
ABSTRACT
Historically, the observation of naturally occurring cases of prion disease led to the classification of different susceptibility grades and to the designation of prion resistant species. However, the development of highly efficient in vitro prion propagation systems and the generation of ad hoc transgenic models allowed determining that leporidae and equidae families have been erroneously considered resistant to prion infection. On the contrary, similar approaches revealed an unexpected high level of resistance of the canidae family. In PLoS Pathogens [Citation1], we describe experiments directed toward elucidating which are the determinants of the alleged prion resistance of this family. Studies based on the sequence of the canine prion protein coupled with structural in silico analysis identified a key residue probably implicated in this resistance. Cell and brain-based PMCA highlighted that the presence of aspartic or glutamic acid at codon 163 of the canid PrP, strongly inhibits prion replication in vitro. Transgenic animals carrying this substitution in mouse PrP were resistant to prion infection after intracerebral challenge with different mouse prion strains. The confirmation of the importance of this substitution and its exclusivity in this family, suggests it could have been evolutionarily favored, due to their diet based on carrion and small ruminants.
To find the first description of a transmissible spongiform encephalopathy (TSE) we have to look back to the 18th century in England, where a rare disease in sheep was reported [Citation2]. Since then, similar pathologies have been described in several mammalians resulting in a growing list of TSEs and species susceptible to this group of disorders. In addition, the bovine spongiform encephalopathy (BSE) outbreak in Europe during the 1990s clearly demonstrated that prions could be transmitted naturally from one species to others, including non-human primates. However, animals belonging to the canidae, leporidae and equidae families, which were presumably exposed to prions equally, have never been reported to succumb to TSEs [Citation3–9]. These data, coupled to the failure of all the experimental interspecies TSE transmission attempts, supported their categorization as prion resistant. Purportedly prion resistant species had long been considered pivotal to elucidate the underlying mechanisms of prion resistance, opening a way to develop therapeutic approaches against these untreatable disorders. However, several recent studies involving species once considered resistant [Citation8,Citation9] indicate that the absence of naturally occurring TSE cases or unsuccessful experimental challenges is not enough to classify any mammalian species as resistant to prion diseases. In fact, several features have to be examined before coining the term prion-resistant, like the population size, uninvestigated sporadic deaths or statistically representative number of individuals in TSE experimental inoculations [Citation7]. Nonetheless, in the case of dog PrP, an extraordinary resistance to misfolding has been undoubtedly proven, revealing a unique amino acid residue that may hide the key to TSE resistance.
The in vitro studies using protein misfolding cyclic amplification (PMCA) have shown it is an effective tool to predict the behavior of prions in vivo [Citation7]. Specifically, its value to push the limits of certain cross-species prion transmission barriers has been proven, overcoming barriers that were thought insuperable in nature. PMCA and subsequent in vivo experiments in its natural host have shown that rabbits are not resistant to prion infection, although they present low susceptibility [Citation8]. In the same direction, PMCA allowed to demonstrate that equids are not completely resistant to TSEs, while they reveal an unusual replicative phenomenon termed nonadaptive prion amplification (NAPA), described in horse-PrP transgenic mice [Citation9]. Although it was argued that other factors besides the PrP sequence could be responsible of the low susceptibility of these species to prion disorders, a study in which transgenic rabbit expressing sheep PrP were shown highly susceptible to scrapie cleared up any doubts about this [Citation10]. However, despite some thorough analysis of rabbit PrP and horse PrP in vitro and in sillico, no clear mechanism arose that could explain their high resistance to misfolding [Citation5,Citation6,Citation8,Citation9,Citation11–15]. However, our recent work published in PLoS Pathogens [Citation1], other studies in canids and the exhaustive analysis of their PrP sequences tell a different story. Although there are reports of suspected TSE cases in canines, none of these studies were conclusive of prion disease in this genus [Citation16]. Further laboratory experiments based on attempts of infection of canine kidney cells or the propensity of dog PrP in forming intermediate β-states support the in vivo data [Citation17,Citation18]. Moreover, during the outbreak of the BSE epidemic during the 1990´s in the UK, several mammalian species, including cheetahs, pumas and cats, became infected with BSE prions [Citation19]. The presumed infection source was prion-contaminated food; however, the phylogenetically close canidae and felidae family members were fed with similar food and no canids have been reported with TSE [Citation19–21].
In the paper published in PLoS Pathogens [Citation1], we describe a series of experiments directed towards elucidating the molecular determinants of the alleged prion resistance of the canidae family. The experiments consisted in an initial in vitro phase in which different normal brain homogenates from two breeds of dogs were subjected to PMCA with prion strains of diverse origins and following distinct propagation procedures. Interestingly, no misfolded dog PrP was detected in any case except when seeded with classical BSE or sheep BSE and with the requirement of modified PMCA conditions, which increased the possibility of misfolding. None of these particular propagation conditions were necessary to convert in vitro other alleged prion resistant species like rabbits or horses [Citation8,Citation9], suggesting that dog PrPC shows a very high resistance to be misfolded. To fully understand this low susceptibility we performed a deep analysis of canid PrP sequences followed by an in silico structural analysis in order to evaluate the determinants of this resistance.
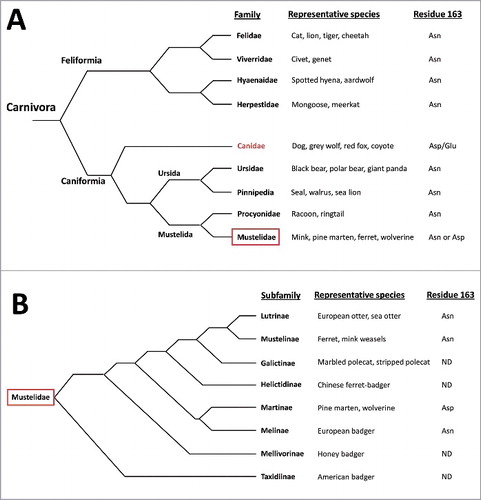
Several interesting findings arose from that set of studies. To start with, the absence in canids of the highly conserved residue Asn at position 163 located in the loop linking helix-α1 and strand-β2, which is replaced by Asp in the canid PrP and Asp/Glu in the canine PrP. The comparison of canid PrPs with several others revealed that this negatively charged residue is highly specific and unique for the members of the canidae family, with the exception of two members of the mustelidae family, wolverine and pine marten that present the same residue. Focusing on the Carnivora order, PrP sequence analysis of PrPs from several species performed by us and others [Citation22] shows the highly conserved Asn at position 163 for all the PrPs analyzed from the suborder Feliformia and most of the families that form the suborder Caniformia whereas all the members of the canidae family present Asp or Glu. This suggests that the substitution of Asn could have occurred early after the divergence of the canids and may have been positively selected during their evolution due to their role as predators of small ruminants and usual carrion consumers. The fact that other carrion consumers from the suborder Feliformia (as the members of the family hyaenidae) do not present this substitution may exclude the possibility of this substitution appearing in an early ancestor of the Carnivora. However, the presence of Asp at codon 163 in two closely related members of the mustelidae family, which are evolutionary quite distant to the canidae would indicate that the substitution could have ocurred in a common ancestor of all the Caniformia and could have been lost in almost all the members of this suborder () with the exception of those most dependent on carrion consumption. Sequencing of grey fox (Urocyon cinereoargenteus) PrP, considered as the most basal of the living canids, could shed some light on this issue. But regrettably its PrP has not been sequenced to date. This is not the first notion that natural selection may have succeeded in making some individuals or a whole species highly or completely resistant to prion diseases. This is the case for the recently described Gly127Val variant of human PrP that was under positive evolutionary selection during the epidemic of kuru and appeared to provide strong protection against disease [Citation23]. In the same direction, variations at codon 171 of sheep PrP appear to be the principal determinants of resistance to classical scrapie [Citation24–26]. Nonetheless, it is unlikely that just two members of the highly diverse mustelidae family (with several species consuming carrion regularly) may have conserved a residue that is present only in the canines amongst all the Caniformia. Therefore, random genetic drifting as an explanation of the presence of Asp at position 163 in the wolverine and pine marten cannot be excluded, which as pointed out by Stewart and collaborators, is supported by the fact that an evolutionary distant species, the insectivorous little brown bat (Myotis lucifugus), also presents an Asp at position 163 [Citation22]. Taking into account that few positions in the PrP sequence may have been evolutionarily selected and that the protein presents many variations among mammalian species, to know whether other unique variants could have been selected through evolution is highly interesting. However, besides the already mentioned examples, there is actually no evidence that other unique polymorphisms could have been favored due to a potential resistance to prion diseases. For this phenomenon to happen, a selection pressure is necessary, what in the case of prions, may involve carrion consumption or living side by side with animals in which prion disorders are highly prevalent. Unfortunately, PrP sequences of several species are lacking and for those species with available PrP sequences, these are based on few individuals, hindering the detection of polymorphisms of interest.
Figure 1. Schematic representation of the relationship of the living members of Carnivora. A) Representation of the phylogenetic relationship of living families of the order Carnivora, including examples of representative members of each family and the PrP residue at position 163 (domestic dog numbering) or equivalent based on the species with their PRNP gene sequenced 1,22. B) Detail on the relationship of the living members of the family Mustelidae based on the analysis of 21 nuclear genes and 1 mitochondrial gene (according to Koepfli et al. BMC Biol. 2008), including examples of representative members of each subfamily and the PrP residue at position 163 (domestic dog numbering) or equivalent based on the species with their PRNP gene sequenced 1,22. ND: non-determined, refers to the ones with no PRNP sequence available for any member of the subfamily.
The in silico analysis of the effects of this substitution revealed a change of the electrostatic potential on the surface of PrP. The acidic properties conferred by this residue disturb this region introducing a positively charged residue that might hamper PrP conversion. To fully evaluate this possibility, mouse cell PMCA studies were conducted based on the expression of the desired Asn158Asp or Asn158Glu substitutions (Asn163Asp or Asn163Glu in domestic dog PrP numbering, respectively). When compared to the wild-type Asn158 mouse PrP it was clearly shown that the presence of an acidic residue in this position hindered its misfolding propagation ability and blocks the propagation over the wild-type mouse protein. In this way, the fact that domestic dogs present a polymorphism at position 163 being in both cases an acidic residue supports the notion that the change in surface electrostatic potential might be directly involved in the resistance to misfolding. Moreover, it suggests that it is an evolutionarily favored element because if was not pivotal, its reversion to the highly conserved Asn or random genetic drift to any other amino acid would be more likely to have occurred.
The in vitro data presented in the work published in PLoS pathogens [Citation1] clearly suggested that one single residue could be the responsible for the absence of reported TSEs in canines and the experiments performed in vivo completely support this observation. A new transgenic mouse model expressing murine PrP with the unique substitution Asn158Asp was generated and in vivo transmission studies using mouse-adapted prion strains failed to cause disease, indicating that this specific amino acid substitution prevented mouse PrP from misfolding. Nonetheless, there are still two key studies required to undoubtedly proof the pivotal role of this substitution in the resistance of canids to TSEs: i) experimental challenge of animal models expressing dog PrP with different prion strains and ii) comparison of the susceptibility of the animal models from the above study with the experimental challenge of an animal model expressing dog PrP with Asn163 inoculated with the same prion strains (both studies are currently ongoing).
One of the main interests of protective mutations is to understand the structural basis of the inability to propagate prions to provide key insights into prion propagation and the development of rational therapies. The potential dominant-negative effect of the mouse Asp158 mutated PrP over the wild-type protein has been further investigated in transgenic mice co-expressing both proteins. The dramatic increase of the survival time of this animal model after inoculation with different mouse-adapted prion strains clearly indicate a dominant-negative effect and makes this naturally occurring mutation a promising candidate for gene therapy strategies for the treatment of TSEs [Citation27]. However, the technology available nowadays does not permit the direct implementation of dominant-negative PrPs as therapeutic strategy since it implicates performing gene therapy on the central nervous system (CNS) or direct injection of recombinant PrP in the brain. Nonetheless, dominant-negative proteins offer a unique opportunity to study the structural characteristics of PrPs that cannot misfold into a pathogenic form allowing the design of small molecules that could mimic the protective effect.
Altogether, these data explain why canines fed with identical food than phylogenetically close species and/or deeply exposed to infectious prions by consuming TSE affected animals such as sheep, cattle or deer are highly resistant to TSEs. And importantly, it may be the second evidence (after the observation of the protective effect of Gly127Val in human PrP) of a prion-driven evolutionary adaptation leading towards prion resistant species.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Fernandez-Borges N, Parra B, Vidal E, et al. Unraveling the key to the resistance of canids to prion diseases. PLoS Pathog. 2017;13:e1006716. doi:10.1371/journal.ppat.1006716. eCollection 2017 Nov. PMID: 29131852
- Liberski PP. Historical overview of prion diseases: a view from afar. Folia Neuropathol. 2012;50:1–12.
- Gibbs CJ, Jr, Gajdusek DC. Experimental subacute spongiform virus encephalopathies in primates and other laboratory animals. Science. 1973;182:67–8. doi:10.1126/science.182.4107.67.
- Barlow RM, Rennie JC. The fate of ME7 scrapie infection in rats, guinea-pigs and rabbits. Res Vet Sci. 1976;21:110–1.
- Vorberg I, Groschup MH, Pfaff E, et al. Multiple amino acid residues within the rabbit prion protein inhibit formation of its abnormal isoform. J Virol. 2003;77:2003–9. doi:10.1128/JVI.77.3.2003-2009.2003.
- Perez DR, Damberger FF, Wuthrich K. Horse prion protein NMR structure and comparisons with related variants of the mouse prion protein. J Mol Biol. 2010;400:121–8. doi:10.1016/j.jmb.2010.04.066.
- Fernandez-Borges N, Chianini F, Erana H, et al. Naturally prion resistant mammals: a utopia? Prion. 2012;6:425–9. doi:10.4161/pri.22057.
- Chianini F, Fernandez-Borges N, Vidal E, et al. Rabbits are not resistant to prion infection. Proc Natl Acad Sci U S A. 2012;109:5080–5. doi:10.1073/pnas.1120076109.
- Bian J, Khaychuk V, Angers RC, et al. Prion replication without host adaptation during interspecies transmissions. Proc Natl Acad Sci USA. 2017;114:1141–6. doi:10.1073/pnas.1611891114.
- Sarradin P, Viglietta C, Limouzin C, et al. Transgenic Rabbits Expressing Ovine PrP Are Susceptible to Scrapie. PLoS Pathog. 2015;11:e1005077. doi:10.1371/journal.ppat.1005077.
- Zhang J, Zhang Y. Molecular dynamics studies on the NMR and X-ray structures of rabbit prion proteins. J Theor Biol. 2014;342:70–82. doi:10.1016/j.jtbi.2013.10.005.
- Zhang J, Wang F, Zhang Y. Molecular dynamics studies on the NMR structures of rabbit prion protein wild type and mutants: surface electrostatic charge distributions. J Biomol Struct Dyn. 2015;33:1326–35. doi:10.1080/07391102.2014.947325.
- Vidal E, Fernandez-Borges N, Pintado B, et al. Transgenic Mouse Bioassay: Evidence That Rabbits Are Susceptible to a Variety of Prion Isolates. PLoS Pathog. 2015;11:e1004977. doi:10.1371/journal.ppat.1004977.
- Vidal E, Fernandez-Borges N, Pintado B, et al. Bovine spongiform encephalopathy induces misfolding of alleged prion-resistant species cellular prion protein without altering its pathobiological features. J Neurosci. 2013;33:7778–86. doi:10.1523/JNEUROSCI.0244-13.2013.
- Erana H, Fernandez-Borges N, Elezgarai SR, et al. In vitro approach to identify key amino acids in low susceptibility of rabbit prion protein to misfolding. J Virol. 2017;91:e01543–17. doi:10.1128/JVI.01543-17.
- Kortz GD, Meier WA, Higgins RJ, et al. Neuronal vacuolation and spinocerebellar degeneration in young Rottweiler dogs. Vet Pathol. 1997;34:296–302. doi:10.1177/030098589703400405.
- Polymenidou M, Trusheim H, Stallmach L, et al. Canine MDCK cell lines are refractory to infection with human and mouse prions. Vaccine. 2008;26:2601–14. doi:10.1016/j.vaccine.2008.03.035.
- Khan MQ, Sweeting B, Mulligan VK, et al. Prion disease susceptibility is affected by beta-structure folding propensity and local side-chain interactions in PrP. Proc Natl Acad Sci USA. 2010;107:19808–13. doi:10.1073/pnas.1005267107.
- Kirkwood JK, Cunningham AA. Epidemiological observations on spongiform encephalopathies in captive wild animals in the British Isles. Vet Rec. 1994;135:296–303. doi:10.1136/vr.135.13.296.
- Imran M, Mahmood S. An overview of animal prion diseases. Virol J. 2011;8:493. doi:10.1186/1743-422X-8-493.
- Wopfner F, Weidenhofer G, Schneider R, et al. Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein. J Mol Biol. 1999;289:1163–78. doi:10.1006/jmbi.1999.2831.
- Stewart P, Campbell L, Skogtvedt S, et al. Genetic predictions of prion disease susceptibility in carnivore species based on variability of the prion gene coding region. PLoS One. 2012;7:e50623. doi:10.1371/journal.pone.0050623.
- Asante EA, Smidak M, Grimshaw A, et al. A naturally occurring variant of the human prion protein completely prevents prion disease. Nature. 2015;522:478–81. doi:10.1038/nature14510.
- Westaway D, Zuliani V, Cooper CM, et al. Homozygosity for prion protein alleles encoding glutamine-171 renders sheep susceptible to natural scrapie. Genes Dev. 1994;8:959–69. doi:10.1101/gad.8.8.959.
- Clouscard C, Beaudry P, Elsen JM, et al. Different allelic effects of the codons 136 and 171 of the prion protein gene in sheep with natural scrapie. J Gen Virol. 1995;76:2097–101. doi:10.1099/0022-1317-76-8-2097.
- Hunter N, Cairns D, Foster JD, et al. Is scrapie solely a genetic disease? Nature. 1997;386:137. doi:10.1038/386137a0.
- Otero A, Bolea R, Hedman C, et al. An amino acid substitution found in animals with low susceptibility to prion diseases confers a protective dominant-negative effect in prion-infected transgenic mice. Mol Neurobiol. 2017. doi:10.1007/s12035-017-0832-8