
ABSTRACT
Introduction. The role of positron emission tomography (PET) in Creutzfeldt-Jakob disease is less defined than in other neurodegenerative diseases. We studied the correlation between the uptake of 18F-florbetaben and 18F-fluorodeoxyglucose with pathological prion protein deposition in histopathology in a case.Methods. A patient with 80 y old with a rapid neurological deterioration with a confirmed diagnosis of CJD was studied. PET and MRI studies were performed between 13–20 d before the death. A region of interest analysis was performed using Statistical Parametric Mapping.Results. MRI showed atrophy with no other alterations. FDG-PET showed extensive areas of hypometabolism including left frontoparietal lobes as well as bilateral thalamus. Correlation between uptake of 18F-florbetaben and pathological prion protein deposition was r = 0.786 (p < 0.05). Otherwise, correlation between uptake of 18F-FDG and pathological prion protein was r = 0.357 (p = 0.385). Immunohistochemistry with β-amyloid did not show amyloid deposition or neuritic plaques.Conclusions. Our study supports the use of FDG-PET in the assessment of CJD. FDG-PET may be especially useful in cases of suspected CJD and negative MRI. Furthermore, this case report provides more evidence about the behavioral of amyloid tracers, and the possibility of a low-affinity binding to other non-amyloid proteins, such as the pathological prion protein, is discussed.
INTRODUCTION
Creutzfeldt-Jakob disease (CJD) is a rapidly progressive neurodegenerative disorder. It is the most common of the human prion diseases, which may be sporadic, inherited or acquired by infection.Citation1 Diagnosis is based on 3 main tests: brain magnetic resonance imaging (MRI), electroencephalogram, and 14–3–3 protein on CSF. The presence of a suggestive clinical picture with at least one of the previously mentioned test positive allows qualifying the diagnosis as “probable”.Citation2 However, definitive diagnosis can only be confirmed by brain tissue examination, usually after death, when the 3 histopathological hallmarks of prion disease, neuronal loss, vacuolation, and gliosis, are observed.
Recently, 18F-fluorodeoxyglucose positron emission tomography (18F-FDG) has been suggested to be useful in the diagnosis of CJD.Citation3-6 A pattern of subcortical and cortical hypometabolism has been reported. This pattern may be variable in the individual cases, but usually impairs large brain regions including more frequently caudate, thalamus and frontal cortex.Citation6 Few studies have been published comparing the diagnostic capacity of MRI and FDG-PET in CJD, and the possible contribution of FDG-PET to clinical diagnosis and to improve the current diagnostic criteria is still under discussion.Citation7 Regarding PET with amyloid tracers, some cases have been communicated, with different results according to the tracer used.Citation8,9 Amyloid prion protein plaques may be present in some variants of CJD and other prion diseases, and experimental studies have shown the binding of some thioflavin and Congo red derivates to plaques of prion protein.Citation10 Overall, these data suggest a potential use of FDG-PET and amyloid tracers in CJD and other prion diseases, although evidence is still scarce.
Amyloid tracers offer the opportunity to detect β-amyloid plaques in vivo. However, these tracers also bind to the normal white matter, a non-amyloid related structure. Thus, we hypothesize that amyloid tracers may be not entirely selective and may bind to other proteins deposited in neurodegenerative diseases. We present the case of a patient with autopsy-confirmed CJD studied with 18F-FDG and 18F-florbetaben PET. In this case, PET imaging was performed very close to death. This permitted to assess the correlation between the uptake of 18F-florbetaben and pathological prion protein in brain tissue examination.
METHODS
Acquisition, Preprocessing and Analysis of PET Imaging
PET images were acquired in a PET-CT Siemens Biograph True Point platform. 18F-FDG was administered at a dose of 185 MBq 30 minutes before images were taken. The patient fasted for 6 hours, and glucose level was checked before the injection. Images were acquired for 15 minutes. CT parameters were 130 kVp, 40 effective mAs, and 1 rotation. Slice thickness was 3 mm, reconstruction interval was 1.5 mm, and pitch was 0.75 mm. The true X method with 2 iterations and 21 subsets was used to reconstruct the images. We used a 30 cm field of view and a Gaussian filter with FWHM of 4 mm.
18F-florbetaben was administered at a dose of 300 MBq 90 minutes before the acquisition of the images. Acquisition time was 15 minutes. The reconstruction was performed using the same methodology as with FDG.
Two nuclear medicine specialists visually analyzed images by consensus. Regarding 18F-florbetaben, we used the validated binary method of reading.Citation11 Images were preprocessed using Statistical Parametric Mapping software version 8 (SPM8) (The Wellcome Trust Centre for Neuroimaging, Institute of Neurology, University College of London).Citation12 Regarding the analysis of amyloid PET, T1-weighted image was co-registered with the 18F-florbetaben image using the normalized mutual information method, with the T1-weighted image as the reference. The T1-weighted was warped to the reference space of the Montreal Neurological Institute using a T1 template to define the parameters of normalization. Then, these parameters were used to normalize the co-registered PET images. Subsequently, we used the MarsBaR software and the Automated Anatomical Labeling atlas to perform a region of interest analyses restricted to the gray matter.Citation13 We extracted the uptake in the following regions in the left hemisphere for the correlation analysis with neuropathology: precuneus, supplementary motor area, calcarine cortex, posterior cingulate, anterior cingulate, frontal orbital gyrus, inferior temporal gyrus and caudate. The same procedure was used for the preprocessing and analysis of 18F-FDG images.
Brain Tissue Examination and Quantification
Brain sections were treated for 1 hour with 98% formic acid before paraffin embedding. Microscopic study was performed with haematoxylin-eosin staining as well as immunohistochemistry for pathological prion protein (3F4 antibody), tau, β-amyloid, α-synuclein and TDP-43 (Table S1).
A total of 22 sections corresponding to the left hemisphere, including areas CERAD primary motor and sensory areas, hippocampus, amygdala, striatum and lenticular nuclei, thalamus and hypothalamus, midbrain, brainstem and cerebellum were studied.
For the study of pathological prion protein expression, 10 fields from each of the brain regions were analyzed. The result was the average of 10 determinations. Data are shown in optical densities, named % immunopositive area per field. Image J software was used to quantify the pathological prion protein deposition.
Statistical Analysis
Statistical analysis was performed using IBM® Statistics version 20 for Mac. Spearman's coefficient was calculated to study the correlation between the uptake of 18F-florbetaben and 18F-FDG and pathological prion protein deposition in each brain region. Pathological prion protein results were represented graphically using GraphPad Prism version 5.0. Data are expressed as mean ± SD.
RESULTS
Case Description
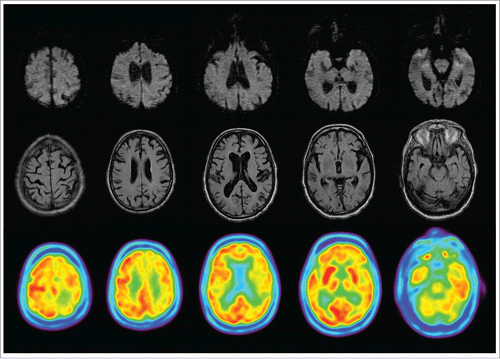
An 80 years-old man with past medical history of hypertension, diabetes and hypercholesterolemia was admitted to our hospital with gait disturbance. The symptoms began 3 weeks earlier with an abrupt onset of unsteadiness and weakness of right limbs. Gait rapidly deteriorated and at the moment of admission the patient needed a wheelchair. Likewise it had involuntary movements of limbs and apathy. In neurological examination, the patient showed an inhibited behavioral, and a disturbance in smooth pursuit and saccadic ocular movements, mild hemiparesis and bilateral ataxia. Furthermore, spontaneous and action myoclonus were present. Initial evaluation, which included biochemical, microbiological and immunological analyses and cranial CT scan, was normal. Cerebrospinal fluid examination was unremarkable aside an increased level of proteins. Cranial MRI showed global atrophy, but without any other alterations. Three electroencephalograms revealed diffuse slowing with no other abnormalities. FDG-PET (20 d before death) demonstrated large regions of hypometabolism including left frontoparietal lobes as well as bilateral thalamus (). 18F-florbetaben PET was also performed (13 d before death). The patient experimented a quickly deterioration associating apraxia, cortical sensory deficits and finally akinetic mutism within the 3 weeks of the patient's hospital stay. A new MRI was unremarkable 15 d before death (). At this time, determination of 14–3–3 protein was positive on CSF. The patient died 2 months since the beginning of the disease. Brain tissue examination was conducted and evidenced spongiform changes, neuronal loss and cortical accumulation of pathological prion protein, confirming the diagnosis of CJD. Sequencing of PRNP gene did not show any pathogenic variant.
FIGURE 1. MRI and FDG-PET. In upper row, diffusion-weighted imaging with no alterations. In middle row, fluid attenuated inversion recovery (FLAIR) sequence showing global atrophy. In lower row, FDG-PET imaging showed a left frontal-parietal, bilateral thalamus and cerebellar hypometabolism.
18F-Florbetaben-PET Analysis
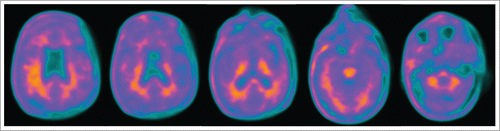
In the visual analysis, uptake in most regions of white matter was higher than the adjacent gray matter. However, in some regions (left parietal and right superior temporal lobes) the uptake in gray matter was almost equal than in the adjacent white matter ().
FIGURE 2. 18F-florbetaben PET imaging. Uptake tracer in white matter is generally higher than in gray matter. In left parietal and right superior temporal lobes, tracer uptake is similar in white and gray matter.
Brain Examination
Spongiform changes, neuronal loss and astrocytosis were observed in all of the areas, with different intensity according to each one. In cortical gray matter these changes were transcortical with mild to moderate spongiosis of small-medium vacuole, mild astrocitosis and moderate neuronal loss. In the other areas the changes were more diffuse. No “kuru type” plaques or neuritic plaques were observed.
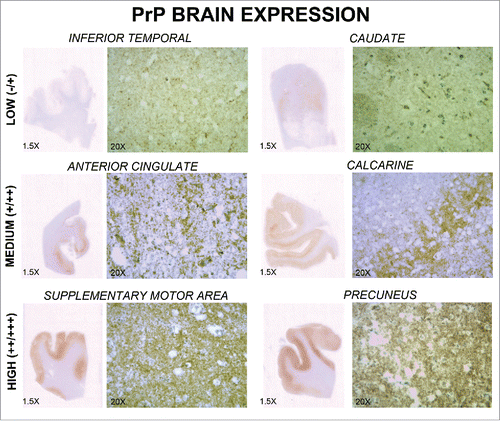
Immunohistochemistry with β-amyloid did not show amyloid deposition or neuritic plaques. In this regard, β-amyloid was negative in CERAD areas, subcortical regions and vessel walls (). α-synuclein immunohistochemistry was also negative. There were occasional neurons with cytoplasmatic granular staining with anti-tau. There was a mainly synaptic pattern of pathological prion protein deposition. In addition, areas with high immunoreactivity (Supplemental motor area, Precuneus) and areas of low immunoreactivity (Caudate, Temporal Inferior) were observed (). None amyloid-prion protein plaques were detected.
FIGURE 3. Frontal cortex showing no deposition of β-amyloid.
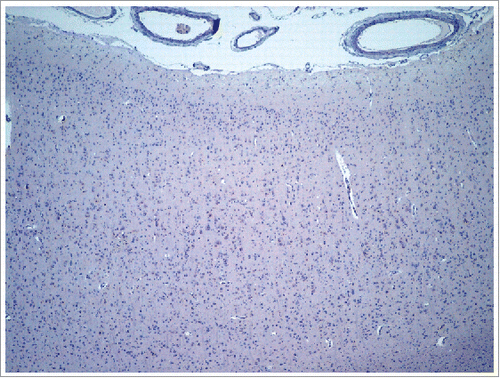
FIGURE 4. Pathological prion protein expression in several brain regions. Immunoreactivity of pathological prion protein in 6 regions (inferior temporal, caudate, anterior cingulate, calcarine, supplementary motor area, and precuneus) is shown. Each region is observed at 1.5x and 20x magnification. Different levels of expression (low, medium, and high) may be observed.
Correlation Between 18F-Florbetaben and 18F-FDG Uptake with Neuropathology
Correlation between uptake of 18F-florbetaben and pathological prion protein was r = 0.786 (p = 0.021). Otherwise, correlation between uptake of 18F-FDG and pathological prion protein was r = 0.357 (p = 0.385).
DISCUSSION
Our study reports the case of a patient with sporadic CJD studied using 18F-FDG and 18F-florbetaben PET. To the best of our knowledge, there are no previous studies with an 18F-labeled amyloid tracer in a prion disease and this is the first to analyze the correlation between amyloid PET and findings in neuropathology. The time between the performance of amyloid PET and the death of the patient was only 13 d. Although CJD often presents an aggressive course, the short time between PET imaging and death allowed us to study the correlation between PET tracers' uptake and neuropathology.
Prion diseases develop because of the conformational conversion of prion protein into pathological prion protein and its propagation. This conformational change is due to several modifications in the protein's secondary and tertiary structure. The pathological prion protein is rich in β-sheet and trends to produce protein aggregates and fibrils. This mechanism also occurs in other proteins, such as β-amyloid.Citation14,15
Amyloid prion plaques may be detected in specific prion diseases, such as variant CJD and Gerstmann-Sträussler-Scheinker disease. However, clinical studies with amyloid tracers have shown different ability to detect amyloid in CJD (amyloid-prion protein plaques) according to the tracers. Indeed, FDDNP and 11C-BF-227 seemed to detect amyloid PrP plaques, but not PiB Citation8,9,16,17,18 (). Hence, it could be hypothesized that fluorine 18-labeled tracers florbetapir, florbetaben and flutemetamol may not be able to detect amyloid PrP plaques because these tracers derive from PiB.Citation19
TABLE 1. Studies of prion diseases using amyloid PET tracers.
Interestingly, in our case immunohistochemistry for β-amyloid in neuropathology was negative. In this regard, pathological prion protein was deposited in a synaptic pattern, but not amyloid plaques were found. 18F-florbetaben uptake, but not 18F-FDG, correlated with the amount of pathological prion protein in the examination of the brain. Current amyloid tracers derive from Congo Red and Thioflavin S, and these dyes may stain deposits of pathological prion protein in certain cases. Citation20 Regarding 18F-FDG, this tracer has been recently associated to neural loss, astrocytosis and spongiosis in a case-series of CJD.Citation21
Therefore, our case suggests the possibility of a low-affinity binding of the tracer to the pathological prion protein. Indeed, amyloid tracers are not totally specific for amyloid plaques, and they also bind, for instance, to another structure such as white matter.Citation22,23 This may explain the increased signal in some specific regions, although the 18F-florbetaben uptake in this case is lower than in Alzheimer disease patients. In Alzheimer disease, all neocortical gray matter regions usually show a high retention of 18F-florbetaben.Citation24 Due to the negativity of amyloid stains in brain examination as well as the correlation with the amount of pathological prion protein, in our case the uptake of 18F-florbetaben may be explained by the binding of the tracer to the pathological prion protein. The hypothetical binding of the amyloid tracer to pathological prion protein could be explained by the fact that both proteins (β-amyloid and pathological prion protein) share a fibrillar structure based on a β-sheet conformation.Citation25,26 These results open the possibility to a low-affinity binding to other non-amyloid proteins in the neurodegenerative diseases, which might explain some false positives of the amyloid PET. However, this is a preliminary study in a single case, so these results should be confirmed in studies including more patients.
Furthermore, although the patient was examined twice using MRI, no specific MRI abnormalities supporting the diagnosis of CJD were observed. MRI demonstrated a sensitivity of 83% and a specificity of 83% in a multicenter study.Citation2 Regarding the other tests, sensitivity and specificity was 86% and 68% for CSF 14–3–3 protein, and 44% and 92% for EEG, respectively. Our case confirms the existence of a subgroup of patients with CJD in which MRI may be repeatedly negative, and supports the use of FDG-PET. In this regard, FDG-PET may detect abnormalities in cortical and subcortical regions without corresponding MRI hyperintensities.Citation27,28 Furthermore, FDG-PET may be more sensitive than MRI, although specific comparative studies between FDG-PET and MRI are necessary to confirm this issue.Citation6,17 In addition, patterns of metabolism may be useful in the differential diagnosis between CJD and other prion disease as well as other neurodegenerative disease that may present with a rapidly progressing dementia.Citation28,29
In conclusion, our study supports the use of FDG-PET in the assessment of CJD. FDG-PET may be especially useful in cases of suspected CJD and negative MRI. Furthermore, this case report provides more evidence about the behavioral of amyloid tracers, and the possibility of a low-affinity binding to other non-amyloid proteins, such as the pathological prion protein. Further studies in Creutzfeldt-Jakob disease and other neurodegenerative diseases with deposition of fibrilar proteins are necessary to better understand the selectivity of amyloid tracers.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
Supplemental_Material.docx
Download MS Word (48.9 KB)Related Research Data
REFERENCES
- Puoti G, Bizzi A, Forloni G, Safar JG, Tagliavini F, Gambetti P. Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol 2012; 11:618-628; PMID:22710755; https://doi.org/10.1016/S1474-4422(12)70063-7
- Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, Heinemann U, Breithaupt M, Varges D, Meissner B, Ladogana A, Schuur M, Haik S, Collins SJ, Jansen GH, Stokin GB, Pimentel J, Hewer E, Collie D, Smith P, Roberts H, Brandel JP, van Duijn C, Pocchiari M, Begue C, Cras P, Will RG, Sanchez-Juan P. Updatec clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain 2009; 132:2659-2668; PMID:19773352; https://doi.org/10.1093/brain/awp191
- Henkel K, Zerr I, Hertel A, Gratz KF, Schröter A, Tschampa HJ, Bihl H, Büll U, Grünwald F, Drzezga A, Spitz J, Poser S. Positron emission tomography with [18F]FDG in the diagnosis of Creutzelft-Jakob disease (CJD). J Neurol 2002; 249:699-705; PMID:12111302; https://doi.org/10.1007/s00415-002-0695-3
- Kim EJ, Cho SS, Jeong BH, Kim YS, Seo SW, Na DL, Geschwind MD, Jeong Y. Glucose metabolism in sporadic Creutzfeldt-Jakob disease: an SPM analysis of 18F-FDG PET. Eur J Neurol 2012; 19:488-493; PMID:22050286; https://doi.org/10.1111/j.1468-1331.2011.03570.x
- Renard D, Vandenbergue R, Collombier L, Kotzki PO, Pouget JP, Boudousq V. Glucose metabolism in nine patients with probable sporadic Creutzfeldt-Jakob disease: FDG-PET study using SPM and individual patient analysis. J Neurol 2013; 260:3055-3064; PMID:24068371; https://doi.org/10.1007/s00415-013-7117-6
- Prieto E, Domínguez-Prado I, Riverol M, Ortega-Cubero S, Ribelles MJ, Luquin MR, de Castro P, Arbizu J. Metabolic patterns in prion diseases: an FDG PET voxel-based analysis. Eur J Nucl Med Mol Imaging 2015; 42:1522-1529; PMID:26041084; https://doi.org/10.1007/s00259-015-3090-x
- Caobelli F, Cobelli M, Pizzocaro C, Pavia M, Magnaldi S, Guerra UP. The role of neuroimaging in evaluating patients affected by Creutzfeldt-Jakob disease: a systematic review of the literatura. J Neuroimaging 2015; 25:2-13; PMID:24593302; https://doi.org/10.1111/jon.12098
- Villemagne VL, McLean CA, Reardon K, Boyd A, Lewis V, Klug G, Jones G, Baxendale D, Masters CL, Rowe CC, Collins SJ. 11C-PiB PET studies in typical sporadic Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry 2009; 80:998-1001; PMID:19332421; https://doi.org/10.1136/jnnp.2008.171496
- Okamura N, Shiga Y, Furumoto S, Tashiro M, Tsuboi Y, Furukawa K, Yanai K, Iwata R, Arai H, Kudo Y, Itovama Y, Doh-ura K. In vivo detection of prion amyloid plaques using [11C]BF-227 PET. Eur J Nucl Med Mol Imaging 2010; 37:934-941; PMID:20016895; https://doi.org/10.1007/s00259-009-1314-7
- Ishikawa K, Doh-ura K, Kudo Y, Nishida N, Murakami-Kubo I, Ando Y, Sawada T, Iwaki T. Amyloid imaging probes are useful for detection of prion plaques and treatment of transmisible spongiform encephalopaties. J Gen Virol 2004; 85:1785-1790; PMID:15166464; https://doi.org/10.1099/vir.0.19754-0
- Barthel H, Gertz HJ, Dresel S, Peters O, Bartenstein P, Buerger K, Hiemeyer F, Wittemer-Rump SM, Seibyl J, Reininger C, Sabri O, for the Florbetaben Study Group. Cerebral amyloid-beta PET with florbetaben (18F) in patients with Alzheimer's disease and healthy controls: a multicentre phase 2 diagnostic group. Lancet Neurol 2011; 10:424-435; PMID:21481640; https://doi.org/10.1016/S1474-4422(11)70077-1
- Ashburner J, Friston KJ. Voxel-based morphometry–the methods. Neuroimage 2000;11:805-21; PMID:10860804; https://doi.org/10.1006/nimg.2000.0582
- Tzourio-Mazoyer N, Landeau D, Papathanassiou D, Crivello F, Etard O, Delcroix N, Mazoyer B, Joliot M. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. Neuroimage 2002; 15:273-89; PMID:11771995; https://doi.org/10.1006/nimg.2001.0978
- Fändrich M, Schmidt M, Grigorieff N. Recent progress in understanding Alzheimer's β-amyloid structures. Trends Biochem Sci 2011; 36:338-345; PMID:21411326; https://doi.org/10.1016/j.tibs.2011.02.002
- Hubin E, Deroo S, Schierle GK, Kaminski C, Serpell L, Subramaniam V, van Nuland N, Broersen K, Raussens V, Sarroukh R. two distinct β-sheet structures in Italian-mutant amyloid-beta fibrils: a potential link to different clinical phenotypes. Cell Mol Life Sci 2015; 72:4899-913; PMID:26190022; https://doi.org/10.1007/s00018-015-1983-2
- Boxer AL, Rabinovici GD, Kepe V, Goldman J, Furst AJ, Huang SC, Baker SL, O'Neil JP, Chui H, Geschwind MD, Small GW, Barrio JR, Jagust W, Miller BL. Amyloid imaging in distinguishing atypical prion disease from Alzheimer disease. Neurology 2007; 69:283-290; PMID:17636066; https://doi.org/10.1212/01.wnl.0000265815.38958.b6
- Kepe V, Ghetti B, Farlow MR, Bresjanac M, Miller K, Huang SC, Wong KP, Murrell JR, Piccardo P, Epperson F, Repovs G, Smid LM, Petric A, Siddarth P, Liu J, Satyamurthy N, Small GW, Barrio JR. PET of brain prion amyloid protein in Gerstmann-Sträussler-Sheinker disease. Brain Pathology 2010; 20:419-430; PMID:19725833; https://doi.org/10.1111/j.1750-3639.2009.00306.x
- Hyare H, Ramlackhansingh A, Gelosa G, Edison P, Rudge P, Brandner S, Brooks DJ, Collinge J, Mead S. 11C-PiB does not detect PrP-amyloid in prion disease patients including variant Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry 2012; 83:340-341; PMID:21478204; https://doi.org/10.1136/jnnp.2010.233692
- Matías-Guiu JA, Oreja-Guevara C, Cabrera-Martín MN, Moreno-Ramos T, Carreras JL, Matías-Guiu J. Amyloid proteins and their role in multiple sclerosis. Considerations in the use of amyloid-PET imaging. Front Neurol 2016; 7:53
- Budka H. Neuropathology of prion diseases. Br Med Bull 2003; 66:121-130; PMID:14522854; https://doi.org/10.1093/bmb/66.1.121
- Mente KP, O'Donnell JK, Jones SE, Cohen ML, Thompson NR, Bizzi A, Gambetti P, Safar JG, Appleby BS. Fluorodeoxyglucose positron emission tomography (FDG-PET) correlation of histopathology and MRI in prion disease. Alzheimer Dis Assoc Disord 2017; 31(1):1-7; PMID:28121634; https://doi.org/10.1097/WAD.0000000000000188
- Matías-Guiu JA, Pytel V, Cabrera-Martín MN, Galán L, Valles-Salgado M, Guerrero A, Moreno-Ramos T, Matías-Guiu J, Carreras JL. Amyloid- and FDG-PET imaging in amyotrophic lateral sclerosis. Eur J Nucl Med Mol Imaging 2016; 43:2050-2060; PMID:27262702; https://doi.org/10.1007/s00259-016-3434-1
- Matías-Guiu JA, Cabrera-Martín MN, Matías-Guiu J, Oreja-Guevara C, Riola-Parada C, Moreno-Ramos T, Arrazola J, Carreras JL. Amyloid PET imaging in multiple sclerosis: an (18)F-Florbetaben study. BMC Neurol 2015; 15:243; PMID:26607782; https://doi.org/10.1186/s12883-015-0502-2
- Barthel H, Gertz HJ, Dresel S, Peters O, Bartenstein P, Buerger K, Hiemeyer F, Wittemer-Rump SM, Seibyl J, Reininger C, Sabri O; Florbetaben Study Group. Cerebral amyloid-beta PET with florbetaben (18F) in patients with Alzheimer's disease and healthy controls: a multicentre phase 2 diagnostic study. Lancet Neurol 2011; 10:424-435; PMID:21481640; https://doi.org/10.1016/S1474-4422(11)70077-1
- Linding R., Schymkowitz J., Rousseau F., Diella F., Serrano L. A comparative study of the relationship between protein structure and beta-aggregation in globular and intrinsically disordered proteins. J. Mol. Biol 2004; 342:345-353; PMID:15313629; https://doi.org/10.1016/j.jmb.2004.06.088
- Chiti F., Dobson C. M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem 2006: 75:333-366; PMID:16756495; https://doi.org/10.1146/annurev.biochem.75.101304.123901
- Xing XW, Zhang JT, Zhu T, Ma L, Yin DY, Jia WQ, Huang XS, Pu CQ, Lang SY, Yu SY. Comparison of diffusion-weighted MRI with 18F-fluorodeoxyglucose-positron emission tomography/CT and electroencephalography in sporadic Creutzfeldt-Jakob disease. J Clin Neurosci 2012; 19:1354-1357; PMID:22795494; https://doi.org/10.1016/j.jocn.2011.11.035
- Ortega-Cubero S, Pagola I, Luquin MR, Viteri C, Pastor P, Gállego Pérez-Larraya J, de Castro P, Domínguez I, Irimia P, Martínez-Vila E, Arbizu J, Riverol M. Clinical and neuroimaging characteristics of 14 patients with prionopathy: a descriptive study. Neurología 2015; 30:144-152; https://doi.org/10.1016/j.nrl.2013.12.004
- Cistaro A, Cassalia L, Ferrara C, Atzori C, Vai D, Quartuccio N, Fania P, Vaudano GP, Imperiale D. Brain 18F-FDG PET/CT findings in a case of genetic Creutzfeldt-Jakob disease due to V203I heterozygous mutation in the PRNP gene. J Neurol 2017; 264:170-173; PMID:27844164; https://doi.org/10.1007/s00415-016-8327-5