
ABSTRACT
Sporadic Creutzfeldt-Jakob disease (sCJD) occurs frequently in the relatively older population, mainly in the groups of 60–69 and 70–79 year-old. Since 2006 when China performed national CJD surveillance, 14 young probable sCJD patients below 40 year-old were identified, counting for 1.93% of all probable sCJD cases. The clinical features of young probable sCJD cases, including the onset feature, the presence of sCJD-associated signs and the clinical duration, are indistinguishable from those of older patients. Special sCJD-associated abnormalities on EEG and MRI were noticed in 7 and 10 cases. CSF 14–3–3 was positive in 7 cases. CSF RT-QuIC showed positive reactive curves in 9 cases, with short lag phases. PRNP sequencing did not find any mutation. Due to low rate of brain autopsy in China, performances of other CJD-associated examinations as much as possible are extremely important for the distinguish diagnosis of young probable sCJD patients.
Human prion diseases (PrD) are a group of rare fatal and transmissible neurodegenerative diseases, such as Creutzfeldt-Jakob disease (CJD), Kuru, Gerstmann–Sträussler–Scheinker syndrome (GSS) and fatal familial insomnia (FFI).Citation1 About 85% of all CJD cases are sporadic (sporadic CJD, sCJD), 10–15% cases are inherited (genetic CJD, gCJD) and less than 1% is infectious (iatrogenic CJD, iCJD).Citation2-4 The majority of sCJD patients develop clinical symptoms at middle-aged and old-aged period, with the incident peak at the age group of 60–69 year-old worldwide.Citation4,5 Young sCJD patients under 40 year-old are extremely rare.
Since 2006 when China conducted national surveillance for CJD,Citation6-8 more than 1000 various PrD cases have been identified and diagnosed, including more than 700 probable sCJD cases. Agreeing well with the data worldwide, most of the sCJD cases are in the age-groups of 60–69 year-old (Shi et al, in preparation). Only 14 probable sCJD patients were under 40 year-old at onset. In this study, the clinical and laboratory characteristics of these 14 young Chinese sCJD patients were discussed.
ETHICS STATEMENT
Usage of the stored patients' information in China CJD Surveillance System has been approved by the Research Ethics Committee of National Institute for Viral Disease Control and Prevention, China CDC. The written informed content of each suspected case has been requested and signed mostly by the family member or the relative of the patient according to the requirement of CJD surveillance.
CLINICAL FEATURES
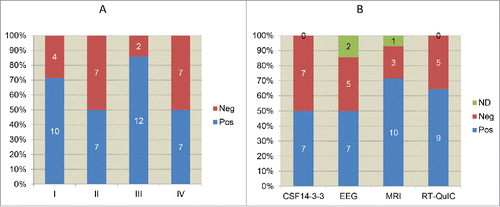
The general clinical and laboratory information of 14 young Chinese sCJD patients were summarized in . 7 cases were female and 7 were male. 2 cases were under 20 year-old, 2 were in the group of 20–29 year-old and 10 were in the group of 30–39 year-old, accounting to 14.28%, 14.28% and 71.43%, respectively (). All 14 cases were geographically distributed widely, without any family linkage. Interviews of the family members did not recall any family-associated disease histories. Most of the patients displayed relatively rapid onsets with various initial symptoms, including progressive dementia, mental symptoms, visual disturbances, gain instability, sleeping disorder, etc. Along with the progression of disease, CJD-associated clinical manifestations gradually appeared. Progressive dementia was noted in all cases during the clinical courses. The other 4 major signs were also frequently recorded. Pyramidal and extrapyramidal symptoms were noted most frequently (85.71%, 12/14), followed by myoclonus (71.43%, 10/14), visual or cerebellar disturbance (50.00%, 7/14) and mutism (50.00%, 7/14) (). All patients were excluded for the possibility of other neural diseases.
TABLE 1. Clinical and laboratory information of 15 young probable sCJD cases.
FIGURE 1. Age-distribution of 14 young probable sCJD patients. The cases numbers and the percentages of each decade group are showed inside and at top of columns.
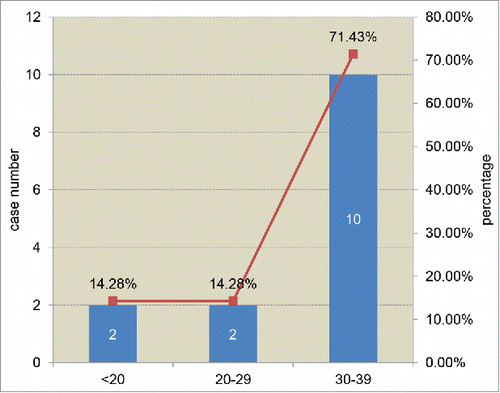
FIGURE 2. The presences of sCJD-associated clinical signs and the relevant examinations. A. Clinical signs. I: myoclonus; II: visual or cerebellar disturbance; III: pyramidal and extrapyramidal symptoms; IV: mutism. B. Clinical examinations and laboratory tests. ND: not done. The case numbers of positive, negative and ND are showed inside of columns, respectively.
CLINICAL EXAMINATIONS AND LABORATORY TESTS
High ratios of the patients received the major clinical examinations and laboratory test during hospitalizations (), including electroencephalogram (EEG), magnetic resonance imaging (MRI) scanning, cerebrospinal fluid (CSF) protein 14–3–3 test and PRNP gene sequencing. The interval times from onset to investigation varied from 1.1 to 5.6 months, with the median of 2.6 months. The peripheral blood leukocytes were used for sequencing analysis of the PRNP gene, as well as the polymorphisms of codon 129 and codon 219, with an automatic genetic analyzer (ABI3130XL).Citation6 All 14 cases were methionine homozygote at codon 129 (M129M). Eight patients had data of codon 219, showing glutamic acid homozygote (E219E) in 7 cases and glutamic acid/lysine heterozygote (E219K) in 1 case. No other mutation in the context of whole PRNP sequence was observed.
As shown in , periodic sharp wave complexes (PSWC) were noted in 7 (58.33%) patients out of 12 who received EEG examinations. sCJD-associated abnormalities (high signal abnormalities in caudate nucleus and putamen or at least 2 cortical regions (temporal-parrietal-occipital) either in DWI or FLAIR were observed in 10 (71.43%) patients out of 14 who received MRI scanning. Western blots for CSF 14–3–3 were performed in all 14 cases according to the protocol described previously.Citation6 Briefly, a total of 20 μl CSF sample was separated in 12% SDS-PAGE and transferred to nitrocellulose (NC) membranes (Whatman, USA). After blocking, the membrane was incubated with 1:1000 diluted 14–3–3 polyclonal antibody (Santa Cruz Biological) at room temperature (RT) for 2 h and subsequently incubated with goat anti-rabbit HRP-conjugated secondary antibody. Reactive signals were visualized using an enhanced chemiluminescence kit (Amersham-Pharmacia Biotech, USA). 7 out of 14 cases (50%) were CSF 14–3–3 positive (). In addition, 4 cases showed positive both in EEG and MRI, 4 were both positive in MRI and CSF 14–3–3, 2 were positive both in EEG and CSF 14–3–3.
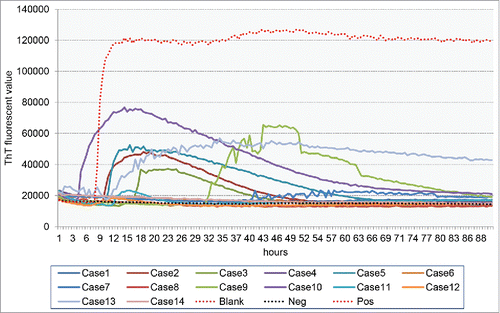
RT-QuIC assays of CSF samples were in all 14 patients. The detailed operating protocol was described elsewhereCitation9-11 with modification (Xiao et al, in preparation). Briefly, 10 µl CSF sample was subjected into RT-QuIC reaction containing 10 µg purified recombinant hamster PrP protein (aa23–230), 10 mM PBS, 170 mM NaCl, 10 µM EDTA, 1 mM Thioflavin T (ThT), 0.001% SDS in final concentration, with a final volume of 100 µl. RT-QuIC was performed on FlUOStar Omega (BMG). The working conditions were temperature: 50°C; vibration speed: 700 rpm; vibration/incubation time: 90/30 sec; total reaction time: 90 h. The cut-off value was set as the average value of the negative controls plus 3 times of SD. Each CSF sample was duplicated in parallel. The sample was considered as positive when the 2 parallel wells revealed positive reactive curves. As shown in , 9 out of 14 cases (64.29%) showed the positive reactions. The positive reactive curves appeared from 5 to 41 h post-reaction (). No statistically significant forrelations were observed between of the positive RT-QuIC reactions and the results in EEG, MRI and CSF 14–3–3.
FIGURE 3. The reactive pattern of CSF RT-QuiC assays. ThT fluorescent values are indicated in Y-axis and the times (h) post-reaction are in X-axis. Pos: positive control containing 100 fg PrPSc prepared from the brain homogenate of scrapie strain 263K-infected hamster. Neg: negative control containing the same amount of the brain homogenate of normal hamster as Pos. Blank: blank control without hamster brain homogenate.
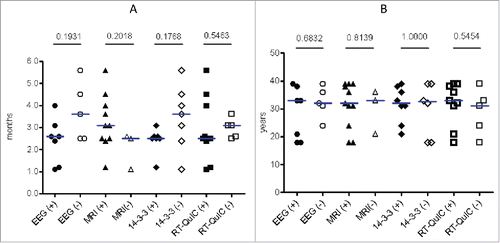
The possible correlations of the results in EEG, MRI, CSF 14–3–3 and RT-QuIC with the intervals from onset to examinations were analyzed. Compared with the groups showing negative results, positive in EEG, CSF 14–3–3 and RT-QuIC appeared to present in the patients with relatively short diseases intervals, while positive MRI was observed in the patients with long intervals (). Statistical analysis did not reveal significant differences. Furthermore analysis of the relationships of the results in EEG, MRI, CSF 14–3–3 and RT-QuIC with the onset ages of the patients did not show any correlation ().
FIGURE 4. Analyses of the associations of the relevant examinations with the interval from onset to examinations (A) and the onset ages (B). P values of t tests between positive and negative groups are indicated above.
COMMENTS
Based on the data of China CJD surveillance network, totally 676 probable sCJD cases have been diagnosed from 2006 to 2015. Among them, 13 cases (1.92%) are under 40 year-old at the onset age time. Another case in this study was reported in 2016. Four out of those 14 cases were under 30 year-old. PRNP sequencing did not detect any mutation and intensive interviews with the family members (mostly parents) did not reveal any potentially associated family disease history or epidemiological clue. Comprehensive clinical examinations, including auto-immunological and infectious assays, do not illustrate any useful clue for the possibility of other diseases. Despite being rare, young sCJD cases have been repeatedly reported since 1980s.Citation12-14 An large-scale study of the CJD surveillance from 1970 to 1996 in UK has proposed 25 (3.8%) young sCJD patients who died under 40 year-old out of 658 cases.Citation15 A prospective study based on the national mortality data from 1979 to 1994 in USA has illustrated 41 young patients (1.13%) below 40 year-old out of 3642 CJD cases.Citation16 Surveillance for CJD in Germany from 1993 to 2005 has also identified 56 sCJD patients (3%) with the onset ages below 50 year-old, with the youngest case of 19 year-old.Citation17 Young sCJD patients are being continually reported in many European countries, Australia, North America, Japan and others, based on the national surveillance data.Citation18-23 The identifying rate of young sCJD patients below 40 year-old in our surveillance system is comparable with that of other countries.
The presences of dementia and other 4 major clinical signs in 14 young Chinese probable sCJD patients during clinical courses are frequently observed, which are quite comparable with the data from all sCJD cases (Shi et al, in preparation). The positive rate (50%) of visual or cerebellar disturbance in young patients is relatively lower than that of total sCJD (69.7%, unpublished). Although less frequent cerebellar signs in young sCJD patients have been described previously,Citation24 the limited case numbers may also produce bias, which needs further analysis with more case numbers. The initial symptoms of young sCJD cases are also undistinguishable from the older patients, predominated by progressive dementia and followed by mental problems and visual disturbances. Boesenberg et al have reported that psychiatric symptoms were common at disease onset in young sCJD patients and 21% the patients contacted psychiatrist as the first specialists.Citation24 We do not observe such pattern in those 14 Chinese sCJD patients. On the other hand, visiting psychiatrist, especially as the first visiting specialist, is uncommon for routine Chinese generally. Some psychiatric symptoms and signs may be ignored at the early stage of disease. That might be a possible reason for relatively uncommon mental problems in young Chinese sCJD patients compared with the western patients.
The positive rates of EEG and MRI in young probable sCJD cases are also comparable with the data of total probable sCJD cases. Similarly, we do not observe the pattern of less sensitive of EEG in young sCJD patients described previously.Citation24 Both special abnormalities in EEG and MRI can be detected during the clinical courses. Although the patients with shorter intervals from onset to examination seem to appear PSWC in EEG and normal MRI images frequently, there is no statistical difference. Since all 14 cases are M129M homozygote, the different distribution of PSWC among various genotypes of codon 129 within PRNP in CaucasianCitation24,25 is undetectable. CSF 14–3–3 positive rate (50%) in 14 young patients is lower than that of total sCJD patients (76.96%, unpublished). The reason and significance of this phenomenon is not clear.
None of those 14 probable sCJD patients underwent neuropathological tests, since the family members refused to perform brain autopsy or biopsy. Recently, a new technique, namely CSF RT-QuIC has been used for sCJD, which shows reliable sensitivity and specificity.Citation10,11,26 Nine patients show positive reaction in CSF RT-QuIC assays. Careful analysis of the patterns of RT-QuIC illustrates that the positive curves occur at relatively early phases post-reaction. In addition, we have CSF RT-QuIC assays for 20 young patients (< 40 year-old) considered as possible sCJD during hospitalization. Three of them display positive reactions, but the positive curves appear at very late phases (> 80 h post-reaction), with relative low ThT fluorescent values. Combined with data of clinical (EEG and MRI) and laboratory (CSF 14–3–3 and RT-QuIC) tests, almost all 14 young probable sCJD patients display at least 2 positive results, which may increase the validity of the diagnosis of sCJD despite of lacking neuropathological assays.
In summary, young probable sCJD cases are rarely, but repeatedly, identified in China since 2006, which counts for less than 2% of all probable sCJD cases. Generally, the clinical and laboratory features of young Chinese sCJD patients are quite comparable with that of older ones. Because of extremely low rate of brain autopsy in China, performances of other CJD-associated clinical examinations (EEG and MRI) and laboratory tests (CSF 14–3–3 and RT-QuIC, PRNP sequencing) as much as possible are highly recommended.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
FUNDING
This work was supported by Chinese National Natural Science Foundation Grants (81301429, 81572048, 81630062), National Key Research and Development Plan (2016YFC1202700) and SKLID Development Grant (2012SKLID102, 2015SKLID503, 2016SKLID603).
REFERENCES
- Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998; 95:13363-83; PMID:9811807; http://dx.doi.org/10.1073/pnas.95.23.13363
- Prusiner SB. The prion diseases. Brain Pathol 1998; 8:499-513; PMID:9669700; http://dx.doi.org/10.1111/j.1750-3639.1998.tb00171.x
- Curcio L, Sebastiani C, Di Lorenzo P, Lasagna E, Biagetti M. Review: A review on classical and atypical scrapie in caprine: Prion protein gene polymorQ6 phisms and their role in the disease. Animal 2016; 10:1585-93; PMID:27109462
- Chen C, Dong XP. Epidemiological characteristics of human prion diseases. Infect Dis Poverty 2016; 5:47; PMID:27251305; http://dx.doi.org/10.1186/s40249-016-0143-8
- Klug GM, Wand H, Simpson M, Boyd A, Law M, Masters CL, Matěj R, Howley R, Farrell M, Breithaupt M, et al. Intensity of human prion disease surveillance predicts observed disease incidence. J Neurol Neurosurg Psychiatry 2013; 84:1372-7; PMID:23965290; http://dx.doi.org/10.1136/jnnp-2012-304820
- Shi Q, Zhou W, Chen C, Zhang BY, Xiao K, Zhang XC, Shen XJ, Li Q, Deng LQ, Dong JH, et al. The features of genetic prion diseases based on chinese surveillance program. PLoS One 2015; 10:e0139552; PMID:26488179; http://dx.doi.org/10.1371/journal.pone.0139552
- Gao C, Shi Q, Tian C, Chen C, Han J, Zhou W, Zhang BY, Jiang HY, Zhang J, Dong XP. The epidemiological, clinical, and laboratory features of sporadic Creutzfeldt-Jakob disease patients in China: surveillance data from 2006 to 2010. PLoS One 2011; 6:e24231; PMID:21904617; http://dx.doi.org/10.1371/journal.pone.0024231
- Shi Q, Zhang XC, Zhou W, Xiao K, Chen C, Zhang HY, Sun JY, Chen LN, Zhang XM, Han J, et al. Analysis of the advantage features of Beijing surveillance network for Creutzfeldt-Jakob disease. Prion 2015; 9:304-14; PMID:26251963; http://dx.doi.org/10.1080/19336896.2015.1075115
- Vascellari S, Orru CD, Hughson AG, King D, Barron R, Wilham JM, Baron GS, Race B, Pani A, Caughey B. Prion seeding activities of mouse scrapie strains with divergent PrPSc protease sensitivities and amyloid plaque content using RT-QuIC and eQuIC. PLoS One 2012; 7:e48969; PMID:23139828; http://dx.doi.org/10.1371/journal.pone.0048969
- Orru CD, Groveman BR, Hughson AG, Zanusso G, Coulthart MB, Caughey B. Rapid and sensitive RT-QuIC detection of human Creutzfeldt-Jakob disease using cerebrospinal fluid. MBio 2015; 6:e02451-14; PMID:25604790
- McGuire LI, Peden AH, Orru CD, Wilham JM, Appleford NE, Mallinson G, Andrews M, Head MW, Caughey B, Will RG, et al. Real time quaking-induced conversion analysis of cerebrospinal fluid in sporadic Creutzfeldt-Jakob disease. Ann Neurol 2012; 72:278-85; PMID:22926858; http://dx.doi.org/10.1002/ana.23589
- Berman PH, Davidson GS, Becker LE. Progressive neurological deterioration in a 14-year-old girl. Pediatr Neurosci 1988; 14:42-9; PMID:3064055; http://dx.doi.org/10.1159/000120361
- Monreal J, Collins GH, Masters CL, Fisher CM, Kim RC, Gibbs CJ Jr, Gajdusek DC.. Creutzfeldt-Jakob disease in an adolescent. J Neurol Sci 1981; 52:341-50; PMID:7031189; http://dx.doi.org/10.1016/0022-510X(81)90015-0
- Packer RJ, Cornblath DR, Gonatas NK, Bruno LA, Asbury AK. Creutzfeldt-Jakob disease in a 20-year-old woman. Neurology 1980; 30:492-6; PMID:6988735; http://dx.doi.org/10.1212/WNL.30.5.492
- Cousens SN, Zeidler M, Esmonde TF, De Silva R, Wilesmith JW, Smith PG, Will RG. Sporadic Creutzfeldt-Jakob disease in the United Kingdom: analysis of epidemiological surveillance data for 1970–96. BMJ 1997; 315:389-95; PMID:9277601; http://dx.doi.org/10.1136/bmj.315.7105.389
- Holman RC, Khan AS, Belay ED, Schonberger LB. Creutzfeldt-Jakob disease in the United States, 1979–1994: using national mortality data to assess the possible occurrence of variant cases. Emerg Infect Dis 1996; 2:333-7; PMID:8969250; http://dx.doi.org/10.3201/eid0204.960409
- Heinemann U, Krasnianski A, Meissner B, Varges D, Kallenberg K, Schulz-Schaeffer WJ, Steinhoff BJ, Grasbon-Frodl EM, Kretzschmar HA, Zerr I. Creutzfeldt-Jakob disease in Germany: a prospective 12-year surveillance. Brain 2007; 130:1350-9; PMID:17472986; http://dx.doi.org/10.1093/brain/awm063
- Ladogana A, Puopolo M, Croes EA, Budka H, Jarius C, Collins S, Klug GM, Sutcliffe T, Giulivi A, Alperovitch A, et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology 2005; 64:1586-91; PMID:15883321; http://dx.doi.org/10.1212/01.WNL.0000160117.56690.B2
- Nozaki I, Hamaguchi T, Sanjo N, Noguchi-Shinohara M, Sakai K, Nakamura Y, Sato T, Kitamoto T, Mizusawa H, Moriwaka F, et al. Prospective 10-year surveillance of human prion diseases in Japan. Brain 2010; 133:3043-57; PMID:20855418; http://dx.doi.org/10.1093/brain/awq216
- Litzroth A, Cras P, De Vil B, Quoilin S. Overview and evaluation of 15 years of Creutzfeldt-Jakob disease surveillance in Belgium, 1998–2012. BMC Neurol 2015; 15:250; PMID:26630984; http://dx.doi.org/10.1186/s12883-015-0507-x
- Gubbels S, Bacci S, Laursen H, Hogenhaven H, Cowan S, Molbak K, Christiansen M. Description and analysis of 12 years of surveillance for Creutzfeldt-Jakob disease in Denmark, 1997 to 2008. Euro Surveill 2012; 17:20142; PMID:22516048
- Belay ED, Gambetti P, Schonberger LB, Parchi P, Lyon DR, Capellari S, McQuiston JH, Bradley K, Dowdle G, Crutcher JM, et al. Creutzfeldt-Jakob disease in unusually young patients who consumed venison. Arch Neurol 2001; 58:1673-8; PMID:11594928; http://dx.doi.org/10.1001/archneur.58.10.1673
- Martindale J, Geschwind MD, De Armond S, Young G, Dillon WP, Henry R, Uyehara-Lock JH, Gaskin DA, Miller BL. Sporadic Creutzfeldt-Jakob disease mimicking variant Creutzfeldt-Jakob disease. Arch Neurol 2003; 60:767-70; PMID:12756143; http://dx.doi.org/10.1001/archneur.60.5.767
- Boesenberg C, Schulz-Schaeffer WJ, Meissner B, Kallenberg K, Bartl M, Heinemann U, Krasnianski A, Stoeck K, Varges D, Windl O, et al. Clinical course in young patients with sporadic Creutzfeldt-Jakob disease. Ann Neurol 2005; 58:533-43; PMID:16037975; http://dx.doi.org/10.1002/ana.20568
- Kovacs GG, Head MW, Bunn T, Laszlo L, Will RG, Ironside JW. Clinicopathological phenotype of codon 129 valine homozygote sporadic Creutzfeldt-Jakob disease. Neuropathol Appl Neurobiol 2000; 26:463-72; PMID:11054187; http://dx.doi.org/10.1046/j.1365-2990.2000.00279.x
- Cramm M, Schmitz M, Karch A, Mitrova E, Kuhn F, Schroeder B, Raeber A, Varges D, Kim YS, et al. Stability and Reproducibility Underscore Utility of RT-QuIC for Diagnosis of Creutzfeldt-Jakob Disease. Mol Neurobiol 2016; 53:1896-904; PMID:25823511; http://dx.doi.org/10.1007/s12035-015-9133-2