
ABSTRACT
We reported the case of a patient with Wernicke-Korsakoff syndrome (WKs) as an early clinical manifestation of sporadic Creutzfeld-Jakob disease (sCJD). The 66-year-old female complained of dizziness and imbalance which mostly occurred while walking. A neurological examination revealed a triad of symptoms characteristic for WKs such as gaze paresis, ataxia of limbs and trunk as well as memory disturbances with confabulations. The disturbances increased during the course of the disease, which led to the death of the patient four months after the appearance of the signs. The patient was finally diagnosed with sCJD disease. The most useful ancillary examination results supporting sCJD diagnosis were brain diffusion DWI MRI (diffusion weighted magnetic resonance imaging) and the presence of 14–3-3 protein in CSF (cerebrospinal fluid). Since that manifestation of sCJD is very unique other causes should be taken into consideration while making a final diagnosis.
1. Introduction
Early clinical symptoms of sporadic Creutzfeldt-Jakob disease (sCJD) may resemble those of other neurodegenerative diseases like Alzheimer disease and frontotemporal dementia (FTD). In this and similar cases, the first manifested symptoms may lead to a misdiagnosis or a delayed diagnosis of sCJD. We are presenting the case of a patient with clinical manifestation of Wernicke-Korsakoff syndrome (WKs) who was finally diagnosed with sCJD. The rare early clinical presentation of sCJD as WKs makes this case exceptional.
2. Case presentation
2.1. History
The 66-year-old female patient was admitted to the Department of Neurology of Medical University of Lublin for diagnostic purposes in June 2015. The woman complained of difficulties in walking because of dizziness and imbalance. The patient did not mention other problems with memory or vision. However, family members observed changes in her personality and mood which deeply disturbed her everyday activities. The patient's relatives also mentioned that the patient had been mentally healthy and all problems had begun about four weeks earlier and had gradually aggravated. Before the appearance of neurological problems the patient had been treated for rheumatoid arthritis with metotrexat. The disease had been diagnosed 6 months earlier. The course of the disease was benign and a rheumatologist described her condition as stable. The patient did not abuse alcohol or drugs, was not addicted to nicotine, and had no history of allergy.
2.2. Examinations on admission
A neurological examination revealed that the patient was not fully orientated, especially with regard to the time and the place. The woman also had difficulty in recalling some events from late and early past, but she covered gaps of memory with confabulations. Her mood was euphoric, which did not correspond to her poor condition. We noticed disturbances in eye movement with the paresis of vertical and horizontal gaze to a milder degree. There were no other abnormalities in cranial nerves. The patient also presented discoordination of upper and lower limbs, which disabled independent walking. We also observed an imbalance of trunk which caused instability of upright position. The strength and reflexes were normal and there were neither sensory disturbances nor pathological signs present. In neuropsychological examination the patient presented corrected autopsychical but uncorrected allopsychical orientation. The slowness of psychical processes was observed together with difficulties in concentration, attention, logical thinking and learning. The fresh and late memory was impaired with a tendency to confabulations. The verbal fluency abated.
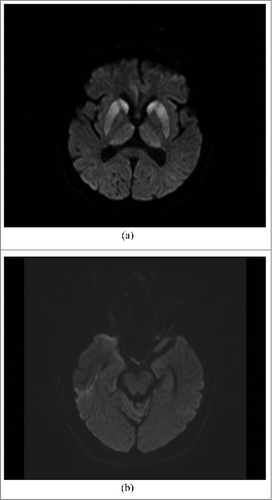
Computed Tomography (CT) was normal. Magnetic Resonance Imaging (MRI) was performed with the use of Siemens Avanto1.5T. The most informative were DWI (diffusion-weighted imaging) sequences which showed a bilateral restriction of diffusion in heads of caudate nuclei, thalami (),and cortical layers of the right temporal lobe (). A very slight, hyperintense signal was also observed in inferior colliculi of midbrain (). There were no abnormalities in laboratory tests, including the level of vitamin B1 whose deficiency could be suggested by clinical and MRI presentation. The general appearance and composition of cerebrospinal fluid (CSF) was normal except for a slight elevation in the protein level (67mg/dl). The electroencephalogram (EEG) showed non-specific diffuse slowing activity with polymorphic delta and alpha waves activity with a predominance of the fronto-centro-parietal part of the brain.
Figure 1. (A). MRI DWI: abnormal high signal intensity in both caudate nuclei, putamina and thalami, particularly prominent in heads of caudate nuclei and anterior parts of putamina. (B). MRI DWI: slight changes in cortex of right temporal lobe and colliculi inferior.
2.3. Examinations during 3 months after admission
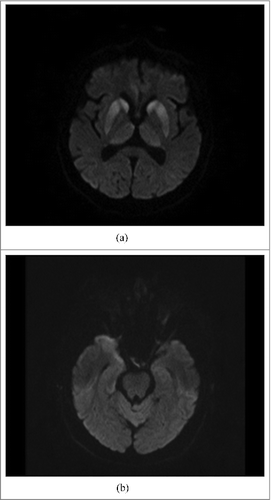
The second DWI MRI, carried out 2 weeks after the first one, showed the same abnormal high signal intensity in both sites of deep structures of the brain (), in the right temporal lobe cortical layers, and the same, very slight signal in inferior colliculi of midbrain. Additionally, analogous abnormal high signal was observed in the cortical layers of the left temporal lobe (). PET (Positron Emission Tomography), which was performed 3 weeks after admission was normal, except for discreet glucose hypometabolism in regions of lentiform nuclei and the right temporal lobe. After 4 weeks the result of Western Blotting test of CSF was obtained and 14-3-3 protein was detected.
Figure 2. (A). MRI DWI: abnormal high signal intensity in both caudate nuclei, putamina and thalami, particularly prominent in heads of caudate nuclei and anterior parts of putamina (the same changes as seen on Fig. A). (B). MRI DWI: abnormal high signal intensity in cortex of anterior pole of the right temporal lobe and slight, linear hyperintensity in cortex of the left temporal lobe.
Repeated EEG performed in the 6th, 8th, and 10th weeks did not significantly change when compared with the first examination. Periodic sharp-wave complexes which are characteristic for sCJD were not present. These changes were not observed even in the final stage of the disease. The condition of the patient gradually worsened during the following weeks. Dementia, which was hardly visible in the early phase because of coexisting confabulations, became apparent after four weeks, when the cognitive dysfunction increased. Imbalance became stronger and got the patient completely bedridden. Finally, in the second month of hospitalization, the patient started to present akinetic mutism, did not move her limbs, was mute and showed a slight eye movement in response to different stimuli. It was then that the diagnosis of probable sCJD was made. In the last stage of the illness, there appeared disturbances of consciousness which made the woman feel sleepy and then go into a coma. The woman died four months after the appearance of the first neurological symptoms and 3 months after hospital admission.
3. Discussion
sCJD is a rare disease with prevalence of 0.5-1/1 mln. It is a fatal neurodegenerative condition which originates spontaneously. Conformational changes of cellular protein (PRPc) into a pathological form (PrPTSE) are crucial for the pathogenesis and formation of the infectious agent or prion [Citation1]. The peak of illness is between the ages of 55 and 70. It affects both men and women.
Currently available criteria for sCJD are published by WHO [Citation2] (World Health Organisation) and NCJDRT [Citation3] (The National CJD Research and Surveillance Unit). According to their guidelines clinical presentation should include rapidly progressive cognitive impairment and at least two clinical features such as myoclonic jerks, visual or cerebellar disturbance, pyramidal or extrapyramidal dysfunction or akinetic mutism, with duration less than 2 years. Probable sCJD should be additionally supported with no less than one feature like a typical MRI brain scan, typical EEG or a positive 14-3-3 assay for CSF. Definite sCJD can be diagnosed only when the neuropathological, immunocytochemical or biochemical tests confirm it. However, initial symptoms may be nonspecific so an initial diagnosis could be challenging.
What makes our case exceptional is its rare early clinical presentation. The primary and the most characteristic signs of the disease were the mobility dysfunctions which resulted from ataxia of limbs and trunk. Dementia was not obvious in the early phase. The features of cognitive impairment were also different from those which have been described so far in sCJD manifestations [Citation4-6]. Memory was disturbed with a tendency to confabulations, which made the cognitive impairment unnoticeable in early stages of the disease. A neurological examination revealed that gaze paresis was prominent. We did not observe myoclonic jerks or pyramidal or extrapyramidal dysfunctions even in the late phase of the disease. There are descriptions of early presentations of sCJDVV2(sCJD with homozygotic kodon 129 Valin/Valin)with rapidly progressive ataxia and oculomotor disturbances without prominent cognitive impairment [Citation7]. There are also other descriptions of unusual clinical manifestations [Citation8,Citation9] of sCJD, but none of them reports such a clear presence of a triad of signs of ataxia, gaze paresis and dementia with confabulations seen in WKs [Citation10]. The patient did not fulfil the current criteria for probable sCJD till the late stage of the disease when akinetic mutism was observed. We were obliged to consider other, potentially curable causes, like vitamin B1 deficiency, to avoid misdiagnosis which can be common in such cases [Citation11]. We considered such an approach beneficial for the patient. That is why the deficiency of vitamin B1 was justified in differential diagnosis even in obvious absence of causes like alcoholism, gastrointestinal diseases, AIDS or anorexia [Citation12]. Also other conditions were taken into account, such as vascular diseases or dementias in the course of other diseases like FTD or Alzheimer, and viral or autoimmune neuroinfections. However, these conditions seemed less probable to us.
The DWI MRI examination was very useful. Diagnostic accuracy of DWI MRI is 97% [Citation13]. The MRI images of our patient were slightly evolving during the course of the disease. We observed in DWI MRI hyperintense signals not only in putamen and caudate nucleus but also in cortical layers of the righttemporal lobe. There was also a weak signal in inferior colliculi. A second DWI MRI examination performed 2 weeks after the first one showed slow progression of the disease. Because of an unusual clinical presentation we considered MRI pictures highly supportive for the diagnosis of sCJD but not fully sufficient. The protein 14-3-3 was present. Its diagnostic accuracy is 70.4% [Citation13] but it can be present in CSF in other conditions connected with acute atrophy of neurons like, for example, viral encephalitis [Citation14].The EEG and PET examinations were not useful as a diagnostic tool in this case. EEG usually has a special role in the diagnosis of some dementing illnesses [Citation15]. Periodic waves in EEG are seen in 90% of cases especially on repeated recordings [Citation14]. EEG of our patient did not show the characteristic changes. PET examination usually demonstrates focal or diffuse hypometabolism, mainly in the temporal lobes [Citation14]. The relationship between FDG-PET hypometabolism and neuropathology region was found, but it was unhelpful for the diagnosis [Citation16]. The individual described above can probably belong to the 16% of patients with the variant VV2 of sCJD according to the genotype [Citation17]. The prominent ataxia and lack of changes in the EEG examination are the most obvious features of that type [Citation14]. Unfortunately, the analysis of the PrP genotype was not performed in this case. The most useful additional examinations in the diagnostic process of our patient were the MRI and CSF. Since the clinical manifestations were not characteristic for the disease, ruling out other mimic conditions and potentially curable illnesses was also significant during a differential diagnosis.
The rare incidence and a wide variety of clinical subtypes of sCJD make an early diagnosis complex and challenging in clinical practice. Our patient met diagnostic criteria for probable sCJD several weeks after the onset of symptoms. The significance of our case lies in the demonstration of a very rare early clinical presentation. This unusual neurological manifestation may help clinicians to take notice of early deficits of sCJD. The awareness of such exceptions can be useful in the interpretation of ancillary tests and in making a quick diagnosis.
Abbreviations
| WKs | = | Wernicke-Korsakoff syndrome |
| sCJD | = | sporadic Creutzfeld-Jakob disease |
| MRI | = | magnetic resonance imaging |
| DWI | = | diffusion weighted imaging |
| CT | = | Computed Tomography |
| CSF | = | cerebrospinal fluid |
| EEG | = | electroencephalogram |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Kovacs KG, Budka H. Prion diseases: from protein to cell pathology. Am J Pathol. 2008;172(3):555-565. doi:10.2353/ajpath.2008.070442. PMID:18245809.
- World Health Organization. http://www.who.int/en/.
- The National CJD Research and Surveillance Unit.http://www.cjd.ed.ac.uk/surveillance.
- Snowden JS, Mann DM, Neary D. Distinct neuropsychological characteristics in Creutzfeld-Jakob disease. J Neurol Neurosurg Psychiatry. 2002;73(6):686-94. doi:10.1136/jnnp.73.6.686. PMID:12438471.
- Gonzalez DA, Soble JR. Corticobasal syndrome due to sporadic Creutzfeldt-Jakob disease: a review and neuropsychological case report. Clin Neuropsychol. 2017;31(3): 676-689. doi:10.1080/13854046.2016.1259434. PMID:27871202.
- Jcghin A, Devamecourt V, Bakchire S, et al. Unusual features of Creutzfeld-Jakob disease followed-up in a memory clinic. J Neurol. 2014;261(4):696-701. doi:10.1007/s00415-014-7246-6. PMID:24477491.
- Baiardi S, Magherini A, Cappellari S, et al. Towards an early clinical diagnosis of sporadic CJDVV2. J Neurol Neurosurg Psychiatry. 2017;88(9):764-772. doi:10.1136/jnnp-2017-315942. PMID:28668775.
- Al Balushi A, Meeks MW, Hayat G, et al. Creutzfeldt-Jakob Disease: Analysis of four cases. Front Neurol. 2016;29(7):138.
- Choi YJ, Kang KW, Lee SY, et al. Creutzfeldt-Jakob disease presenting with and gaze-evoked nystagmus: a case report. Medicine (Baltimore). 2016;95(7):e2766. doi:10.1097/MD.0000000000002766. PMID:26886621.
- Scalzo SJ, Bowden SC, Amrose ML, et al. Wernicke-Korsakoff syndrome not related to alcohol use: a systematic review. J Neurol Neurosurg Psychiatry. 2015;86(12):1362-8. PMID:25589780.
- Chitronas N, Yung RS, Kofiskey DM, et al. Treatable neurological disorders misdiagnosed as Creutzfeldt-Jakob disease. Ann Neurol. 2011;70(3):437-44. doi:10.1002/ana.22454. PMID:21674591.
- Lough ME. Wernicke's encephalopathy: expanding the diagnostic toolbox. Neuropsychol Rev. 2012;22(2): 181-94. doi:10.1007/s11065-012-9200-7. PMID:22577001.
- Forner SA, Takada LT, Bettcher BM, et al. Comparing CSF biomarkersbrain MRI in the diagnosis of sporadic Creutzfeldt-Jakob disease. Neurol Clin Pract. 2015;5(2):116-125. doi:10.1212/CPJ.0000000000000111. PMID:26137420.
- Liberski PP. Choroba Creutzfeldta i inne choroby wywołane przez priony – pasażowalne encefalopatie gąbczaste człowieka. Lublin: Wydawnictwo CZELEJ Sp.z o.o; 2003.
- Malek N, Baker MR, Mann C, et al. Electroencepalographic markers in dementia. Acta Neurol Scand. 2017;135(4):388-393. doi:10.1111/ane.12638. PMID:27430350.
- Mente KP, O'Donnell JK, Jones SE, et al. Fluorodeoxyglucose positron emission tomography (FDG-PET) correlation of histopathology and MRI in prion diseases. Alzheimer Dis Assoc Disord. 2017;31(1):1-7. doi:10.1097/WAD.0000000000000188. PMID:28121634.
- Parchi P, Castellani R, Capellari S, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1996;39(6):767-78. doi:10.1002/ana.410390613. PMID:8651649.