
ABSTRACT
We report a Japanese patient with spinocerebellar ataxia type 31 (SCA31) and sporadic Creutzfeldt-Jakob disease (sCJD). A 52-year-old man developed progressive cognitive impairment after the appearance of cerebellar symptoms. Brain MR diffusion-weighted imaging (DWI) demonstrated a slowly expanding hyperintense lesion in the cerebral cortex. The patient was finally diagnosed as having both SCA31 and sCJD by identification of genetic mutations and by real-time quaking-induced conversion (RT-QUIC) analysis of the cerebrospinal fluid (CSF), respectively. Here, we report the clinical details of this rare combined case, with particular reference to the association between prion protein and the early onset of SCA31.
Introduction
Spinocerebellar ataxia type 31 (SCA31) is an autosomal-dominant spinocerebellar ataxia caused by a penta-nucleotide (TGGAA) repeat expansion in chromosome 16 [Citation1]. No association between SCA31 and sporadic Creutzfeldt-Jakob disease (sCJD) has been reported previously. Here we report a patient with both SCA31 and sCJD, with special reference to the process of SCA31 gene expression and abnormal prion protein (PrPsc) in the two diseases.
Case presentation
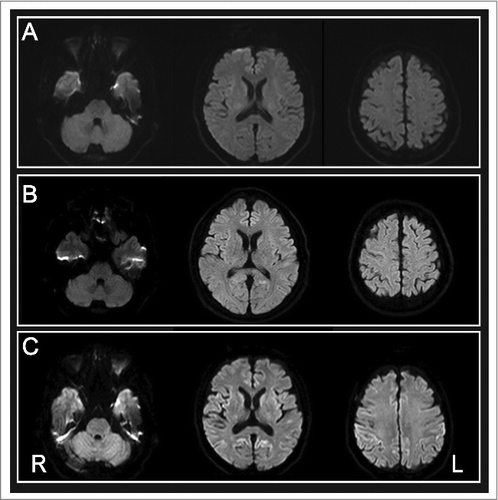
A 52-year-old man presented with mild dysarthria and an unsteady gait. He had no history of any form of surgery or travel abroad, and no family history of similar symptoms. One month after onset, he developed saccadic eye movement and truncal-dominant ataxia. Diffusion-weighted brain MRI (DWI) demonstrated slightly hyperintense areas in the cingulate gyri (). Although the patient was still able to work, his symptoms gradually worsened. Seven months later, his Mini-Mental State Examination (MMSE) score was 26/30, and DWI revealed abnormal areas of laminar hyperintensity distributed more widely in the frontal cortex (), suggesting the possibility of prion disease. However, electroencephalography (EEG) demonstrated no remarkable findings, and CSF assay including 14-3-3 protein and real-time quaking-induced conversion (RT-QUIC) analysis for prion protein yielded negative results [Citation2]. PrP gene analysis demonstrated no mutations, and examination of polymorphism revealed methionine/methionine (MM) at codon 129. For differential diagnosis, we investigated genetic mutations in several SCA-related genes and found a penta-nucleotide (TGGAA) repeat expansion in chromosome 16q22.1 using repeat-primed PCR [Citation3]. We diagnosed the patient as having SCA31. His cognitive impairment and gait disturbance worsened, and at 11 months after onset, use of a walker became necessary. By 18 months, he had become mostly bedridden, and his MMSE score had decreased to 7/30. He showed dropped head, bradykinesia and rigidity of the limbs, but no involuntary movements including myoclonus. The results of CSF examination remained normal and EEG showed no periodic synchronous discharges (PSD). The basic activity was a diffuse alpha-wave pattern that indicated brain hypo-activity. However, DWI demonstrated symmetrical areas of cortical hyperintensity, being more obvious in the bilateral cingulate gyri (). The rapid clinical progression and MRI findings were atypical for SCA31, and suggested the coexistence of sCJD. Therefore, CSF RT-QUIC was performed again, and this time a positive result was obtained. This allowed us to make a diagnosis of combined SCA31 and sCJD. Twenty months after onset, the patient's MMSE score became 4/30. At 32 months, oral food intake was still possible with total assistance, and the patient was admitted to another hospital to receive supportive care.
Figure 1. Brain diffusion-weighted magnetic resonance images obtained at (A) one month, (B) 7 months and (c) 18 months after onset.
Discussion
The mean age at onset of SCA31 is 59.1 years, and the disease is manifested as pure and slowly progressive cerebellar ataxia without dementia [Citation1,Citation4] (). Meanwhile, the clinical course of sCJD depends on PrP polymorphism at codon 129 and the PrP Western blot pattern (type 1 or 2). The MM1 type is characterized by rapidly progressive dementia and death within several months [Citation5] (). The MM2 cortical (MM2C) type begins with dementia, then ataxia develops within about 6 months after onset, death occurring at around 24 months [Citation6,Citation7] (). In our patient, the initial cerebellar symptoms would have been attributable to SCA31 because the clinical progression was very slow for sCJD. Also, as SCA31 rarely causes dementia, the dementia in this case would have been due to MM2C-type sCJD.
Table 1. Comparison of SCD31, sCJD and this case.
In our present patient, symptom onset was earlier than that for typical SCA31. Longer TGGAA repeats accelerate the onset of SCA31 [Citation1]. The repeat length in our patient could not be determined precisely because the method we employed was semi-quantitative [Citation3]. In this case, the effect of prion protein on SCA31 onset was also assumed. The issue of whether sCJD appearing as a complication of SCA31 has a pathophysiological impact on the clinical manifestation is an important consideration. The relevance of PrPsc to SCA has been examined in an in vitro study [Citation8] and one further report [Citation9]. In the in vitro study, forced expression of human prion protein in HEK293 cells caused marked upregulation of mRNA for the SCA12 causative gene, PPP2R2B [Citation8]. A previous postmortem pathological investigation of a 57-year-old man diagnosed as having both CJD and SCA12 revealed elevation of the CSF level of 14-3-3 protein and a 49 CAG-repeat expansion in the PPP2R2B gene [Citation9]. The number of CAG repeats can range from normal (7-31) to abnormal (55-78), and the number of repeat expansions in that case was intermediate [Citation9]. The cerebellar symptoms might have been promoted by PrPsc, which was found by histological investigation of the cerebellum [Citation9]. The mutation responsible for SCA31 is located in an untranslated region. Among the untranslated-region repeat diseases, repeat-associated non-AUG (RAN) translation has attracted attention. Recently, the effect of RAN translation on SCA31 was also reported [Citation10]. If PrP also promotes mRNA expression of TGGAA-containing genes, then the toxic protein derived from RAN translation might increase and affect the course of SCA31.
Although the exact causative mechanism in the present case remains unclear, examination of further patients would help to throw further light on the pathophysiological mechanisms involved.
Conclusion
We have reported the first case of SCA31 combined with sCJD. Although the relationship between the two remains unclear, there is a possibility that onset of SCA31 became clinically evident earlier because of the complicating CJD.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed
Author roles
Conception and design: Ishihara T, Nishizawa M.
Data acquisition and analysis: Mana N, Ishiguro T, Kasuga K, Nozaki H, Onodera O. Drafting: Saito N, Ishihara T.
Editing: Kasuga K, Shimohata T.
Revision: Nishizawa M.
Acknowledgments
We are grateful to Prof. K. Sato, Nagasaki University, for technical help with analysis of 14-3-3 protein, total-tau protein, and RT-QUIC in CSF. We also thank Prof. T. Kitamoto, Tohoku University, for examation of the prion protein gene.
References
- Sato N, Amino T, Kobayashi K, et al. Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA) n. Am J Hum Genet. 2009;85:544–557.
- Atarashi R, Satoh K, Sano K, et al. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat Med. 2011;17:175–178.
- Ishige T, Sawai S, Itoga S, et al. Pentanucleotide repeat-primed PCR for genetic diagnosis of spinocerebellar ataxia type 31. J Hum Genet. 2012;57:807–808.
- Nozaki H, Ikeuchi T, Kawakami A, et al. Clinical and genetic characterizations of 16q‐linked autosomal dominant spinocerebellar ataxia (AD‐SCA) and frequency analysis of AD‐SCA in the Japanese population. Mov Dis. 2007;22:857–862.
- Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–233.
- Krasnianski A, Meissner B, Schulz-Schaeffer W, et al. Clinical features and diagnosis of the MM2 cortical subtype of sporadic Creutzfeldt-Jakob disease. Arch Neurol. 2006;63:876–880.
- Nozaki I, Hamaguchi T, Sanjo N, et al. Prospective 10-year surveillance of human prion diseases in Japan. Brain. 2010;133:3043–3057.
- Satoh J-i, Yamamura T. Gene expression profile following stable expression of the cellular prion protein. Cell Mol Neurobiol. 2004;24:793–814.
- Hellenbroich Y, Schulz-Schaeffer W, Nitschke MF, et al. Coincidence of a large SCA12 repeat allele with a case of Creutzfeld-Jacob disease. J Neurol Neurosurg Psychiat. 2004;75:937–938.
- Ishiguro T, Sato N, Ueyama M, et al. Regulatory role of RNA chaperone TDP-43 for RNA misfolding and repeat-associated translation in SCA31. Neuron. 2017;94:108–124.