
ABSTRACT
A misfolded version of the prion protein represents an essential component in the pathophysiology of fatal neurodegenerative prion diseases, which affect humans and animals alike. They may be of sporadic origin, acquired through exogenous introduction of infectious misfolded prion protein, or caused by genetic alterations in the prion protein coding gene. We have recently described a novel pathway linking retention of mutant prion protein in the early secretory pathway to activation p38-MAPK and a neurodegenerative phenotype in transgenic mice. Here we review the consequences that mutations in prion protein have on intracellular transport and stress responses focusing on protein quality control. We also discuss the neurotoxic signaling elicited by the accumulation of mutant prion protein in the endoplasmic reticulum and the Golgi apparatus. Improved knowledge about these processes will help us to better understand complex pathogenesis of prion diseases, a prerequisite for therapeutic strategies.
INTRODUCTION
The prion protein (PrPC) is a glycosylphosphatidylinositol (GPI)-anchored protein located at the plasma membrane in lipid-enriched microdomains commonly known as lipid rafts or detergent resistant membranes.Citation1 A plethora of functions ranging from neurodevelopment to neuroprotection have been proposed for PrPC but the fact that it is enriched at the neuronal synapse suggests a potential role in neuromodulation, possibly by its involvement in neuronal signal transduction cascades.Citation2,3 In its misfolded state, it plays a fundamental role in neurodegenerative prion diseases.Citation4,5 In humans, prion diseases can be sporadic, acquired or genetic.Citation6 In the latter, mutations in the PRNP gene encoding the prion protein lead to a broad spectrum of prion diseases with a wide variation in penetrance.Citation7,8 Interestingly, PRNP mutations present in the structured C-terminal domain of PrPC may result in either a fully penetrant prion disease (E200K), a dementia clinically not resembling a prion disease (R208C) or a variant rarely associated with dementia (R208H).Citation8 Some mutations leading to genetic human prion diseases are also present in the C-terminal signal sequence for the GPI-anchor attachment, implying that the GPI-anchor signal sequence itself, though not being present in the mature protein, can also play a role in neurodegeneration.Citation9
Plasma membrane attachment via a GPI-anchor is a unique and evolutionary highly conserved mean to bestow lipid raft localization. Thus, GPI-anchored proteins are able to carry out specific functions related to signal transduction and membrane trafficking.Citation10 The GPI-anchor is composed of a highly conserved glycan core of three mannose residues, a glucosamine and a phosphatidylinositol (PI) group attached en block through an amide bond to the ethanolamine phosphate of the nascent protein at the ER. This core GPI-anchor undergoes several remodeling steps through the passage to the ER and Golgi with the protein finally localizing in lipid rafts at the outer leaflet of the plasma membrane.
Protein homeostasis or proteostasis is fundamental for the viability of cells. It is estimated that about 10% of total PrPC is improperly folded when synthesizedCitation11 and, therefore, strict protein quality control (PQC) is of outstanding importance. In the case of secreted and membrane proteins, quality control takes place at the ER (ERQC) and Golgi apparatus.Citation12 Misfolded proteins are retained at the ER and those that repeatedly fail to fold properly are directed to the cytosol where they are ubiquitinated to be degraded by the proteasome (the so-called ER-associated degradation (ERAD) pathway).Citation13 Regarding degradation, GPI-anchored proteins can behave differently than other membrane proteins. In some instances, misfolded GPI-anchored proteins are not targeted to ERAD but seem to follow alternative PQC pathways instead, such as the rapid ER stress-induced export (RESET) pathway.Citation14 Our own studies investigating a PrP version with a C-terminal deletion indicate that in stress conditions, either artificially induced in cell culture or caused by age-associated decay, this pathway cannot be fully executed, and leads to activation of p38-MAP kinase with subsequent neurodegeneration.Citation15,16 Here we briefly summarize new findings related to PQC of GPI-anchored proteins with special attention to PrPC and the implicated signaling pathways that could play a role in neurodegeneration.
Quality Control at the ER
Once the protein folding capacity of the ER is overwhelmed, misfolded proteins accumulate in the ER lumen and induce ER stress. This activates the unfolded protein response (UPR), a signaling network that tries to re-establish homeostasis by increasing the folding capacity, transiently attenuating the translation of proteins while up-regulating ERAD components.Citation17,18 If homeostasis cannot be restored, ER stress becomes chronic leading to cell death. Investigations of ER stress, UPR and ERAD in prion disease is an active field of research and has been extensively reviewed.Citation18,19 It has been shown that PrPC and some mutants of PrP (e.g. PrP with an unprocessed GPI-anchor or PrP expressing the D177N mutation) follow the ERAD pathway.Citation11,20-22 Moreover, ERAD and the proteasome system influence prion diseases as evidenced by the fact that variations in the hectd2 gene (encoding E3 ligases that targets proteins to the proteasome) are linked to alterations in prion disease incubation time in mice and humans.Citation23 UPR activation can likewise participate in the progression of prion disease in miceCitation24-26 although this issue remains controversial since in other instances markers of the UPR could not be observed in human prion diseases and prion disease mouse models.Citation27,28 More recently, it has been shown in vitro that expression of PrP mutants disables retrotranslocation not only of PrPC but also of other ERAD substrates but does not lead to activation of the UPR.Citation29 As described elsewhere, induction of the UPR pathway in disease is complex and context-dependent.Citation18,30
Apart from the variability of the different models and the ways of inducing ER stress, it seems that the cell possesses several pathways to ensure elimination of misfolded PrP (and probably other GPI-anchored proteins as well). Nevertheless, it has been repeatedly shown that certain PrP mutants can escape this ERQCCitation31,32 and, more recently, Ashok et al. observed that when they expressed different PrPC mutants bearing C-terminal mutations found in human prion diseases, ERQC was bypassed.Citation20,33 They demonstrated that GPI-anchored mutants could passage from ER to Golgi albeit being obviously misfolded. Once at the Golgi there was a rerouting to lysosomes where proteins were finally degraded.Citation33 In a more detailed study, Satpute-Krishnan et al.Citation14 described the novel RESET pathway, in which GPI-anchored PrP lacking its disulfide bond follows the secretory pathway, transiently reaching the plasma membrane to finally get degraded in the lysosome. Calnexin was identified as the retention factor to the ER and Tmp21 as the receptor that mediates the egress from ER to Golgi. Factors facilitating the transport to the plasma membrane and from there to the lysosome are currently unknown. The fact that a PrP mutant (H187R) with an unprocessed GPI-anchor is degraded via ERAD while the same protein with a functional GPI-anchor is degraded in the lysosomeCitation20 made the authors hypothesize that the GPI-anchor could sterically impair the ERAD pathway. This question has recently been addressed by Sikorska et al.Citation34 When a mutated version of the GPI-anchored Gas1 (Gas1*) was expressed in a cell line, in which lipid remodeling of the GPI-anchor is blocked, Gas1* underwent ERAD degradation, whereas when the lipid remodeling and maturation of the GPI-anchor was complete, ER-to-Golgi transport was rescued. Thus, in the case of misfolded GPI-anchored proteins it is the presence of a lipid remodeled GPI-anchor that prevents the ERAD pathway and promotes an alternative quality control pathway (such as RESET).
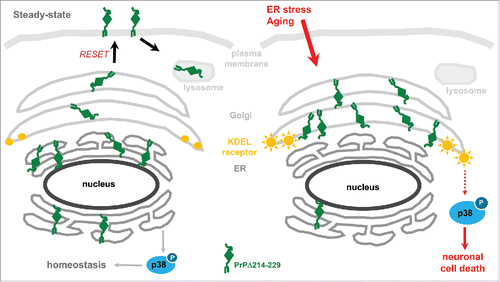
Our own studies demonstrated that misfolded GPI-anchored PrP is directed to the secretory pathway in vivo and that, under stress conditions (in cells) or aging (in transgenic mice), it leads to the activation of neurotoxic signaling cascades.Citation15 Aged transgenic mice expressing PrP partially lacking the C-terminal portion (Tg(PrPΔ214-229)) presented with a fatal neurodegenerative disease. Neuropathological analysis showed neuronal loss especially in the hippocampus and cerebellum and, strikingly, we could observe a considerable concomitant activation of p38-MAPK without obvious activation of UPR pathways. In a cell model expressing this C-terminally deleted PrP, we observed that, at steady state, PrPΔ214-229 was mainly retained at the ER and that, under stress conditions, it reached the Golgi where it remained, not being able to travel further. The latter retention at the Golgi led to p38-MAPK activation. As shown in , we hypothesize that PrPΔ214-229 follows the RESET pathway under steady-state conditions. Under ER stress or during aging, however, PrPΔ214-229 accumulates at the Golgi, which results in activation of p38-MAPK and cell death.
FIGURE 1. Schematic representation illustrating mechanisms of the neurotoxic effect of PrPΔ214-229. (A) In steady-state conditions, PrPΔ214-229 is found mainly at the ER but also slightly at the Golgi and plasma membrane probably following the RESET pathway. In our cell model (but not in the transgenic mice) we see activation of p38-MAPK at steady state levels. We hypothesize that, in this case, this activation leads to protein homeostasis. (B) After applying external stress conditions to cells or, in the case of Tg(PrPΔ214-229) mice due to aging, PrPΔ214-229 leaves the ER in order to follow the RESET pathway for degradation but, due to the mutation, is retained at the Golgi. We hypothesize that this leads to activation of Golgi-resident receptors (e.g., KDEL receptor) leading to increased p38-MAPK phosphorylation and cell death.
Other mouse models of genetic human prion diseases showing retention of mutant PrP at the ER or Golgi have been described that likewise did not present with activation of the UPR. For example, Tg(PG14) mice expressing PrP with a N-terminal octapeptide insertionCitation35-38 or other transgenic mouse models for fatal familial insomnia (FFI; Tg(FFI))Citation39 showed retention at the secretory pathway and decreased plasma membrane expression without signs of UPR activation. Remarkably, in these transgenic mice, mutant PrP accumulates in different organelles of the secretory pathway supporting the idea that phenotypic variation seen in genetic prion diseases could be caused by organelle-specific retention of mutant PrP.Citation40,41
How could ER and Golgi retention lead to cell death in these mice? In Tg(PG14) mice mentioned above, cell death has been linked to a defect in Ca2+ homeostasis caused by inefficient membrane delivery of voltage-gated calcium channels promoted by retention of the mutant PrP.Citation42 In our study, where PrPΔ214-229 is retained at the ER, we did not investigate Ca2+ dynamics or delivery of voltage-gated calcium channels to membranes. Thus, it is conceivable that alterations in these aspects and transport deficit of other proteins at the secretory pathway may contribute to neurodegeneration in our mice. In addition, it would be interesting to investigate if Tg(PG14) or Tg(FFI) mice likewise present with activation of p38-MAPK seen in our PrPΔ214-229 mouse model.
Golgi Quality Control
Why is PrPΔ214-229 retained at the Golgi? How can retention of PrPΔ214-229 at the Golgi lead to cell death? The Golgi apparatus plays a central role as a sorting station and in post-translational modification of cargos in the secretory pathway. But it has recently become obvious that the Golgi apparatus has additional functions, such as quality control and in integrating signaling events.Citation43
Golgi quality control is clearly less understood than ERQC. The fact that mutations in the C-terminal part of PrP cause lysosomal degradation lead to the concept that the Golgi apparatus behaves as a quality control organelle for GPI-anchored proteins (as shown previously for some yeast proteins or for the incomplete T-cell receptor complex in eukaryotes).Citation33,44 Golgi quality control can route misfolded proteins either back to the ER or forward to the endosomal system for degradation.Citation45 Hence it has been demonstrated that non-remodeled GPI-anchors that reach the Golgi are redirected to the ER by the p24 cargo receptor complexCitation46 and misfolded proteins with a strong ER exit signal or lacking determinants recognizable by the ERQC (in the case of transmembrane proteins) can bypass ERAD and become a substrate for Golgi quality control leading to lysosomal degradation.Citation47 It is noteworthy that one principle of quality control in sorting proteins from the Golgi to the lysosome (among others proposed “signals”Citation45) is based on the state of protein oligomerization/aggregation rather than the exact folding state of a monomeric protein. Thus, for some Golgi-resident proteins, oligomerization leads to lysosomal targeting and subsequent degradation.Citation48 This may have implications for PrPC as, in cell culture, the drug Suramin leads to oligomerization of PrPC at the Golgi and is then re-routed to lysosomes.Citation49,50 Suramin also showed protective effects in prion disease, although the mechanism is unclear. Whether this sorting mechanism also applies for the re-routing of PrPΔ214-229 to lysosomes needs further investigation. In our studies, we observed that the presence of PrPΔ214-229 at the ER and Golgi after induction of cellular stress leads to p38-MAPK activation. This kinase is also activated in aged Tg(PrPΔ214-229) mice. At this point, we can only speculate about the mechanism of p38-MAPK, activation in our mice, but it seems likely that different factors are involved. It is well known that ER stress can activate p38-MAPK, playing an important role in UPR.Citation51 We did not observe activation of UPR in the transgenic mice and in cells we observed that induction of ER stress with Thapsigargin was leading to accumulation of PrPΔ214-229 at the Golgi co-incidental with activation of p38-MAPK. What leads to ER-independent with activation of p38-MAPK? As pointed out above, the Golgi apparatus has several functions as an integrator but also as an initiator of signaling cascades. On the one hand, it has been shown that under severe ER-stress conditions, the KDEL-receptor, a cis-Golgi resident and key regulator of signaling in ER-to-Golgi trafficking,Citation51-54 is able to activate both p38- and JNK-MAPK. Although in the latter studies, p38-MAPK activation was linked to a protective role, this may likely be cell- and context-dependent and, in fact, p38-MAPK activation can also be associated with pro-inflammatory and cell death responses.Citation55,56 On the other hand, it has also been shown that when ER-to-Golgi trafficking is arrested by treatment with Brefeldin A, this leads to activation of p38- and JNK-MAPK and subsequent cell death.Citation57 Whether accumulation of PrPΔ214-229 leads to impaired ER-to-Golgi communication thus leading to p38-MAPK activation, deserves further studies.
Consequences of PrP Accumulation in the Golgi Apparatus
In prion infected mice expressing a PrP-green fluorescent protein fusion protein, accumulation of this protein can be seen in the Golgi apparatus long before onset of prion disease, thus raising the question if this may causally be involved in disease pathogenesis.Citation58 In another mouse model, expression of a transmembrane version of the prion protein leads to accumulation of this protein in the Golgi apparatus and subsequent neurodegeneration.Citation59 This is reminiscent of our Tg(PrPΔ214-229) mice and, as we do, Stewart et al. discuss the possibility that Golgi stress may directly or indirectly be involved in neurotoxicity. Again, it would be interesting to see if p38-MAPK activation may be a common pathway. The issue that prion infection itself may impair post-Golgi trafficking was recently highlighted by Uchiyama et al. when they investigated cell surface expression of PrPC and other neuronal membrane-bound proteins.Citation60 In prion-infected mice, they observed reduced expression of PrPC and other GPI-anchored proteins at the outer leaflet of neuronal membranes while these proteins accumulated in the Golgi well before onset of clinical symptoms. This reinforces the concept that misfolded GPI-anchored proteins use diverse transport routes and raises the question if a Golgi stress response may involve translational shutdown of distinct proteins. The molecular mechanisms underlying defective post-Golgi transport in prion diseases are only now being appreciated. It is suggested that the Rab GDP dissociation inhibitor α (GDI), which regulates the function of those Rabs involved in intracellular vesicular trafficking, might regulate this.Citation61 In fact, cells accumulating mutant PrP at the Golgi upregulate GDI and silencing of GDI rescues post-Golgi transport of reporter proteins.
Implications in Human Prion Diseases and Future Directions
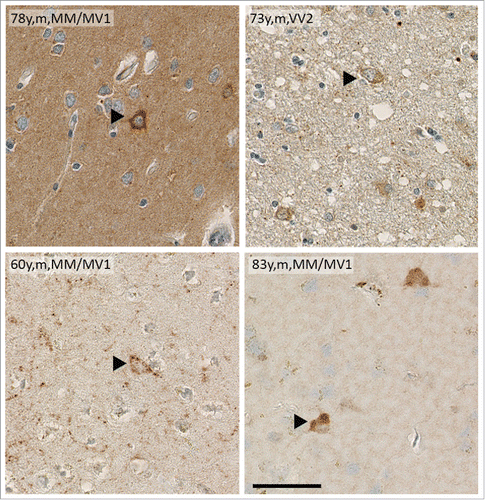
Little is known about the signaling cascades underlying neurodegeneration in human prion diseases.Citation62-64 In sporadic CJD brains, intracellular PrPSc deposits similar to those seen in Tg(PrPΔ214-229) mice can be observed () raising the question if p38-MAPK activation may also be relevant in human prion diseases.
FIGURE 2. Brain samples of sCJD patients show intracellular deposits of PrPSc. Immunohistochemistry of human cortical brain samples of sCJD showing intracellular PrPSc deposition similar to that seen in Tg(PrPΔ214-229). Age, gender and sCJD type are indicated in the picture. Scale bar is 100 μm.
Our knowledge on the molecular basis of prion disease has rapidly grown over the last years and it has become obvious that misfolding of PrPC and the generation of infectious “prions” represent only one out of several facets in the complex pathogenesis of prion diseases. Here we have presented our point of view regarding transport pathways and degradation of misfolded PrP. It seems that in our model misfolded PrP does not lead to UPR induction but rather follows the so-called RESET pathway. However, instead of being degraded in the lysosome, PrPΔ214-229 does not fully complete the RESET pathway but accumulates at the Golgi and leads to activation p38-MAPK and subsequent neurodegeneration. Future work will have to concentrate on establishing if these 2 features are causally linked and if p38-MAPK activation may represent a further mechanistic aspect of Golgi stress.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
ACKNOWLEDGMENTS
The authors would like to apologize to all authors whose important contributions to this field of prion research could not be cited due to space limitations.
FUNDING
The work of the authors is supported by the Deutsche Forschungsgemeinschaft (DFG) (SFB877, project A12 (to MG), GRK1459 (to MG) and by the Werner-Otto-Stiftung, Hamburg, Germany and the Creutzfeldt-Jakob Disease Foundation, Inc. (to HCA).
REFERENCES
- Mayor S, Riezman H. Sorting GPI-anchored proteins. Nat Rev Mol Cell Biol 2004; 5(2):110-20; PMID:15040444; http://dx.doi.org/10.1038/nrm1309
- Aguzzi A, Baumann F, Bremer J. The prion's elusive reason for being. Annu Rev Neurosci 2008; 31:439-77; PMID:18558863; http://dx.doi.org/10.1146/annurev.neuro.31.060407.125620
- Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. Signal transduction through prion protein. Science 2000; 289(5486):1925-8; PMID:10988071; http://dx.doi.org/10.1126/science.289.5486.1925
- Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. Prion protein biology. Cell 1998; 93(3):337-48; PMID:9590169; http://dx.doi.org/10.1016/S0092-8674(00)81163-0
- Aguzzi A, Polymenidou M. Mammalian prion biology: one century of evolving concepts. Cell 2004; 116(2):313-27; PMID:14744440; http://dx.doi.org/10.1016/S0092-8674(03)01031-6
- Geissen M, Krasemann S, Matschke J, Glatzel M. Understanding the natural variability of prion diseases. Vaccine 2007; 25(30):5631-6; PMID:17391814; http://dx.doi.org/10.1016/j.vaccine.2007.02.041
- Kovacs GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, van Duijn C, Collins SJ, Boyd A, Giulivi A, Coulthart M, et al. Genetic prion disease: the EUROCJD experience. Hum Genet 2005; 118(2):166-74; PMID:16187142; http://dx.doi.org/10.1007/s00439-005-0020-1
- Minikel EV, Vallabh SM, Lek M, Estrada K, Samocha KE, Sathirapongsasuti JF, McLean CY, Tung JY, Yu LP, Gambetti P, et al., Quantifying prion disease penetrance using large population control cohorts. Sci Transl Med 2016; 8(322):322ra9; PMID:26791950
- Puig B, Altmeppen H, Glatzel M. The GPI-anchoring of PrP: Implications in sorting and pathogenesis. Prion 2014; 8(1):11-8; PMID:24509692; http://dx.doi.org/10.4161/pri.27892
- Zurzolo C, Simons K. Glycosylphosphatidylinositol-anchored proteins: Membrane organization and transport. Biochim Biophys Acta 2016; 1858(4):632-9; PMID:26706096; http://dx.doi.org/10.1016/j.bbamem.2015.12.018
- Yedidia Y, Horonchik L, Tzaban S, Yanai A, Taraboulos A. Proteasomes and ubiquitin are involved in the turnover of the wild-type prion protein. Embo J 2001 20(19):5383-91; PMID:11574470; http://dx.doi.org/10.1093/emboj/20.19.5383
- Anelli T, Sitia R. Protein quality control in the early secretory pathway. EMBO J 2008; 27(2):315-27; PMID:18216874; http://dx.doi.org/10.1038/sj.emboj.7601974
- Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat Cell Biol 2005; 7(8):766-72; PMID:16056268; http://dx.doi.org/10.1038/ncb0805-766
- Satpute-Krishnan P, Ajinkya M, Bhat S, Itakura E, Hegde RS, Lippincott-Schwartz J. ER stress-induced clearance of misfolded GPI-anchored proteins via the secretory pathway. Cell 2014; 158(3):522-33; PMID:25083867; http://dx.doi.org/10.1016/j.cell.2014.06.026
- Puig B, Altmeppen HC, Ulbrich S, Linsenmeier L, Krasemann S, Chakroun K, Acevedo-Morantes CY, Wille H, Tatzelt J, Glatzel M. Secretory pathway retention of mutant prion protein induces p38-MAPK activation and lethal disease in mice. Sci Rep 2016; 6:24970; PMID:27117504; http://dx.doi.org/10.1038/srep24970
- Schipanski A, Lange S, Segref A, Gutschmidt A, Lomas DA, Miranda E, Schweizer M, Hoppe T, Glatzel M. A novel interaction between ageing and ER overload in a protein conformational dementia. Genetics 2013; 193(3):865-76; PMID:23335331; http://dx.doi.org/10.1534/genetics.112.149088
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011; 334(6059):1081-6; PMID:22116877; http://dx.doi.org/10.1126/science.1209038
- Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci 2014; 15(4):233-49; PMID:24619348; http://dx.doi.org/10.1038/nrn3689
- Hetz CA, Soto C. Stressing out the ER: a role of the unfolded protein response in prion-related disorders. Curr Mol Med 2006; 6(1):37-43; PMID:16472111; http://dx.doi.org/10.2174/156652406775574578
- Ashok A, Hegde RS. Retrotranslocation of prion proteins from the endoplasmic reticulum by preventing GPI signal transamidation. Mol Biol Cell 2008; 19(8):3463-76; PMID:18508914; http://dx.doi.org/10.1091/mbc.E08-01-0087
- Jin T, Gu Y, Zanusso G, Sy M, Kumar A, Cohen M, Gambetti P, Singh N. The chaperone protein BiP binds to a mutant prion protein and mediates its degradation by the proteasome. J Biol Chem 2000; 275(49):38699-704; PMID:10970892; http://dx.doi.org/10.1074/jbc.M005543200
- Ma J, Lindquist S. Wild-type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. Proc Natl Acad Sci U S A 2001; 98(26):14955-60; http://dx.doi.org/10.1073/pnas.011578098
- Lloyd SE, Maytham EG, Pota H, Grizenkova J, Molou E, Uphill J, Hummerich H, Whitfield J, Alpers MP, Mead S, Collinge J. HECTD2 is associated with susceptibility to mouse and human prion disease. PLoS Genet 2009; 5(2):e1000383; http://dx.doi.org/10.1371/journal.pgen.1000383
- Hetz C, Russelakis-Carneiro M, Wälchli S, Carboni S, Vial-Knecht E, Maundrell K, Castilla J, Soto C. The disulfide isomerase Grp58 is a protective factor against prion neurotoxicity. J Neurosci 2005; 25(11):2793-802; PMID:15772339; http://dx.doi.org/10.1523/JNEUROSCI.4090-04.2005
- Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori CA, et al. Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature 2012; 485(7399):507-11; PMID:22622579
- Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA, et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med 2013; 5(206):206ra138; PMID:24107777
- Unterberger U, Höftberger R, Gelpi E, Flicker H, Budka H, Voigtländer T. Endoplasmic reticulum stress features are prominent in Alzheimer disease but not in prion diseases in vivo. J Neuropathol Exp Neurol 2006; 65(4):348-57; PMID:16691116
- Quaglio E, Restelli E, Garofoli A, Dossena S, De Luigi A, Tagliavacca L, Imperiale D, Migheli A, Salmona M, Sitia R, et al. Expression of mutant or cytosolic PrP in transgenic mice and cells is not associated with endoplasmic reticulum stress or proteasome dysfunction. PLoS One 2011; 6(4):e19339; PMID:21559407
- Peters SL, Dery MA, LeBlanc AC. Familial prion protein mutants inhibit Hrd1-mediated retrotranslocation of misfolded proteins by depleting misfolded protein sensor BiP. Hum Mol Genet 2016; 25(5):976-88; PMID:26740554
- Torres M, Matamala JM, Duran-Aniotz C, Cornejo VH, Foley A, Hetz C. ER stress signaling and neurodegeneration: At the intersection between Alzheimer disease and Prion-related disorders. Virus Res 2015; 207:69-75; PMID:25556124
- Kang S, Kim MH, Park IA, Kim JW, Park NH, Kang D, Yoo KY, Kang SB, Lee HP, Song YS. Elevation of cyclooxygenase-2 is related to lymph node metastasis in adenocarcinoma of uterine cervix. Cancer Lett 2006; 237(2):305-11; PMID:16111807
- Drisaldi B, Stewart RS, Adles C, Stewart LR, Quaglio E, Biasini E, Fioriti L, Chiesa R, Harris DA. Mutant PrP is delayed in its exit from the endoplasmic reticulum, but neither wild-type nor mutant PrP undergoes retrotranslocation prior to proteasomal degradation. J Biol Chem 2003; 278(24):21732-43; PMID:12663673
- Ashok A, Hegde RS. Selective processing and metabolism of disease-causing mutant prion proteins. PLoS Pathog 2009; 5(6):e1000479; PMID:19543376
- Sikorska, N, Lemus L, Aguilera-Romero A, Manzano-Lopez J, Riezman H, Muñiz M, Goder V. Limited ER quality control for GPI-anchored proteins. J Cell Biol 2016; 213(6):693-704; PMID:27325793
- Chiesa, R, Piccardo P, Ghetti B, Harris DA. Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron 1998; 21(6):1339-51; PMID:9883727
- Chiesa R, Drisaldi B, Quaglio E, Migheli A, Piccardo P, Ghetti B, Harris DA. Accumulation of protease-resistant prion protein (PrP) and apoptosis of cerebellar granule cells in transgenic mice expressing a PrP insertional mutation. Proc Natl Acad Sci U S A, 2000; 97(10):5574-9
- Ivanova L, Barmada S, Kummer T, Harris DA. Mutant prion proteins are partially retained in the endoplasmic reticulum. J Biol Chem 2001; 276(45):42409-21; PMID:11527974
- Biasini E, Medrano AZ, Thellung S, Chiesa R, Harris DA. Multiple biochemical similarities between infectious and non-infectious aggregates of a prion protein carrying an octapeptide insertion. J Neurochem 2008; 104(5):1293-308; PMID:18034781
- Bouybayoune I, Mantovani S, Del Gallo F, Bertani I, Restelli E, Comerio L, Tapella L, Baracchi F, Fernández-Borges N, Mangieri M, et al. Transgenic fatal familial insomnia mice indicate prion infectivity-independent mechanisms of pathogenesis and phenotypic expression of disease. PLoS Pathog 2015; 11(4):e1004796
- Senatore A, Restelli E, Chiesa R. Synaptic dysfunction in prion diseases: a trafficking problem? Int J Cell Biol 2013; 2013:543803; PMID:24369467
- Chiesa R, Restelli E, Comerio L, Del Gallo F, Imeri L. Transgenic mice recapitulate the phenotypic heterogeneity of genetic prion diseases without developing prion infectivity: Role of intracellular PrP retention in neurotoxicity. Prion 2016; 10(2):93-102
- Senatore A, Colleoni S, Verderio C, Restelli E, Morini R, Condliffe SB, Bertani I, Mantovani S, Canovi M, Micotti E, et al. Mutant PrP suppresses glutamatergic neurotransmission in cerebellar granule neurons by impairing membrane delivery of VGCC α(2)delta-1 Subunit. Neuron 2012; 74(2):300-13; PMID:22542184
- Wilson C, Venditti R, Rega LR, Colanzi A, D'Angelo G, De Matteis MA. The Golgi apparatus: an organelle with multiple complex functions. Biochem J 2011; 433(1):1-9
- Li Y, Kane T, Tipper C, Spatrick P, Jenness DD. Yeast mutants affecting possible quality control of plasma membrane proteins. Mol Cell Biol 1999; 19(5):3588-99; PMID:10207082
- Arvan P, Zhao X, Ramos-Castaneda J, Chang A. Secretory pathway quality control operating in Golgi, plasmalemmal, and endosomal systems. Traffic 2002; 3(11):771-80; PMID:12383343
- Castillon GA, Aguilera-Romero A, Manzano-Lopez J, Epstein S, Kajiwara K, Funato K, Watanabe R, Riezman H, Muñiz M. The yeast p24 complex regulates GPI-anchored protein transport and quality control by monitoring anchor remodeling. Mol Biol Cell 2011; 22(16):2924-36; PMID:21680708
- Wang S, Ng DT. Evasion of endoplasmic reticulum surveillance makes Wsc1p an obligate substrate of Golgi quality control. Mol Biol Cell 2010; 21(7):1153-65; PMID:20130083
- Tewari R, Bachert C, Linstedt AD. Induced oligomerization targets Golgi proteins for degradation in lysosomes. Mol Biol Cell 2015; 26(24):4427-37; PMID:26446839
- Gilch S, Winklhofer KF, Groschup MH, Nunziante M, Lucassen R, Spielhaupter C, Muranyi W, Riesner D, Tatzelt J, Schätzl HM. Intracellular re-routing of prion protein prevents propagation of PrP(Sc) and delays onset of prion disease. Embo J 2001; 20(15):3957-66; PMID:11483499
- Nunziante M, Kehler C, Maas E, Kassack MU, Groschup M, Schätzl HM. Charged bipolar suramin derivatives induce aggregation of the prion protein at the cell surface and inhibit PrPSc replication. Journal of Cell Science 2005; 118: http://dx.doi.org/10.1242/jcs.02609
- Darling NJ, Cook SJ. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim Biophys Acta 2014; 1843(10):2150-63; PMID:24440275
- Cancino J, Capalbo A, Di Campli A, Giannotta M, Rizzo R, Jung JE, Di Martino R, Persico M, Heinklein P, Sallese M, et al. Control systems of membrane transport at the interface between the endoplasmic reticulum and the Golgi. Dev Cell 2014; 30(3):280-94; PMID:25117681
- Giannotta M, Ruggiero C, Grossi M, Cancino J, Capitani M, Pulvirenti T, Consoli GM, Geraci C, Fanelli F, Luini A, et al. The KDEL receptor couples to Galphaq/11 to activate Src kinases and regulate transport through the Golgi. Embo J 2012; 31(13):2869-81; PMID:22580821
- Pulvirenti T, Giannotta M, Capestrano M, Capitani M, Pisanu A, Polishchuk RS, San Pietro E, Beznoussenko GV, Mironov AA, Turacchio G, et al. A traffic-activated Golgi-based signalling circuit coordinates the secretory pathway. Nat Cell Biol 2008; 10(8):912-22; PMID:18641641
- Yamamoto K, Fujii R, Toyofuku Y, Saito T, Koseki H, Hsu VW, Aoe T. The KDEL receptor mediates a retrieval mechanism that contributes to quality control at the endoplasmic reticulum. Embo J 2001; 20(12):3082-91; PMID:11406585
- Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res 2005; 15(1):11-8; PMID:15686620
- Yamamoto K, Hamada H, Shinkai H, Kohno Y, Koseki H, Aoe T. The KDEL receptor modulates the endoplasmic reticulum stress response through mitogen-activated protein kinase signaling cascades. J Biol Chem 2003; 278(36):34525-32; PMID:12821650
- Murakami Y, Aizu-Yokota E, Sonoda Y, Ohta S, Kasahara T. Suppression of endoplasmic reticulum stress-induced caspase activation and cell death by the overexpression of Bcl-xL or Bcl-2. J Biochem 2007; 141(3):401-10; PMID:17301078
- Barmada SJ, Harris DA. Visualization of prion infection in transgenic mice expressing green fluorescent protein-tagged prion protein. J Neurosci 2005; 25(24):5824-32; PMID:15958749
- Stewart RS, Piccardo P, Ghetti B, Harris DA. Neurodegenerative illness in transgenic mice expressing a transmembrane form of the prion protein. J Neurosci 2005; 25(13):3469-77; PMID:15800202
- Uchiyama K, Muramatsu N, Yano M, Usui T, Miyata H, Sakaguchi S. Prions disturb post-Golgi trafficking of membrane proteins. Nat Commun, 2013; 4:1846
- Massignan T, Biasini E, Lauranzano E, Veglianese P, Pignataro M, Fioriti L, Harris DA, Salmona M, Chiesa R, Bonetto V. Mutant prion protein expression is associated with an alteration of the Rab GDP dissociation inhibitor α (GDI)/Rab11 pathway. Mol Cell Proteomics 2010; 9(4):611-22; PMID:19996123
- Puig B, Ferrer I. Cell death signaling in the cerebellum in Creutzfeldt-Jakob disease. Acta Neuropathol 2001; 102(3):207-15; PMID:11585244
- Rodriguez A, Ferrer I. Expression of transcription factors CREB and c-Fos in the brains of terminal Creutzfeldt-Jakob disease cases. Neurosci Lett 2007; 421(1):10-5; PMID:17548164
- Zafar S, Schmitz M, Younus N, Tahir W, Shafiq M, Llorens F, Ferrer I, Andéoletti O, Zerr I. Creutzfeldt-Jakob disease subtype-specific regional and temporal regulation of ADP Ribosylation Factor-1-dependent Rho/MLC pathway at pre-clinical stage. J Mol Neurosci 2015; 56(2):329-48; PMID:25896910