
ABSTRACT
Considerable advances in understanding the protein features favoring prion formation in yeast have facilitated the development of effective yeast prion prediction algorithms. Here we discuss a recent study in which we systematically explored the utility of the yeast prion prediction algorithm PAPA for designing mutations to modulate the aggregation activity of the human prion-like protein hnRNPA2B1. Mutations in hnRNPA2B1 cause multisystem proteinopathy in humans, and accelerate aggregation of the protein in vitro. Additionally, mutant hnRNPA2B1 forms cytoplasmic inclusions when expressed in Drosophila, and the mutant prion-like domain can substitute for a portion of a yeast prion domain in supporting prion activity in yeast. PAPA was quite successful at predicting the effects of PrLD mutations on prion activity in yeast and on in vitro aggregation propensity. Additionally, PAPA successfully predicted the effects of most, but not all, mutations in the PrLD of the hnRNPA2B1 protein when expressed in Drosophila. These results suggest that PAPA is quite effective at predicting the effects of mutations on intrinsic aggregation propensity, but that intracellular factors can influence aggregation and prion-like activity in vivo. A more complete understanding of these intracellular factors may inform the next generation of prion prediction algorithms.
Numerous prions in yeast result from the structural conversion of proteins into an insoluble amyloid form (for review, seeCitation1). Most of the yeast prion proteins contain a glutamine/asparagine (Q/N) rich prion domain that drives prion formation. Hundreds of human proteins contain prion-like domains, defined as regions with high amino acid compositional similarity to the yeast prion domains.Citation2 Recently, mutations in several of these PrLD-containing proteins have been linked to various degenerative disorders, including amyotrophic lateral sclerosis (ALS), frontotemporal dementia, and inclusion body myopathy (for review, seeCitation2,3) Therefore, there has been considerable interest in understanding how mutations in prion and prion-like proteins affect aggregation propensity.
Prion formation by Q/N-rich yeast prion proteins appears to be driven primarily by amino acid composition, not primary sequence.Citation4,5 We previously developed a quantitative mutagenesis method to empirically estimate the prion propensities of the 20 canonical amino acids within the context of a Q/N-rich prion domain.Citation6 Amino acids with similar physicochemical properties tend to have similar prion propensity scores, suggesting that basic physicochemical properties of the amino acid groups are largely responsible for their effects on prion formation. These prion propensity scores allowed us to develop a prion prediction algorithm (which we named the Prion Aggregation Prediction Algorithm, or PAPA).Citation6,7 PAPA is reasonably effective at predicting the prion-like activity of Q/N-rich yeast PrLDs,6,7 and at predicting the effects of mutations on yeast prion domains.Citation8,9
In a recent study, we examined whether PAPA would be similarly proficient at predicting the effects of mutations in human prion-like proteins.Citation10 The human heterogeneous nuclear ribonucleoprotein hnRNPA2B1, which is expressed as 2 isoforms (A2 and B1), provides a useful system to examine this question. An aspartic acid to valine substitution in the prion-like domain of hnRNPA2B1 causes mislocalization and aggregation of the protein, resulting in multisystem proteinopathy in humans.Citation11 When expressed in Drosophila, the wild-type protein is predominantly detergent-soluble and localized to the nucleus, while the mutant protein is largely detergent-insoluble, forms cytoplasmic inclusions, and causes muscle degeneration. In vitro, the hnRNPA2 protein (as well as the related hnRNPA1 protein) can undergo liquid-liquid phase separation,Citation12,13 while the disease-associated mutations in both proteins accelerate conversion to an insoluble amyloid form.Citation11,12 Finally, in yeast, the core PrLD from mutant hnRNPA2 can support prion activity when substituted into the prion nucleation domain of the yeast prion protein Sup35.Citation11 Thus, hnRNPA2 offers a variety of experimental systems to examine the effects of mutation on aggregation activity.
RATIONALLY MANIPULATING THE AGGREGATION ACTIVITY OF A HUMAN PRION-LIKE DOMAIN
PAPA accurately predicts that the disease-associated D290V mutation in hnRNPA2 should increase aggregation propensity. While this is encouraging, a single mutant is clearly too small a sample size to draw any broad conclusions. According to our prion propensity estimates, aspartic acid is not expected to be uniquely prion inhibiting, and valine is not expected to be uniquely prion-promoting. Additionally, since prion formation is predominantly composition-dependent, the precise location of a given mutation within a PrLD should not dramatically alter its effects. To test these predictions, and to more comprehensively investigate PAPA's prediction accuracy, we designed a range of single and double mutations categorized as prion-promoting, neutral, or prion-inhibiting based on PAPA's predictions.Citation10
Because of the ease of assaying prion activity in yeast, we initially tested a set of 25 mutants in yeast. As demonstrated previously, when the wild-type hnRNPA2 PrLD was inserted in the place of the portion of Sup35 responsible for prion nucleation (amino acids 1–40),Citation14 the resulting fusion displayed little to no prion activity; by contrast, the disease-associated D290V mutation dramatically enhanced its prion formation in yeast.Citation11 We tested 8 additional substitutions at the 290 position, and found in all cases that PAPA accurately predicted their effects on prion formation. Furthermore, PAPA successfully predicted the combinatorial effects of various mutations at alternative positions. This demonstrates that PAPA is a good predictor of the prion propensity of human PrLDs when expressed in yeast, even when the mutations differ from those observed to cause disease in humans.
Additionally, PAPA accurately predicted the effects on aggregation and associated cellular pathology of most hnRNPA2 mutations in Drosophila. However, in a few cases, mutations resulted in unexpected deviation from PAPA predictions, and from our results in yeast. For example, PAPA predicts that tyrosine and isoleucine will have similar effects on prion formation. As predicted, the “neutral” Y283I substitution (in combination with the D290V mutation) maintained high rates of prion formation in yeast. However, the Y283I mutation completely restored the solubility of hnRNPA2(D290V) in Drosophila.
We used in vitro experiments to examine the mutants that showed divergent behavior between yeast and Drosophila. A protein's aggregation in vivo will depend on both its intrinsic aggregation propensity and its interactions with other cellular factors. In each case where the Drosophila and yeast results diverged, the in vitro aggregation kinetics were consistent with the yeast results (and with PAPA predictions). This suggests that both PAPA and our yeast system accurately predict intrinsic aggregation propensity, but that additional cellular factors may influence aggregation in Drosophila.
It should be noted that this ability to design mutations to modulate intrinsic aggregation propensity may not be unique to PAPA. A subsequent reanalysis of our data set showed that PrionW is also reasonably effective at predicting prion activity in yeast for our mutants, and that combining PrionW and PAPA may further improve prediction accuracy.Citation15 PrionW differs from PAPA in that it combines amino acid composition analysis with a position-specific matrix to identify predicted amyloid-prone segments.Citation16 Other algorithms show varying degrees of predictive success. ZipperDB uses structure-based modeling to identify 6-amino-acid peptides with a high predicted propensity to form steric zipper segments.Citation17 For all aggregation-promoting mutations at the disease-associated position in hnRNPA2, ZipperDB predicted that the mutations would create a strong steric zipper.Citation10,11 However, when we designed subsequent mutations to specifically test the importance of these predicted steric zippers, we found that the presence of predicted steric zippers was neither necessary nor sufficient for strong aggregation activity in yeast or in vitro.Citation10 ArchCandy predicts the probability that a sequence will adopt a β-arch.Citation18 Prion-promoting mutations tended to increase the number of predicted β-arches, or the scores for the predicted β-arches, although in some cases these effects are only seen by adjusting the default scoring threshold (data not shown). Other algorithms were less effective. For example, PLAACCitation19 correctly identifies the PrLD in hnRNPA2,Citation11 but was not effective at predicting the effects of mutations;Citation10,11 AggrescanCitation20 fails to identify aggregation hot spots in most of the aggregation-prone mutants (data not shown); and TANGOCitation21 shows a relatively poor correlation between predicted and observed aggregation activity (data not shown).
FUTURE CHALLENGES IN THE PREDICTION OF PRION-LIKE AGGREGATION
The ability to rationally design mutations to modulate aggregation activity will provide a powerful tool for examining functional and pathogenic aggregation by PrLDs. An emerging theory suggests that many PrLDs have evolved to form dynamic, reversible interactions that mediate the formation of functional assemblies, including stress granules and P-bodies, and that mutations may cause disease by disrupting the dynamics of these assemblies.Citation3 Consistent with this theory, the cytoplasmic inclusions observed for disease-associated hnRNPA1 and A2 mutants are RNA-protein granules.Citation11,22 However, the limited number of naturally occurring pathogenic mutations makes this theory difficult to test. Additionally, most mutations experimentally designed to test the role of PrLDs in functional or pathogenic aggregation have involved substantial changes in protein sequence, such as deletion or substitution of the entire PrLD (for example, seeCitation23,24). Such mutations may have other effects on the protein. By contrast, we observed dramatic changes in aggregation with just single and double point mutations.Citation10 Thus, it should be possible to design a variety of mutations in other PrLDs to more rigorously explore the role of PrLDs in functional and pathogenic aggregation.
However, our results also demonstrate a fundamental challenge in predicting protein aggregation: although methods like PAPA can be quite effective at predicting the intrinsic aggregation propensity of proteins (Pathway 1 in ), in a complex cellular environment, a variety of interactions may influence aggregation (). These other factors are not currently incorporated into any prion prediction algorithm. It is worth noting that some prediction algorithms were derived from analysis of sequences that demonstrate bona fide prion activity in yeast.Citation6,19,25 Therefore, to some degree, they indirectly integrate the cumulative influence of an intracellular environment into their predictions; however, this may also limit their adaptability to other organisms.
FIGURE 1. Intracellular processes commonly affecting PrLD aggregation. Mutations, changes in protein concentration, or post-translational modifications in PrLDs can affect protein aggregation by altering intrinsic aggregation propensity (1); liquid-liquid phase separation/stress granule dynamics (2); conversion of granules to a pathological state (3); organism-, tissue-, or compartment-specific intermolecular interactions (4); proteasome-mediated degradation (5); or nucleocytoplasmic transport (6).
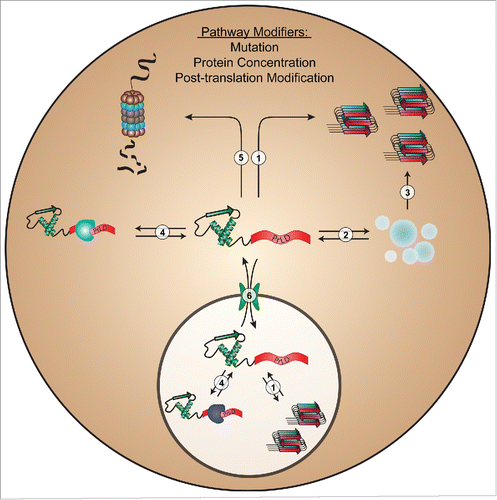
Indeed, while it is encouraging that most mutants showed similar behavior in yeast, Drosophila and in vitro, the rare deviations among these systems highlight a limitation of using any single model system to study protein aggregation. Without understanding the basis for these differences, it is unclear whether the yeast or Drosophila results will be more predictive of human disease.
Here we discuss some of the factors that may influence in vivo protein aggregation, many of which were evident among our panel of mutants. While it may not be feasible to take into account all of these factors on a protein-by-protein basis, a better understanding of how these factors influence aggregation activity could lead to better-informed predictions.
Protein Expression and Abundance
Aggregation is a concentration-dependent process,Citation26 so intracellular protein concentrations could affect the probability of self-association and prion formation. By extension, the free intracellular concentration of a given protein might be affected by the concentrations and affinities of molecular interacting partners, such as RNA or other proteins. For example, β-thalassemia results from mutations that decrease β-globin levels, resulting in an imbalance in the ratio of α-globin to β-globin, and aggregation of the excess α-globin.Citation27 Thus, the aggregation is not caused by a change in α-globin's aggregation propensity, but instead by a change in the concentration of its binding partner. Protein levels and molecular interacting partners vary from organism to organism, from tissue to tissue, and even between subcellular compartments, potentially explaining why many aggregation-based diseases only affect specific tissues. Mutations could therefore affect aggregation by altering protein expression or stability, or by disrupting stabilizing protein-protein interactions.
Distinct Steps in Protein Aggregation
The formation of amyloid aggregates involves at least 2 steps: nucleation and fiber growth.Citation26 Additionally, because amyloid fibers grow from their ends, fragmentation of fibers allows for the exponential amplification of amyloid aggregates by creating new ends for fiber growth.Citation28 Each of these steps likely has distinct sequence/compositional requirements,Citation14,29 yet existing prediction algorithms do not separately examine the effects of mutations on each step. Furthermore, because each of these steps is likely influenced by distinct sets of chaperone proteins (for review, seeCitation1,30), the different chaperone environments in different cells types or organisms may further affect the sequence requirements for prion-like aggregation.
One of our double-mutants, D276V/D290V, appeared to show unique aggregate dynamics in Drosophila.Citation10 As predicted, these prion-promoting mutations had an additive effect on the rate of prion formation in yeast, and resulted in the formation of cytoplasmic aggregates in Drosophila. However, the aggregates in Drosophila were greater in number and smaller in size compared with those formed by the D290V single mutant, suggesting that the dynamics of aggregate formation or fragmentation had been fundamentally altered.
Liquid-Liquid Phase Separation
A number of recent studies demonstrate that many proteins with PrLDs undergo concentration-dependent liquid-liquid phase separation (Pathway 2 in ) under a variety of conditions, including changes in salt concentration, RNA concentration, poly(ADP)-ribose, pH, and temperature (for review, seeCitation31). In many cases, the PrLDs play a direct role in liquid-liquid de-mixing, or affect de-mixing indirectly. This biophysical process has been proposed as the basis for the formation of various non-membrane-bound organelles, and can occur in both the cytoplasm (e.g., stress granules and P-bodies) and the nucleus (e.g., nucleolus, nucleolar subcompartments, cajal bodies, nuclear paraspeckles, etc.).Citation32 Protein de-mixing greatly enhances the local concentrations of incorporated proteins, which may increase the likelihood of forming stable amyloid-like aggregates. Mutations within PrLDs may also perturb the dynamics of these assemblies, which can result in the formation of off-pathway pathological aggregates (Pathway 3 in ). It is also unclear whether differences in subcellular environments (particularly, between the nucleus and the cytoplasm) can affect PrLDs differently based on their sequence and composition. Further investigation is necessary to develop a more complete mechanistic understanding of the biophysical conditions and protein features that govern liquid-liquid phase separation, which may ultimately inform prion prediction methods.
Aggregation, Proteostasis, and Aging
For most proteins, misfolding and aggregation represent a cytotoxic threat to cells. Accordingly, cells possess an arsenal of protein quality control factors to maintain proper proteostasis. The ability to evade the proteostasis machinery may be an important protein trait when considering aggregation propensity in vivo. Furthermore, cellular proteostasis typically declines with age (for review, seeCitation33), so aggregation-promoting mutations in PrLDs coupled with an age-related decline in cellular proteostasis may contribute to the late-onset nature of many neurodegenerative diseases. Mutations could affect aggregation by altering interactions with the proteostasis machinery, which includes protein chaperones and protein degradation systems (Pathways 4 and 5 in ). A deeper understanding of the rules governing cellular recognition and handling systems of aggregation-prone proteins may facilitate improvements in prion prediction methods.
Nucleocytoplasmic Shuttling of Prion-Like Proteins
Many of the disease-linked PrLD-containing proteins are predominantly nuclear, but form cytoplasmic inclusions in diseased patients. This has led some to propose that disruption of nucleocytoplasmic transport is a key mediator of disease pathology (for review, seeCitation34). Aberrant transport dynamics of PrLD-containing proteins could lead to accumulation of the protein in the cytosol, which may increase aggregation risk (Pathway 6 in ). A particularly well-characterized example is the PrLD-containing protein, fused in sarcoma (FUS); many ALS-associated mutations in FUS directly impair its nuclear import and lead to accumulation in cytoplasmic aggregates.Citation35 Although it is clear that nucleocytoplasmic transport is affected in disease, it is currently unclear whether subcellular localization and trafficking dynamics directly influence or simply coincide with protein aggregation. Understanding how mutations alter intracellular trafficking dynamics may shed light on why many of these proteins form cytoplasmic inclusions.
Intriguingly, for one of our mutants, aggregation and cytoplasmic mislocalization were uncoupled. Consistent with PAPA predictions, a D290F mutation enhanced prion formation in yeast and formation of detergent insoluble aggregates in Drosophila. However, while hnRNPA2(D290V) formed cytoplasmic inclusions, hnRNPA2(D290F) aggregates appeared to localize to the nucleus, suggesting that while both mutations increase aggregation propensity, the mutations have different effects on nucleocytoplasmic transport dynamics. Such mutants may also provide insight into the relative roles of aggregation and mislocalization in cellular pathology.
Post-Translational Regulation of Prion-Like Proteins
Post-translational modifications within or near PrLDs could affect intrinsic aggregation propensity, liquid-liquid phase separation, intracellular trafficking, targeted degradation, or a variety of other processes. The human PrLDs tend to contain a high proportion of glutamine, asparagine, serine, and tyrosine residues (for review, seeCitation36), which can all be modified post-translationally. Additionally, the hnRNPA1 and hnRNPA2 PrLDs (as well as other PrLD-containing proteins, including FUS, EWSR1, and TAF15; for review, seeCitation37) possess a small number of arginine residues within or near the PrLDs that are asymmetrically di-methylated.Citation38,39 Methylation of these residues in the hnRNPs or similar PrLD-containing proteins has been shown to regulate nucleocytoplasmic shuttling,Citation40,41 liquid-liquid phase separation,Citation42 stress granule assembly,Citation43 and pathological protein aggregation.Citation35,40
Therefore, post-translational modifications can affect directly or indirectly a multitude of intracellular processes that relate to protein aggregation. Mutations may affect aggregation by altering these post-translational modifications. Indeed, in Drosophila the insoluble fraction of hnRNPA2(D290V) appears as 2 distinct bands by western blot, suggesting a post-translational modification. This double-band was not observed for any of the other hnRNPA2 mutants (including the D276V/D290V double mutant), and this modification is not detectable when the D290V PrLD was expressed in yeast. It is unclear whether this modification affects aggregation or pathology.
CONCLUSION
Our intention is to highlight the interplay between intrinsic prion domain features and prion formation in a cellular context. A generalized understanding of the features governing these prion-modifying pathways may offer ways to explicitly weight prion predictions with additional intracellular considerations. This will be especially important when extending prion predictions to multicellular eukaryotic organisms, in which organism-specific and tissue-specific intracellular conditions may differ substantially.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
FUNDING
This work was supported by the National Institutes of Health under Grant GM105991 to EDR.
REFERENCES
- Liebman SW, Chernoff YO. Prions in yeast. Genetics. 2012; 191:1041-72. doi:10.1534/genetics.111.137760 PMID:22879407
- King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012; 1462:61-80. doi:10.1016/j.brainres.2012.01.016 PMID:22445064
- Taylor JP, Brown RH, Jr., Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016; 539:197-206. doi:10.1038/nature20413 PMID:27830784
- Afsar Minhas FUA, Ross ED, Ben-Hur A. Amino acid composition predicts prion activity. PLoS Comput Biol. 2017; 13:e1005465. doi:10.1371/journal.pcbi.1005465 PMID:28394888
- Ross ED, Edskes HK, Terry MJ, Wickner RB. Primary sequence independence for prion formation. Proc. Natl. Acad. Sci. USA. 2005; 102:12825-30; doi:10.1073/pnas.0506136102
- Toombs JA, McCarty BR, Ross ED. Compositional determinants of prion formation in yeast. Mol Cell Biol. 2010; 30:319-32. doi:10.1128/MCB.01140-09 PMID:19884345
- Toombs JA, Petri M, Paul KR, Kan GY, Ben-Hur A, Ross ED. De novo design of synthetic prion domains. Proc Natl Acad Sci USA. 2012; 109:6519-24. doi:10.1073/pnas.1119366109 PMID:22474356
- Gonzalez Nelson AC, Paul KR, Petri M, Flores N, Rogge RA, Cascarina SM, Ross ED. Increasing prion propensity by hydrophobic insertion. PLoS One. 2014; 9:e89286. doi:10.1371/journal.pone.0089286 PMID:24586661
- Paul KR, Hendrich CG, Waechter A, Harman MR, Ross ED. Generating new prions by targeted mutation or segment duplication. Proc Natl Acad Sci USA. 2015; 112:8584-9. doi:10.1073/pnas.1501072112 PMID:26100899
- Paul KR, Molliex A, Cascarina S, Boncella AE, Taylor JP, Ross ED. The effects of mutations on the aggregation propensity of the human prion-like protein hnRNPA2B1. Mol Cell Biol. 2017:e00652-16. doi:10.1128/MCB.00652-16. PMID:28137911
- Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013; 495:467-73. doi:10.1038/nature11922 PMID:23455423
- Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 2015; 163:123-33. doi:10.1016/j.cell.2015.09.015 PMID:26406374
- Xiang S, Kato M, Wu LC, Lin Y, Ding M, Zhang Y, Yu Y, McKnight SL. The LC domain of hnRNPA2 adopts similar conformations in hydrogel polymers, liquid-like droplets, and nuclei. Cell. 2015; 163:829-39. doi:10.1016/j.cell.2015.10.040 PMID:26544936
- Osherovich LZ, Cox BS, Tuite MF, Weissman JS. Dissection and design of yeast prions. PLoS Biol. 2004; 2:E86. doi:10.1371/journal.pbio.0020086 PMID:15045026
- Batlle C, Fernandez MR, Iglesias V, Ventura S. Perfecting prediction of mutational impact on the aggregation propensity of the ALS-associated hnRNPA2 prion-like protein. FEBS Lett. 2017; 591:1966-1971. doi:10.1002/1873-3468.12698 PMID: 28542905
- Zambrano R, Conchillo-Sole O, Iglesias V, Illa R, Rousseau F, Schymkowitz J, Sabate R, Daura X, Ventura S. PrionW: a server to identify proteins containing glutamine/asparagine rich prion-like domains and their amyloid cores. Nucleic Acids Res. 2015; 43:W331-7. doi:10.1093/nar/gkv490 PMID:25977297
- Goldschmidt L, Teng PK, Riek R, Eisenberg D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. USA. 2010; 107:3487-92; doi:10.1073/pnas.0915166107
- Ahmed AB, Znassi N, Chateau MT, Kajava AV. A structure-based approach to predict predisposition to amyloidosis. Alzheimers Dement. 2015; 11:681-90. doi:10.1016/j.jalz.2014.06.007 PMID:25150734
- Lancaster AK, Nutter-Upham A, Lindquist S, King OD. PLAAC: a web and command-line application to identify proteins with prion-like amino acid composition. Bioinformatics. 2014; 30:2501-2. doi:10.1093/bioinformatics/btu310 PMID:24825614
- Sanchez de Groot N, Pallares I, Aviles FX, Vendrell J, Ventura S. Prediction of “hot spots” of aggregation in disease-linked polypeptides. BMC Struct Biol. 2005; 5:18. doi:10.1186/1472-6807-5-18 PMID:16197548
- Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol. 2004; 22:1302-6. doi:10.1038/nbt1012 PMID:15361882
- Li S, Zhang P, Freibaum BD, Kim NC, Kolaitis RM, Molliex A, Kanagaraj AP, Yabe I, Tanino M, Tanaka S, et al. Genetic interaction of hnRNPA2B1 and DNAJB6 in a Drosophila model of multisystem proteinopathy. Hum Mol Genet. 2016; 25:936-50. doi:10.1093/hmg/ddv627 PMID:26744327
- Decker CJ, Teixeira D, Parker R. Edc3p and a glutamine/asparagine-rich domain of Lsm4p function in processing body assembly in Saccharomyces cerevisiae. J Cell Biol. 2007; 179:437-49. doi:10.1083/jcb.200704147 PMID:17984320
- Gilks N, Kedersha N, Ayodele M, Shen L, Stoecklin G, Dember LM, Anderson P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol Biol Cell. 2004; 15:5383-98. doi:10.1091/mbc.E04-08-0715 PMID:15371533
- Espinosa Angarica V, Ventura S, Sancho J. Discovering putative prion sequences in complete proteomes using probabilistic representations of Q/N-rich domains. BMC Genomics. 2013; 14:316. doi:10.1186/1471-2164-14-316 PMID:23663289
- Ferrone F. Analysis of protein aggregation kinetics. Methods Enzymol. 1999; 309:256-74. doi:10.1016/S0076-6879(99)09019-9 PMID:10507029
- Goodman MA, Malik P. The potential of gene therapy approaches for the treatment of hemoglobinopathies: achievements and challenges. Ther Adv Hematol. 2016; 7:302-15. doi:10.1177/2040620716653729 PMID:27695619
- Collins SR, Douglass A, Vale RD, Weissman JS. Mechanism of prion propagation: amyloid growth occurs by monomer addition. PLoS Biology. 2004; 2:e321. doi:10.1371/journal.pbio.0020321 PMID:15383837
- MacLea KS, Paul KR, Ben-Musa Z, Waechter A, Shattuck JE, Gruca M, Ross ED. Distinct amino acid compositional requirements for formation and maintenance of the [PSI(+)] prion in yeast. Mol Cell Biol. 2015; 35:899-911. doi:10.1128/MCB.01020-14 PMID:25547291
- Chernova TA, Wilkinson KD, Chernoff YO. Prions, chaperones, and proteostasis in yeast. Cold Spring Harb Perspect Biol. 2017:a023663. doi:10.1101/cshperspect.a023663 PMID:27815300
- Aumiller WM, Jr., Keating CD. Experimental models for dynamic compartmentalization of biomolecules in liquid organelles: Reversible formation and partitioning in aqueous biphasic systems. Adv Colloid Interface Sci. 2017; 239:75-87. doi:10.1016/j.cis.2016.06.011 PMID:27401136
- Hyman AA, Weber CA, Julicher F. Liquid-liquid phase separation in biology. Annu Rev Cell Dev Biol. 2014; 30:39-58. doi:10.1146/annurev-cellbio-100913-013325 PMID:25288112
- Labbadia J, Morimoto RI. The biology of proteostasis in aging and disease. Annu Rev Biochem. 2015; 84:435-64. doi:10.1146/annurev-biochem-060614-033955 PMID:25784053
- Jovicic A, Paul JW, 3rd, Gitler AD. Nuclear transport dysfunction: a common theme in amyotrophic lateral sclerosis and frontotemporal dementia. J Neurochem. 2016; 138:134-44. doi:10.1111/jnc.13642 PMID:27087014
- Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B, et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010; 29:2841-57. doi:10.1038/emboj.2010.143 PMID:20606625
- Cascarina SM, Ross ED. Yeast prions and human prion-like proteins: sequence features and prediction methods. Cell Mol Life Sci. 2014; 71:2047-63. doi:10.1007/s00018-013-1543-6 PMID:24390581
- Thandapani P, O'Connor TR, Bailey TL, Richard S. Defining the RGG/RG motif. Mol Cell. 2013; 50:613-23. doi:10.1016/j.molcel.2013.05.021 PMID:23746349
- Beyer AL, Christensen ME, Walker BW, LeStourgeon WM. Identification and characterization of the packaging proteins of core 40S hnRNP particles. Cell. 1977; 11:127-38. doi:10.1016/0092-8674(77)90323-3 PMID:872217
- Wilk HE, Werr H, Friedrich D, Kiltz HH, Schafer KP. The core proteins of 35S hnRNP complexes. Characterization of nine different species. Eur J Biochem. 1985; 146:71-81. doi:10.1111/j.1432-1033.1985.tb08621.x PMID:3881256
- Fujii S, Takanashi K, Kitajo K, Yamaguchi A. Treatment with a global methyltransferase inhibitor induces the intranuclear aggregation of ALS-Linked FUS mutant in vitro. Neurochem Res. 2016; 41:826-35. doi:10.1007/s11064-015-1758-z PMID:26603295
- Nichols RC, Wang XW, Tang J, Hamilton BJ, High FA, Herschman HR, Rigby WF. The RGG domain in hnRNP A2 affects subcellular localization. Exp Cell Res. 2000; 256:522-32. doi:10.1006/excr.2000.4827 PMID:10772824
- Nott TJ, Petsalaki E, Farber P, Jervis D, Fussner E, Plochowietz A, Craggs TD, Bazett-Jones DP, Pawson T, Forman-Kay JD, et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol Cell. 2015; 57:936-47. doi:10.1016/j.molcel.2015.01.013 PMID:25747659
- Wall ML, Lewis SM. Methylarginines within the RGG-Motif region of hnRNP A1 affect its IRES trans-acting factor activity and are required for hnRNP A1 stress granule localization and formation. J Mol Biol. 2017; 429:295-307. doi:10.1016/j.jmb.2016.12.011 PMID:27979648