prion diseases are now less frequent, as awareness of transmission risk has led to prevent such occurrences.
The tenet of experimental transmissibility has been fulfilled for most of the known human neurodegenerative conditions accompanied by misfolded PrP. We have focused our interest on a remarkable exception, represented by cases of a hereditary neurodegenerative syndrome, Gerstmann-Sträussler-Scheinker disease (GSS), induced by pathogenic mutations in the prion protein gene and accompanied by aggregates of misfolded PrP with peculiar biochemical properties.
The recognition of human inherited prion diseases predates molecular diagnosis. Historically, families with inherited prion disease have been categorized on clinical grounds in 3 syndromes: GSS, Fatal Familial Insomnia and CJD. GSS is an autosomal dominant disorder with a wide spectrum of clinical presentations including ataxia, spastic paraparesis, extrapyramidal signs, and dementia. A single mutation at codon 102 of the PRNP gene, resulting in an amino acid change from proline to leucine (P102L), was first linked to the typical ataxic presentation of GSS.Citation2 GSS has later been associated with several rarer PrP mutations at codons 84, 105, 117, 131, 132, 133, 176, 187, 198, 202, 211, 212, 217, 218 and 226, and with insertional mutations in the octapeptide repeat.Citation3 The variety of PrP mutations conceivably contributes to the widely recognized clinical and pathological diversity in GSS. Still, GSS is characterized by considerable heterogeneity in age at onset, disease duration, clinical manifestations and neuropathological phenotypes both between and within families, pointing to other genetic or epigenetic factors as additional drivers of phenotypic heterogeneity.
Common pathological denominators in GSS are cerebral PrP amyloid plaques and accumulation of misfolded PrPSc conformers characterized by an atypical protease-resistant core of ∼8 kDa. Plaque deposits isolated from different GSS-associated mutations (P102L, A117V, F198S, Q217R) appear to be composed of PrP fragments nearly overlapping with the ∼8 kDa PrPres and which originate only from the mutant allele.Citation4 However, there is significant variability among reported families in the pattern of amyloid deposition in regions of the central nervous system and in the conformation of PrPSc. Amyloidosis is usually accompanied by mild or absent spongiform changes, but coexists with severe spongiform degeneration in some patients with P102L mutation.
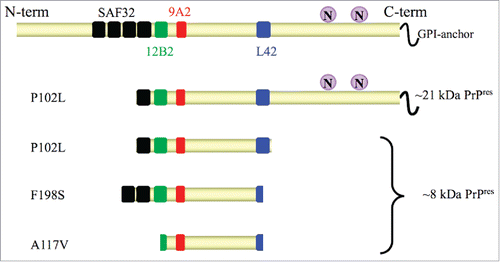
N-terminal and C-terminal truncated, nonglycosylated, PK-resistant PrP peptides of ∼8 kDa (8 kDa PrPres) are considered the molecular signature of GSS. However, in patients with P102L mutation and severe spongiform degeneration, 8 kDa PrPres coexists with classical PrPSc,Citation5 characterized by N-terminal truncated, glycosylated PrPres of ∼21 kDa (21 kDa PrPres). Furthermore, remarkable conformational heterogeneity characterizes PrPSc conformers with 8 kDa PrPres derived from different PrP mutants, as shown by their PK-cleavage siteCitation6,7,8 and conformational stabilityCitation8 (). Owing to the rarity of these inherited neurodegenerative diseases, a comprehensive analysis of PrPSc conformers across the full spectrum of the PrP mutations causing GSS is still lacking.
FIGURE 1. Schematic representation of PrPres fragments in GSS with P102L, F198S and A117V mutations. In full length PrP (upper cartoon) the glycosylation sites “N” (aa 181 and 197) and the epitopes of monoclonal antibodies SAF32 (black, octarepeat), 12B2 (green, aa 89–93), 9A2 (red, aa 99–101), L42 (blue, aa 145–150) used for epitope mapping are shown. Cleavage sites of protease resistant PrPres fragments after digestion with protease K are depicted according to Pirisinu et al., 2013. PrPres from brain of P102L patients is characterized by ∼8 kDa PrPres cleaved at N-terminus (aa ∼78–82) and C-terminus (aa ∼150), which is accompanied in some cases by a classical ∼21 kDa PrPres. F198S PrPres spans aa ∼74 to ∼146–150, while A117V PrPres is characterized by a smaller fragment spanning from aa ∼90–96 to 146.
Different animal models have been used to study GSS with the aims to model the disease in mice and to assess the potential transmissibility of this heritable prion disease.
Genetic Modeling of GSS in Mice
Attempts to model the disease involved the generation of transgenic mice expressing human or mouse mutant PrP. The first demonstration that a neurodegenerative process similar to GSS can be modeled by transgenesis in mice was reported by Hsiao et alCitation9 with the generation of mice overexpressing mouse PrP with a leucine substitution at codon 101 (homologous to codon 102 in humans). These mice were reported to develop spontaneously clinical and pathological features of prion diseases, but no PK-resistant PrP was identified in their brains. Several other lines of transgenic mice expressing different levels of murine P101L PrP were then generated,Citation10,11,12,13 and it was shown that co-expression of wild-type murine PrP was able to modulate the age of disease onset and the severity of neurodegeneration.Citation11 Studies aimed to investigate the transmissibility of the disease from spontaneously sick mice showed that transmission occurred in recipient tg196 mice, which express lower levels of the mutant transgene.Citation11 However, further observations led to consider this phenomenon as disease acceleration rather than prion transmission.Citation13 Indeed, Tg196 mice were reported to develop spontaneous neurodegeneration at advanced ageCitation14,15 and transgenic mice expressing mutant PrP at low levels or wild-type mice failed to develop disease either spontaneously or following inoculation.Citation13 Accordingly, knock-in mice in which endogenous wild-type mouse PrP was replaced with a mutant P101L mouse PrP did not develop a spontaneous illness.Citation16 Finally, transgenic mice overexpressing human PrP with P102L mutation did not develop spontaneous disease,Citation17 suggesting that the mouse PrP background might have played a role in the occurrence of the spontaneous disease described in previous studies. However, spontaneous neurodegenerative disease was also reported in transgenic mice overexpressing mutant 113L bovine prion protein (homologous to human P102L and mouse P101L), but not in those expressing physiological levels of the mutant PrP.Citation18 Although PrPres was not detected in the brain of these mice, the disease was transmissible, with low efficiency, to transgenic mice expressing wild-type bovine PrP (BoPrP-Tg110).
Other mouse models support the tenet that PrP mutations associated to GSS might induce spontaneous neurodegeneration. Transgenic mice overexpressing mouse PrP carrying an A116V mutation (the homologous of the GSS-associated A117V mutation) developed a spontaneous neurological illness with some neuropathological hallmarks of prion diseases, although obvious PrPres was not observed and the spontaneously sick mice accumulated a faint and weakly PK-resistant PrP fragment.Citation19 The transmissibility of this spontaneous disease has not been reported. As previously observed with P102L, transgenic mice overexpressing human PrP with A117V mutation instead of mouse PrP with the homologous mutation did not develop a spontaneous neurological disease.Citation20
Transgenic mice expressing the mouse homolog of a mutant human PrP containing a 9 octapeptide insertion associated with GSS were reported to spontaneously develop a fatal neurodegenerative disorder characterized by ataxia, neuronal apoptosis, and accumulation in the brain of an aggregated and weakly protease-resistant form of mutant PrP.Citation21 However, attempts to transmit the disease in recipient mice were unsuccessful.Citation22
Finally, it has been shown that transgenic mice overexpressing GPI-anchorless mouse PrP, that somewhat resembles the rare mutations of GSS which generate C-terminally truncated PrP molecules lacking the GPI-anchor, develop a spontaneous prion disease characterized by a GSS-like protease-resistant PrP fragment.Citation23
Collectively, these studies suggest that the neurotoxicity can depend at least in part by the expression of mutant PrP, even in the absence of classic PrPres, that there might be a direct effect of the mutation in defining the phenotype of spontaneous disease and that there may be a distinction between neurotoxic and infectious isoforms.
Transmissibility of GSS in Animal Models
Although GSS with 8 kDa PrPres is considered poorly or not transmissible, evidences for this are rather limited. Indeed, the first studies attempting the transmission of GSS in mice were performed before being aware that GSS with P102L mutation might contain a classical 21 kDa PrPres, besides the atypical 8 kDa PrPres. Furthermore, many mutations that induce GSS are extremely rare and their transmissibility has not been assessed.
GSS with P102L mutation is the most studied form of GSS. In 1981 Masters et alCitation24 reported that the inoculation in non-human primates of brain homogenates from GSS-affected patients, later identified as carrying P102L mutation,Citation25 induced spongiform encephalopathy in some recipient animal. Then, transmission studies were also reported in marmosetsCitation25 and mice.Citation26,27 It has been later shown that most of the transmitted cases were characterized by severe spongiform degeneration and by the accumulation of both, 21 and 8 kDa PrPres.Citation5 Accordingly, no transmission occurred in mice, hamster and marmosets after inoculation with brain homogenate from patients with F198S mutationCitation28,29 and studies of infectivity of GSS with A117V mutation showed unsuccessful transmissions in wild-type mice.Citation30 Indeed, both F198S and A117V GSS are characterized by the accumulation of the only 8 kDa PrPres.
More recently, the issue of the transmissibility of GSS has been addressed in knock-in or transgenic mice carrying the homologous GSS-associated mutations and not developing a spontaneous disease. Overall, these studies confirmed previous findings showing variable transmissibility of GSS. Indeed, GSS P102L with 21 kDa PrPres could be transmitted in knock-in mice expressing endogenous levels of mouse PrP 101LCitation31 and in transgenic mice overexpressing mutant human 102L PrP,Citation17 while inocula derived from GSS P102L patients with 8 kDa PrPres induced PrP-amyloid deposition but not clinical disease and neurodegeneration, and did not lead to the accumulation of a TSE infectious agent.Citation31 Finally, A117V GSS cases transmitted with low clinical attack rates and after long incubation periods in transgenic mice overexpressing the human 117V PrP.Citation20 In this study, the disease was accompanied by the accumulation of inherently unstable PrPSc aggregates, and the accumulation of a transmissible agent in the brain of recipient mice was not addressed.
Taken together, transmission experiments suggest that P102L GSS cases with 21 kDa PrPres and possibly with extensive spongiform degeneration are associated with classical transmissibility of PrP misfolding and prion disease, while these studies fall short in providing definitive proof that GSS forms characterized by 8 kDa PrPres actually contain transmissible agents. In keeping with evidences from transgenic mouse models of spontaneous disease, this might lead to hypothesize that the atypical PrPSc conformers which characterize most GSS cases could be associated with neurotoxicity, but not with “true” infectivity. Some support to this hypothesis derives from the study of a newly described sporadic prion disease, Variably Protease Sensitive Prionopathy (VPSPr). VPSPr is characterized by an atypical PrPSc isoform with a ∼7 kDa PK-resistant core, similar to GSS, suggesting that VPSPr might be the long-sought sporadic form of GSS.Citation32 Interestingly, it has been difficult so far to provide evidence that VPSPr is a transmissible prion disease, as transmission in transgenic mice expressing human PrP showed low penetrance of disease on primary transmission and did not lead to the accumulation of an infectious agent, as shown by the lack of disease transmission on subpassage into the same transgenic mice.Citation33 Thus GSS and other TSEs characterized by 8 kDa PrPres might provide an opportunity to investigate the pathogenic roles played by different conformers of misfolded protein aggregates, an issue which attracts increasing interest of the scientific community in the field of neurodegenerative diseases accompanied by protein misfolding, such as Alzheimer disease and Parkinson disease.
Experimental Transmission of GSS in Bv109I
Although transmissibility is the prototypical property of prions, the efficiency of experimental transmissions is highly variable and mostly depends on the prion strain and the recipient host involved. We established bank voles as a rodent model able to propagate most of the known TSEs,Citation34,35,36 a property later shown to depend on the primary structure of bank vole PrP.Citation37,38 Bank vole PrP is polymorphic at codon 109, coding for methionine or isoleucine, and this amino acid variation further modulates the susceptibility of bank voles to various prion strains,Citation36,39 including atypical scrapie, which is characterized by GSS-like PrPSc conformers.Citation8 We thus reasoned that bank voles could have been a convenient animal model to challenge the hypothesis that neurotoxicity and transmissibility might be uncoupled in the atypical PrPSc conformers which characterize GSS. Seven GSS cases with P102L, A117V and F198S mutations were inoculated in bank voles carrying isoleucine at codon 109 of PrP (Bv109I). The overall results of this study are summarized in . In our experiments, all GSS cases induced in voles a prion disease characterized by spongiform degeneration and PrPSc deposition, most of them between 3 and 7 months post-challenge.Citation40 Furthermore, second passages in voles were highly efficient proving that in all cases (ref. 40 and unpublished data) a transmissible agent propagated in the brain of recipient voles. Notably, all GSS inocula induced the accumulation of 7–8 kDa PrPres, although a P102L case with 21 kDa PrPres led to the accumulation of 21 kDa PrPres in some recipient voles with long incubation period. It is somehow unexpected that 21 kDa PrPres transmitted with longer incubation time than 8 kDa PrPres. This finding could suggest that in Bv109I 8 kDa PrPres is more rapidly infectious than 21 kDa PrPres; alternatively, as 8 and 21 kDa PrPres were both present in the human inoculum, the 8 kDa PrPres could have slowed down the replication rate of 21 kDa PrPres due to strain interference. Second passages will help in discriminating between these 2 hypotheses.
FIGURE 2. Primary transmission of GSS A117V, F198S and P102L cases in bank voles. Schematic representation of the electrophoretic patterns of PrPres accumulated in brains of patients used as inocula (left panels) and in the inoculated bank voles (middle panels). Different PrPres types are represented by different colors. For each PrPres type the denaturation-resistant multimers (for ∼8 kDa PrPres types) and the glycosylated isoforms (∼21 kDa PrPres) are represented as color gradient bands. Molecular weights (kDa) are indicated on the left. The representation of PrPres profiles mimics the profiles observed by western blot with 9A2 mAb. The right panels show a schematic representation of the neuropathological phenotypes observed in terminally ill bank voles which accumulated the PrPres type reported within each box. Each box shows 2 groups of standard coronal sections of the vole brain which depict the spongiform changes (tonal values of red) and the PrPSc deposition (tonal values of blue). Arrows, colored according to the PrPres types, indicate the groups of voles in which the depicted neuropathological phenotype was observed. Note that the neuropathological phenotypes which characterize 7 kDa PrPres (red box) and 8 kDa PrPres (black box) are similar, while that of 21 kDa PrPres (green box) is clearly distinct from the others. The survival time of voles varied significantly depending on the PrPres type, and was between 83 and 266 d post-inoculation (dpi) in voles showing 7 kDa PrPres, between 168 and 375 dpi in voles with 8 kDa PrPres and between 644 and 804 dpi in voles with 21 kDa PrPres.
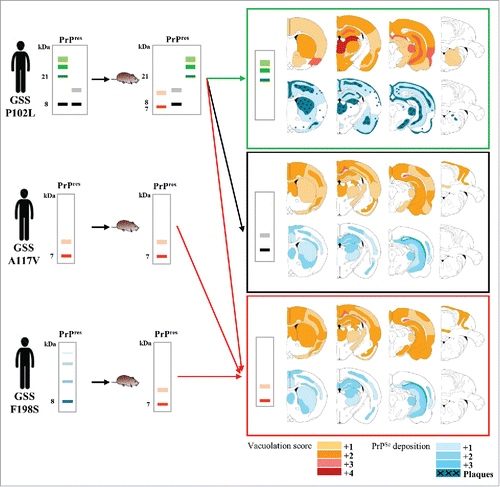
Interestingly, P102L, A117V and F198S GSS cases displayed varying transmission efficiencies and phenotypes of disease (). The two cases of GSS A117V induced a rapid and aggressive TSE in voles, which faithfully reproduced the PrPSc molecular feature of the human counterpart and propagated without an obvious transmission barrier. Although the transmission efficiency was lower with GSS F198S cases than with A117V cases, both F198S cases were transmitted in bank voles, providing the first evidence of transmissibility of this disease. Despite previous evidences that the infectivity of GSS cases with P102L was associated with the 21 kDa PrPres, we found that cases characterized by the accumulation of 8 kDa PrPres were transmissible in voles and produced all the neuropathological and molecular features of prion diseases. Interestingly, the P102L cases selected on their different PrPSc patterns resulted in different transmission efficiencies and disease phenotypes. Indeed, within the limited set of P102L GSS patients used, 3 different molecular and pathological phenotypes were derived in voles, and the resulting PrPSc types partially matched those present in the respective inocula ().
Overall, these findings prove that GSS with 8 kDa PrPres is a transmissible prion disease and elicit new questions which can be now experimentally addressed.
The Elusive Nature of the Experimental Transmissibility of GSS
The notion of the low transmissibility of GSS dates back to experiments using wild-type mice, so that it could be argued that this might depend on the limited susceptibility of wild-type mice to several human TSEs.Citation34,41 A systematic investigation of the transmissibility of GSS in transgenic mice carrying human PrP is still lacking. However, transgenic mice expressing mutated PrP have been challenged with GSS cases with the homologous mutations, with limited success. The failure of transmitting P102L cases with 8 kDa PrPres in transgenic mice which express murine PrP P101LCitation31 could still be tentatively explained by amino acid mismatches between human and mouse PrP sequences. However, it would be difficult to explain on this ground why A117V GSS cases have been transmitted more efficiently in voles than in transgenic mice which express human PrP with the A117V mutation.
These findings suggest that the full propagation of PrP misfolding and prion-induced neurodegeneration of GSS with 8 kDa PrPres might require a combination of peculiar factors in the recipient host, such as the amount and distribution of PrP expression, the conformational features of PrPC or other host factors. For the transmission of GSS in bank voles, the peculiar features of the vole PrP amino acid sequence might have been one of these factors. It has been found that the presence of asparagines in or near the β2-α2 loop might create a permissive PrP sequence able to overcome some species barriers,Citation42 and that recombinant vole PrP with methionine at residue 109 is an apparently universal substrate for RT-QuIC-based in vitro replication of disease-linked PrPSc isoforms.Citation38 However, ongoing studies in bank voles carrying methionine instead of isoleucine at residue 109 of PrP (Bv109M) show that, at variance with Bv109I, Bv109M are rather resistant to GSS with 8 kDa PrPres (unpublished data). This implies that having a “permissive” β2-α2 loop in the vole PrP is not sufficient per se for being susceptible to GSS and that the presence of isoleucine at PrP residue 109 might have been an additional key factor. Interestingly, residue 109 might be implicated in GSS-specific PrPSc misfolding pathways as it lies in a region of PrP enriched in GSS-linked mutations, i.e. P102L, P105L, P105T and A117V. Studies with transgenic mice overexpressing vole PrP with isoleucine at codon 109 support this hypothesis, as these mice spontaneously develop a transmissible neurological disease,Citation43 which is characterized by a GSS-like atypical 8 kDa PrPres.Citation44 Ongoing experiments aimed at investigating the relationships between the spontaneous disease in mice overexpressing bank vole PrP containing isoleucine at codon 109 and the disease induced in wild-type Bv109I by the inoculation of GSS cases might thus provide significant clues for explaining the susceptibility of Bv109I to GSS and the nature of the elusive transmissibility of GSS.
Heterogeneity of Clinical and Pathological Features of GSS: The Strain Hypothesis
Although GSS is most frequently associated with prominent ataxia, the clinical presentation can be highly heterogeneous in different patients and families. Similarly, large multicentric PrP amyloid plaques and atypical 8 kDa PrPres are prototypical pathological hallmarks of GSS, still other neuropathological changes, such as more or less severe spongiosis, white matter degeneration, synaptic deposition of PrPSc and tau pathology, may be present with a strong inter-individual variability. Importantly, these variations cannot be solely explained by the different pathogenic mutations which cause the disease, as different clinical and pathological features often characterize members of the same family.Citation45 Either genetic or environmental factors might play a role in driving different neuropathological pathways by i) affecting the pathological consequences of PrP misfolding, i.e., the neurotoxicity of PrPSc, or ii) affecting PrP misfolding itself, leading to different PrPSc conformers, i.e. different strains, which in turn elicit distinct neuropathological cascades. The findings that different PrPSc conformers can be associated with different GSS cases (), and that cases associated with different PrPSc conformers may induce different phenotypes of disease in voles (), support the latter hypothesis. In our experiments with voles we have found that most, but not all, GSS transmissions resulted in vole PrPSc with conformational properties similar to the human PrPSc in the inoculum, clearly suggesting that the human PrPSc conformation acted by templating vole PrPC in a likeness of itself. A convincing paradigm was provided by the transmission of 2 different brain areas from the same P102L GSS case (case #6 in ref. 40). This case was peculiar because 2 distinct isoforms of PrPSc accumulated in the cerebral cortex, namely 8 kDa and 21 kDa PrPres, but only one of them, 8 kDa PrPres, was found in other brain areas such as the cerebellum. Upon transmission in voles, the cerebellar inoculum induced 8 kDa PrPres only, while the frontal cortex inoculum induced 2 distinct phenotypes, with 8 kDa PrPres in an individual vole after a short survival period, and of 21 kDa PrPres in the remaining voles after much longer survival times. This finding strongly suggests that PrPSc conformers associated with 21 kDa or 8 kDa PrPres might behave as independent transmissible PrPSc species. Most importantly, the 2 PrPSc isoforms were accompanied by distinct pathologies (). In voles with 8 kDa PrPres spongiform degeneration was prominent in white matter tracts, and was accompanied by coarse and granular PrPSc deposits. In contrast voles with 21 kDa PrPres had severe spongiform degeneration in several areas of gray matter, accompanied by synaptic and punctuate PrPSc deposits and, in some voles, by florid plaques. As a similar association between 21 kDa PrPres and spongiform degeneration with synaptic PrPSc deposits has been previously reported in P102L GSS cases,Citation5 it is tempting to speculate that different GSS strains, encoded by distinct PrPSc isoforms, could cause at least in part the pathological and clinical heterogeneity observed in GSS cases. Ongoing experiments of subpassage in Bv109I and transmission of these GSS cases in other susceptible animal models will establish if different GSS strains can be isolated upon experimental transmission in animal models.
Can Atypical PrPSc with ∼8 kDa Protease-Resistant Core Advance Our Understanding of Infectious Prions?
Previous findings that GSS forms with only 8 kDa PrPres were characterized by low or absent spongiform degeneration and by lack of transmissibility in mice led to hypothesize that this atypical PrPSc could be endowed with low neurotoxicity and/or low infectivity. Our findings challenge this view, as they show that the intracerebral inoculation of A117V, F198S and P102L GSS cases with only 8 kDa PrPres induced in voles seeded misfolding, spongiform degeneration, fatal neurological disease and the replication of a TSE infectious agent. Notably, the brain of recipient voles contained only 8 kDa PrPres, which argues for atypical PrPSc as being the misfolded conformer responsible for neurotoxicity and infectivity. Similarly, it has been recently shown that transgenic mice expressing bank vole PrP carrying isoleucine at codon 109 spontaneously develop neurodegeneration and fatal neurological disease accompanied by the accumulation of infectious prions and atypical, GSS-like, PrPSc conformers.Citation44 Evidences supporting this view also derive from atypical scrapie or Nor98, a natural TSE of sheep and goats. Atypical scrapie has a sporadic occurrence and is mainly diagnosed in old animals carrying rather rare polymorphic PrP variants.Citation46 PrPSc from atypical scrapie is characterized by GSS-like N-terminal and C-terminal truncated PK-resistant PrP fragments.Citation8 Still, brain tissue from animals with atypical scrapie is characterized by prominent cortical and cerebellar spongiform degenerationCitation47 and contain infectious prions which are experimentally transmissible in natural hostsCitation47,48 and transgenic mice.Citation49,50,51,52
These data cannot however exclude that other PrPSc conformers, sensitive to proteases, might have played a role in driving the observed phenotypes. Indeed, the identification of PrPSc is usually accomplished by treatment with PK of brain homogenates, and the isolation of native PrPSc conformers has not been routinely attempted. Thus, definitive proof of the infectious and/or neurotoxic roles of 8 kDa PrPres conformers is still lacking, and could be experimentally pursued by studies of infectivity in PK-treated brain homogenates.
The identification of the 3-dimensional structure of infectious PrPSc conformers is one of the major unmet needs in the prion field. The insoluble, non-crystalline nature of PrPSc precluded so far to determine their structure by high resolution methods such as NMR and crystallography. Current structural models of PrPSc mainly derived their constraints from data obtained with a variety of techniques, such as electron microscopy, circular dicroism, Fourier transform infrared spectroscopy, X-ray fiber diffraction and hydrogen/deuterion exchange among the others.Citation53 Finally, attempts to obtain pure preparations of synthetic prions failed so far to provide highly infectious material. Further studies with 8 kDa PrPres will hopefully lead to the purification of atypical PrPres aggregates associated with high levels of infectivity and made up of short, nonglycosylated and non GPI-anchored PrP fragments, which might represent a convenient “miniprion” for structural studies, potentially able to overcome some of the experimental obstacles posed by the highly glycosylated and GPI-anchored conventional PrPSc conformers.
Concluding Remarks
Following the lack of replication of a TSE infectious agent in mouse models of GSS, it has been recently proposed that there are three potential pathways associated with transmission of protein misfolding, resulting in i) neurodegenerative disease and replication of an infectious agent; ii) the induction of a non-infectious proteinopathy with toxic effects in the brain; and iii) the seeding of protein misfolding that is neither infectious nor toxic.Citation54 The finding that cases of GSS which are not obviously transmissible in transgenic mice carrying the homologous PrP could be easily transmitted in wild type voles calls for caution in ascribing solely to the PrPSc conformers in the donor tissue the different proposed degrees of transmissibility. Indeed our findings imply that these different degrees of transmissibility might rather represent the outcome of the interaction of the pathological tissues with the recipient host species. This suggests that the boundary between experimental transmissibility of disease and mere induction of misfolding might be subtle. We believe that these evidences might put in a new perspective the ongoing debate on the nature of other prion-like neurodegenerative diseases, such as Alzheimer disease and Parkinson disease, for which it has been difficult so far to demonstrate the transmissibility of the neurodegenerative condition per se, including the induction of protein misfolding and of the associated neurodegenerative changes leading to clinical disease and death.Citation55
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
REFERENCES
- Head MW. Human prion diseases: molecular, cellular and population biology. Neuropathology 2013; 33:221-36; PMID:23331517; http://dx.doi.org/10.1111/neup.12016
- Hsiao K, Baker HF, Crow TJ, Poulter M, Owen F, Terwilliger JD, Westaway D, Ott J, Prusiner SB. Linkage of a prion protein missense variant to Gerstmann-Straussler syndrome. Nature 1989; 338:342-5; PMID:2564168; http://dx.doi.org/10.1038/338342a0
- Schmitz M, Dittmar K, Llorens F, Gelpi E, Ferrer I, Schulz-Schaeffer WJ, Zerr I. Hereditary human prion diseases: an update. Mol Neurobiol 2016; http://dx.doi.org/10.1007/s12035-016-9918-y
- Ghetti B, Tagliavini F, Takao M, Bugiani O, Piccardo P. Hereditary prion protein amyloidoses. Clin Lab Med 2003; 23:65-85, viii; PMID:12733425; http://dx.doi.org/10.1016/S0272-2712(02)00064-1
- Parchi P, Chen SG, Brown P, Zou W, Capellari S, Budka H, Hainfellner J, Reyes PF, Golden GT, Hauw JJ, et al. Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann-Straussler-Scheinker disease. Proc Natl Acad Sci U S A 1998; 95:8322-7; PMID:9653185; http://dx.doi.org/10.1073/pnas.95.14.8322
- Piccardo P, Dlouhy SR, Lievens PM, Young K, Bird TD, Nochlin D, Dickson DW, Vinters HV, Zimmerman TR, Mackenzie IR, et al. Phenotypic variability of Gerstmann-Straussler-Scheinker disease is associated with prion protein heterogeneity. J Neuropathol Exp Neurol 1998; 57:979-88; PMID:9786248; http://dx.doi.org/10.1097/00005072-199810000-00010
- Piccardo P, Liepnieks JJ, William A, Dlouhy SR, Farlow MR, Young K, Nochlin D, Bird TD, Nixon RR, Ball MJ, et al. Prion proteins with different conformations accumulate in Gerstmann-Straussler-Scheinker disease caused by A117V and F198S mutations. Am J Pathol 2001; 158:2201-7; PMID:11395398; http://dx.doi.org/10.1016/S0002-9440(10)64692-5
- Pirisinu L, Nonno R, Esposito E, Benestad SL, Gambetti P, Agrimi U, Zou WQ. Small ruminant nor98 prions share biochemical features with human gerstmann-straussler-scheinker disease and variably protease-sensitive prionopathy. PLoS One 2013; 8:e66405; PMID:23826096; http://dx.doi.org/10.1371/journal.pone.0066405
- Hsiao KK, Scott M, Foster D, Groth DF, DeArmond SJ, Prusiner SB. Spontaneous neurodegeneration in transgenic mice with mutant prion protein. Science 1990; 250:1587-90; PMID:1980379; http://dx.doi.org/10.1126/science.1980379
- Hsiao KK, Groth D, Scott M, Yang SL, Serban H, Rapp D, Foster D, Torchia M, Dearmond SJ, Prusiner SB. Serial transmission in rodents of neurodegeneration from transgenic mice expressing mutant prion protein. Proc Natl Acad Sci U S A 1994; 91:9126-30; PMID:7916462; http://dx.doi.org/10.1073/pnas.91.19.9126
- Telling GC, Haga T, Torchia M, Tremblay P, DeArmond SJ, Prusiner SB. Interactions between wild-type and mutant prion proteins modulate neurodegeneration in transgenic mice. Genes Dev 1996; 10:1736-50; PMID:8698234; http://dx.doi.org/10.1101/gad.10.14.1736
- Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, DeArmond SJ, Prusiner SB. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 1995; 83:79-90; PMID:7553876; http://dx.doi.org/10.1016/0092-8674(95)90236-8
- Nazor KE, Kuhn F, Seward T, Green M, Zwald D, Purro M, Schmid J, Biffiger K, Power AM, Oesch B, et al. Immunodetection of disease-associated mutant PrP, which accelerates disease in GSS transgenic mice. EMBO J 2005; 24:2472-80; PMID:15962001; http://dx.doi.org/10.1038/sj.emboj.7600717
- Kaneko K, Ball HL, Wille H, Zhang H, Groth D, Torchia M, Tremblay P, Safar J, Prusiner SB, DeArmond SJ, et al. A synthetic peptide initiates Gerstmann-Straussler-Scheinker (GSS) disease in transgenic mice. J Mol Biol 2000; 295:997-1007; PMID:10656806; http://dx.doi.org/10.1006/jmbi.1999.3386
- Tremblay P, Ball HL, Kaneko K, Groth D, Hegde RS, Cohen FE, DeArmond SJ, Prusiner SB, Safar JG. Mutant PrPSc conformers induced by a synthetic peptide and several prion strains. J Virol 2004; 78:2088-99; PMID:14747574; http://dx.doi.org/10.1128/JVI.78.4.2088-2099.2004
- Manson JC, Jamieson E, Baybutt H, Tuzi NL, Barron R, McConnell I, Somerville R, Ironside J, Will R, Sy MS, et al. A single amino acid alteration (101L) introduced into murine PrP dramatically alters incubation time of transmissible spongiform encephalopathy. EMBO J 1999; 18:6855-64; PMID:10581259; http://dx.doi.org/10.1093/emboj/18.23.6855
- Asante EA, Gowland I, Grimshaw A, Linehan JM, Smidak M, Houghton R, Osiguwa O, Tomlinson A, Joiner S, Brandner S, et al. Absence of spontaneous disease and comparative prion susceptibility of transgenic mice expressing mutant human prion proteins. J Gen Virol 2009; 90:546-58; PMID:19218199; http://dx.doi.org/10.1099/vir.0.007930-0
- Torres JM, Castilla J, Pintado B, Gutiérrez-Adan A, Andréoletti O, Aguilar-Calvo P, Arroba AI, Parra-Arrondo B, Ferrer I, Manzanares J, Espinosa JC. Spontaneous generation of infectious prion disease in transgenic mice. Emerg Infect Dis 2013; 19:1938-47; PMID:24274622; http://dx.doi.org/10.3201/eid1912.130106
- Yang W, Cook J, Rassbach B, Lemus A, DeArmond SJ, Mastrianni JA. A New Transgenic Mouse Model of Gerstmann-Straussler-Scheinker Syndrome Caused by the A117V Mutation of PRNP. J Neurosci 2009; 29:10072-80; PMID:19675240; http://dx.doi.org/10.1523/JNEUROSCI.2542-09.2009
- Asante EA, Linehan JM, Smidak M, Tomlinson A, Grimshaw A, Jeelani A, Jakubcova T, Hamdan S, Powell C, Brandner S, et al. Inherited prion disease A117V is not simply a proteinopathy but produces prions transmissible to transgenic mice expressing homologous prion protein. PLoS Pathog 2013; 9:e1003643; PMID:24086135; http://dx.doi.org/10.1371/journal.ppat.1003643
- Chiesa R, Piccardo P, Ghetti B, Harris DA. Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron 1998; 21:1339-51; PMID:9883727; http://dx.doi.org/10.1016/S0896-6273(00)80653-4
- Chiesa R, Piccardo P, Quaglio E, Drisaldi B, Si-Hoe SL, Takao M, Ghetti B, Harris DA. Molecular distinction between pathogenic and infectious properties of the prion protein. J Virol 2003; 77:7611-22; PMID:12805461; http://dx.doi.org/10.1128/JVI.77.13.7611-7622.2003
- Stöhr J, Watts JC, Legname G, Oehler A, Lemus A, Nguyen HO, Sussman J, Wille H, DeArmond SJ, Prusiner SB, Giles K. Spontaneous generation of anchorless prions in transgenic mice. Proc Natl Acad Sci U S A 2011; 108:21223-8; PMID:22160704; http://dx.doi.org/10.1073/pnas.1117827108
- Masters CL, Gajdusek DC, Gibbs CJ, Jr. Creutzfeldt-Jakob disease virus isolations from the Gerstmann-Straussler syndrome with an analysis of the various forms of amyloid plaque deposition in the virus-induced spongiform encephalopathies. Brain 1981; 104:559-88; PMID:6791762; http://dx.doi.org/10.1093/brain/104.3.559
- Baker HF, Duchen LW, Jacobs JM, Ridley RM. Spongiform encephalopathy transmitted experimentally from Creutzfeldt-Jakob and familial Gerstmann-Straussler-Scheinker diseases. Brain 1990; 113(Pt 6):1891-909; PMID:2276050; http://dx.doi.org/10.1093/brain/113.6.1891
- Manuelidis EE, Fritch WW, Kim JH, Manuelidis L. Immortality of cell cultures derived from brains of mice and hamsters infected with Creutzfeldt-Jakob disease agent. Proc Natl Acad Sci U S A 1987; 84:871-5; PMID:3543937; http://dx.doi.org/10.1073/pnas.84.3.871
- Tateishi J, Kitamoto T, Hoque MZ, Furukawa H. Experimental transmission of Creutzfeldt-Jakob disease and related diseases to rodents. Neurology 1996; 46:532-7; PMID:8614527; http://dx.doi.org/10.1212/WNL.46.2.532
- Hsiao K, Dlouhy SR, Farlow MR, Cass C, Da Costa M, Conneally PM, Hodes ME, Ghetti B, Prusiner SB. Mutant prion proteins in Gerstmann-Straussler-Scheinker disease with neurofibrillary tangles. Nat Genet 1992; 1:68-71; PMID:1363810; http://dx.doi.org/10.1038/ng0492-68
- Gambetti P, Petersen RB, Parchi P, Chen SG, Capellari S, Goldfarb L, Gabizon R, Montagna P, Lugaresi E, Piccardo P, et al. Inherited prion diseases. In: Prusiner SB, ed., Prion biology diseases. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 1999; 509–583.
- Tateishi J, Kitamoto T, Doh-ura K, Sakaki Y, Steinmetz G, Tranchant C, Warter JM, Heldt N. Immunochemical, molecular genetic, and transmission studies on a case of Gerstmann-Straussler-Scheinker syndrome. Neurology 1990; 40:1578-81; PMID:1699173; http://dx.doi.org/10.1212/WNL.40.10.1578
- Piccardo P, Manson JC, King D, Ghetti B, Barron RM. Accumulation of prion protein in the brain that is not associated with transmissible disease. Proc Natl Acad Sci U S A 2007; 104:4712-7; PMID:17360589; http://dx.doi.org/10.1073/pnas.0609241104
- Gambetti P, Dong Z, Yuan J, Xiao X, Zheng M, Alshekhlee A, Castellani R, Cohen M, Barria MA, Gonzalez-Romero D, et al. A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol 2008; 63:697-708; PMID:18571782; http://dx.doi.org/10.1002/ana.21420
- Notari S, Xiao X, Espinosa JC, Cohen Y, Qing L, Aguilar-Calvo P, Kofskey D, Cali I, Cracco L, Kong Q, et al. Transmission characteristics of variably protease-sensitive prionopathy. Emerg Infect Dis 2014; 20:2006-14; PMID:25418590; http://dx.doi.org/10.3201/eid2012.140548
- Nonno R, Di Bari MA, Cardone F, Vaccari G, Fazzi P, Dell'Omo G, Cartoni C, Ingrosso L, Boyle A, Galeno R, et al. Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog 2006; 2:e12; PMID:16518470; http://dx.doi.org/10.1371/journal.ppat.0020012
- Di Bari MA, Chianini F, Vaccari G, Esposito E, Conte M, Eaton SL, Hamilton S, Finlayson J, Steele PJ, Dagleish MP, et al. The bank vole (Myodes glareolus) as a sensitive bioassay for sheep scrapie. J Gen Virol 2008; 89:2975-85; PMID:19008382; http://dx.doi.org/10.1099/vir.0.2008/005520-0
- Di Bari MA, Nonno R, Castilla J, D'Agostino C, Pirisinu L, Riccardi G, Conte M, Richt J, Kunkle R, Langeveld J, et al. Chronic wasting disease in bank voles: characterisation of the shortest incubation time model for prion diseases. PLoS Pathog 2013; 9:e1003219; PMID:23505374; http://dx.doi.org/10.1371/journal.ppat.1003219
- Watts JC, Giles K, Patel S, Oehler A, DeArmond SJ, Prusiner SB. Evidence that bank vole PrP is a universal acceptor for prions. PLoS Pathog 2014; 10:e1003990; PMID:24699458; http://dx.doi.org/10.1371/journal.ppat.1003990
- Orru CD, Groveman BR, Raymond LD, Hughson AG, Nonno R, Zou W, Ghetti B, Gambetti P, Caughey B. Bank Vole Prion Protein As an Apparently Universal Substrate for RT-QuIC-Based Detection and Discrimination of Prion Strains. PLoS Pathog 2015; 11:e1004983; PMID:26086786; http://dx.doi.org/10.1371/journal.ppat.1004983
- Cartoni C, Schinina ME, Maras B, Nonno R, Vaccari G, Di Baria MA, Conte M, Liu QG, Lu M, Cardone F, et al. Identification of the pathological prion protein allotypes in scrapie-infected heterozygous bank voles (Clethrionomys glareolus) by high-performance liquid chromatography-mass spectrometry. J Chromatogr A 2005; 1081:122-6; PMID:16013608; http://dx.doi.org/10.1016/j.chroma.2005.04.035
- Pirisinu L, Di Bari MA, D'Agostino C, Marcon S, Riccardi G, Poleggi A, Cohen ML, Appleby BS, Gambetti P, Ghetti B, et al. Gerstmann-Straussler-Scheinker disease subtypes efficiently transmit in bank voles as genuine prion diseases. Sci Rep 2016; 6:20443; PMID:26841849; http://dx.doi.org/10.1038/srep20443
- Bruce M, Chree A, McConnell I, Foster J, Pearson G, Fraser H. Transmission of bovine spongiform encephalopathy and scrapie to mice: strain variation and the species barrier. Philos Trans R Soc Lond B Biol Sci 1994; 343:405-11; PMID:7913758; http://dx.doi.org/10.1098/rstb.1994.0036
- Kurt TD, Jiang L, Fernandez-Borges N, Bett C, Liu J, Yang T, Spraker TR, Castilla J, Eisenberg D, Kong Q, et al. Human prion protein sequence elements impede cross-species chronic wasting disease transmission. J Clin Invest 2015; 125:2548; PMID:25961458; http://dx.doi.org/10.1172/JCI82647
- Watts JC, Giles K, Stohr J, Oehler A, Bhardwaj S, Grillo SK, Patel S, DeArmond SJ, Prusiner SB. Spontaneous generation of rapidly transmissible prions in transgenic mice expressing wild-type bank vole prion protein. Proc Natl Acad Sci U S A 2012; 109:3498-503; PMID:22331873; http://dx.doi.org/10.1073/pnas.1121556109
- Watts JC, Giles K, Bourkas ME, Patel S, Oehler A, Gavidia M, Bhardwaj S, Lee J, Prusiner SB. Towards authentic transgenic mouse models of heritable PrP prion diseases. Acta Neuropathol 2016; 132:593–610; PMID:27350609; http://dx.doi.org/10.1007/s00401-016-1585-6
- Hainfellner JA, Brantner-Inthaler S, Cervenakova L, Brown P, Kitamoto T, Tateishi J, Diringer H, Liberski PP, Regele H, Feucht M, et al. The original Gerstmann-Straussler-Scheinker family of Austria: divergent clinicopathological phenotypes but constant PrP genotype. Brain Pathol 1995; 5:201-11; PMID:8520719; http://dx.doi.org/10.1111/j.1750-3639.1995.tb00596.x
- Benestad SL, Arsac JN, Goldmann W, Noremark M. Atypical/Nor98 scrapie: properties of the agent, genetics, and epidemiology. Vet Res 2008; 39:19; PMID:18187032; http://dx.doi.org/10.1051/vetres:2007056
- Simmons MM, Konold T, Simmons HA, Spencer YI, Lockey R, Spiropoulos J, Everitt S, Clifford D. Experimental transmission of atypical scrapie to sheep. BMC Vet Res 2007; 3:20; PMID:17725818; http://dx.doi.org/10.1186/1746-6148-3-20
- Simmons MM, Moore SJ, Konold T, Thurston L, Terry LA, Thorne L, Lockey R, Vickery C, Hawkins SA, Chaplin MJ, et al. Experimental oral transmission of atypical scrapie to sheep. Emerg Infect Dis 2011; 17:848-54; PMID:21529394; http://dx.doi.org/10.3201/eid1705.101654
- Le Dur A, Beringue V, Andreoletti O, Reine F, Lai TL, Baron T, Bratberg B, Vilotte JL, Sarradin P, Benestad SL, et al. A newly identified type of scrapie agent can naturally infect sheep with resistant PrP genotypes. Proc Natl Acad Sci U S A 2005; 102:16031-6; PMID:16239348; http://dx.doi.org/10.1073/pnas.0502296102
- Arsac JN, Betemps D, Morignat E, Feraudet C, Bencsik A, Aubert D, Grassi J, Baron T. Transmissibility of atypical scrapie in ovine transgenic mice: major effects of host prion protein expression and donor prion genotype. PloS One 2009; 4:e7300; PMID:19806224; http://dx.doi.org/10.1371/journal.pone.0007300
- Griffiths PC, Spiropoulos J, Lockey R, Tout AC, Jayasena D, Plater JM, Chave A, Green RB, Simonini S, Thorne L, et al. Characterization of atypical scrapie cases from Great Britain in transgenic ovine PrP mice. J Gen Virol 2010; 91:2132-8; PMID:20392900; http://dx.doi.org/10.1099/vir.0.018986-0
- Espinosa JC, Herva ME, Andreoletti O, Padilla D, Lacroux C, Cassard H, Lantier I, Castilla J, Torres JM. Transgenic mice expressing porcine prion protein resistant to classical scrapie but susceptible to sheep bovine spongiform encephalopathy and atypical scrapie. Emerg Infect Dis 2009; 15:1214-21; PMID:19751582; http://dx.doi.org/10.3201/eid1508.081218
- Requena JR, Wille H. The structure of the infectious prion protein: experimental data and molecular models. Prion 2014; 8:60-6; PMID:24583975; http://dx.doi.org/10.4161/pri.28368
- Barron RM, King D, Jeffrey M, McGovern G, Agarwal S, Gill AC, Piccardo P. PrP aggregation can be seeded by pre-formed recombinant PrP amyloid fibrils without the replication of infectious prions. Acta Neuropathol 2016; 132:611–624; PMID:27376534; http://dx.doi.org/10.1007/s00401-016-1594-5
- Beekes M, Thomzig A, Schulz-Schaeffer WJ, Burger R. Is there a risk of prion-like disease transmission by Alzheimer- or Parkinson-associated protein particles? Acta Neuropathol 2014; 128:463-76; PMID:25073522; http://dx.doi.org/10.1007/s00401-014-1324-9