
ABSTRACT
Gerstmann-Sträussler-Scheinker disease (GSS) with the P102L mutation in PRNP gene is characterized with progressive cerebellar dysfunction clinically and PrPSc plaques neurologically. Due to the cerebellar ataxia in the early stage, GSS P102L is often misdiagnosed as other neurodegenerative disorders. We presented here a 49-year-old female patient with proven P102L PRNP mutation, and three heterologous mutations in hereditary ataxias associated gene SYNE1, including p.V3643L, p.M3376V and p.T2860A. The patient appeared progressive unsteady gait in early stage and developed the Creutzfeldt-Jacob disease (CJD) – associated clinical manifestations, including progressive dementia, myoclonus, pyramidal and extrapyramidal signs. She is still alive but with akinetic mutism 21 months after onset. Observation of intense signal changes in cortical regions (cortical ribboning) in diffusion weighted imaging (DWI) MRI scanning and positive protein 14-3-3 in cerebrospinal fluid (CSF) proposed the diagnosis of sporadic CJD. The final diagnosis of P102L GSS was made after PRNP sequencing.
Introduction
Gerstmann–Sträussler–Scheinker disease (GSS) is a rare autosomal dominant hereditary human prion disease [Citation1]. Clinically, GSS is characterized with progressive cerebellar dysfunction. Neuropathologically, PrPSc specific amyloid plaques are primarily localized to the cerebellum in the GSS patients [Citation2]. Up to now, more than 15 different point mutations and several octarepeat insertions within PRNP gene are linked with GSS, among them P102L, P105L, A117V, F198S and M232T are reported relatively frequently [Citation3,Citation4]. GSS associated mutations in the prion protein do not appear to overlap with genetic Creutzfeldt-Jakob disease (gCJD) [Citation4]. Additionally, although GSS also has transmissible capacity, its transmission seems to be extremely inefficient [Citation5,Citation6]. However, recent study has demonstrated that human GSS with P102L, A117V and F198S mutations induce efficiently transmission onto bank vole [Citation7].
Based on the surveillance data from China National CJD Surveillance Network, P102L mutation GSS is the predominant subtype of GSS among Chinese [Citation8]. Here we reported a 49 year-old female patient of P102L GSS with other disease-associated mutations in the gene of SYNE1. The progressive unsteady gait was the main onset symptom, which made the suspected diagnosis of cerebellar ataxia. Later, the neurological manifestations of CJD appeared. The diagnosis of P102L GSS was made after PRNP sequencing.
Case report
A 49-year old woman having progressive cerebellum disturbance and dementia for about 17 months was referred to the China National CJD Surveillance Network as a suspected CJD case. Her general process of the disease progression, main treatment and diagnosis was summarized in . Initially, the patient described unsteady gait with unknown reason accompanying with dizzy and sleeping disturbance. MRI scanning indicated enlarged sulci in cerebellum. Treatment for improving neuronutrition and circulation (including vitamin B1, vitamin B2, Mecobalamin, Butyphthalide and Compound piracetam and Cerebroprotein Hydrolysate Tablets) relieved dizzy somehow, but did not show any improvement for unsteady gait. 7 months later, the patient displayed sensory abnormalities, such as numbness and band-feeling in distal limbs. She had bucking occasionally, especially having liquid food. CSF examination revealed GQ1b antibody positive. Clinical suspicion was Miller-Fisher syndrome. During the hospitalization, she received gamma globulin pulse therapy and neurotrophy medicament and her unsteady gait was recorded being slightly improved. She had visited several hospitals and received the therapy of traditional Chinese medicine and acupuncture afterwards, without certain improvement. 13 months after onset, she was hospitalized again, showing positive in heel-knee-tibia test and Romberg sign neurologically. Brain MRI scanning suggested MELAS possible. EEG recorded a mild abnormality, with dispersedly distributed medium waves (4-6C/θs) together with sharp waves that discharged paroxysmally. The unsteady gait and sleeping disturbance aggravated gradually. Meanwhile, her recognition ability worsened markedly. Myoclonus was repeatedly observed. High signal intensities in bilateral frontal, parietal, temporal and occipital cortices were observed in diffusion weighted imaging (DWI) during the latest hospitalization (). She was referred to China National CJD Surveillance Network as possible sCJD. Her blood routine and blood chemistry tests have been repeated for many times, without any obvious abnormality during the disease progression. The elements of routine CSF biochemistry assays were also at the normal ranges.
Figure 1. Chronologic timeline of the clinical manifestations, the results of examinations, main therapeutics and diagnosis of this P102 GSS case.
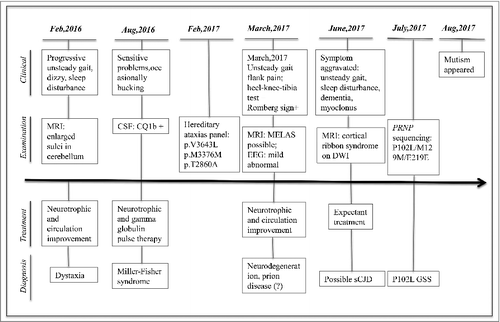
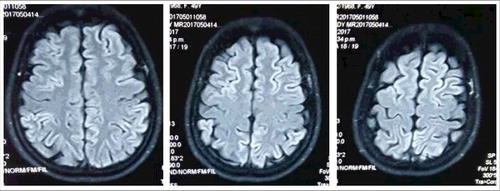
Figure 2. Diffusion weighted imaging (DWI) MRI. High intense signal changes in bilateral frontal, parietal, temporal and occipital cortices.
The CSF sample and blood sample were taken again during the latest hospitalization and transferred to the reference laboratory of China National CJD Surveillance Network in China CDC. The CSF protein 14-3-3 was tested by Western blot according our SOP described elsewhere [Citation9,Citation10]. A 26 KD-large band was observed in her CSF sample (). CSF RT-QuIC assays were also performed with the recombinant truncated hamster PrP protein (HaPrP90-231) as the substrate. Briefly, RT-QuIC was conducted with a black 96-well, optical-bottomed plate (Nunc, 265301) on a BMG FLUOstar plate reader (BMG LABTECH). 10 µl of CSF sample were mixed with 10 µg HaPrP90-231 in the buffer containing 10 mM PBS, 170 mM NaCl, 10 µM Thioflavin T (ThT), 10µM EDTA and 1 ng SDS in final. The final reaction volume was 100µl. Each reaction contained the controls of blank (reaction buffer), negative (1 µl of 10% brain homogenates from normal hamster) and positive (1 µl of 10% brain homogenates from scrapie agent 263K-infected hamster). The working conditions were temperature: 50ºC; vibration speed: 900 rpm; vibration/incubation time: 90/30 sec; total reaction time: 90 h. ThT fluorescence (450 nm excitation and 480 nm emission) were measured every 30 min automatically as relative fluorescence units (rfu). The cutoff value was set as the average value of the negative controls plus 3 times of SD (mean+3SD). The sample was considered as positive when the two parallel wells revealed positive reactive curves. Besides of positive control, no increased ThT curve was noticed in the tested CSF sample, as well as in the negative and blank controls (data not shown).
Figure 3. Western blot for CSF 14-3-3 and PRNP gene sequencing. A. Western blot. The tested CSF samples were separated in 12% SDS-PAGE and immunobloted with 14-3-3 specific polyclonal antibody. Positive Ctrl: 10% goat brain homogenate; M: molecular weight; sCJD: CSF sample of a sCJD case; P102L GSS: CSF sample of the case in this study. B. Graphic presentation of the sequencing analysis of PRNP. A missense mutation at codon 102 (CCG to CTG) causing the substitution of Pro (P) to Leu (L) (left panel), Met (M) homozygote at codon 129 (middle panel) and Glu (E) homozygote at codon 219 (right panel).
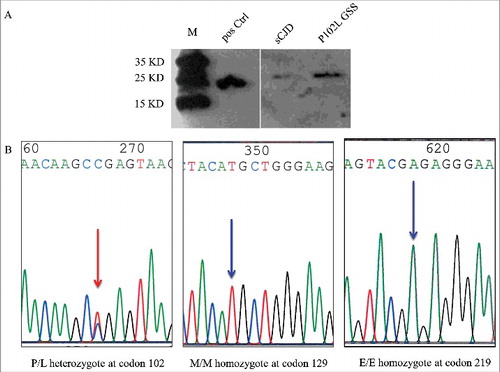
PRNP gene assays were conducted based on the issued SOP [Citation9,Citation10]. Direct sequencing of the PCR product identified a missense mutation at codon 102 (CCG to CTG), leading to a substitution from proline (P) to leucine (L) (). The polymorphism of codon 129 was methionine (M) homozygote and that of codon 219 was glutamate (E) homozygote, respectively (). No other nucleotide exchanges were detected in the rest of the PRNP sequence. Furthermore, her mother and her two adult children agreed to donate the blood for PRNP sequencing. It was verified that the PRNP sequences of her mother and her son were wild type, but her asymptomatic daughter contained a P102L mutation ().
Figure 4. Medical history, P102L mutation in PRNP and the mutation in SYNE1 in the patient's family. Open square: male; open circle: female; square or circle with prolonged diagonal lines: deceased individuals; red circle with arrow: proband case with P102L mutation in PRNP and p.V3643L, p.M3376V, p.T2860A mutations in SYNE1; Dark brown circle: with p.V3643L, p.M3376V mutations in SYNE1, no mutation in PRNP; Blue circle: no mutation in SYNE1, PRNP sequencing not done; Dark blue circle: no mutation in PRNP, SYNE1 sequencing not done; Pink circle: with P102L mutation in PRNP, SYNE1 sequencing not done.
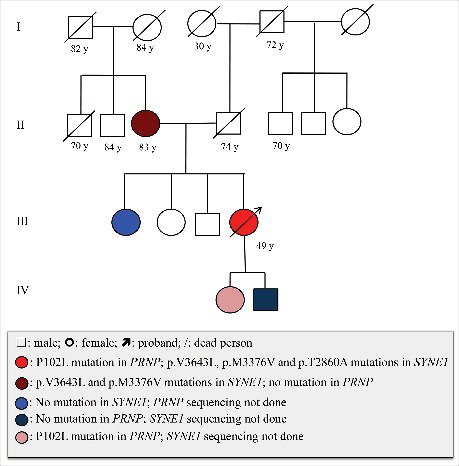
During last hospitalization, she and her mother and one of the sisters had also donated their blood samples for a commercial gene assay of hereditary ataxias panel, which consisted 197 genes. Three heterologous mutations were found in SYNE1, including p.V3643L, p.M3376V and p.T2860A. Those mutations were not normal polymorphism and may relate with autosomal recessive hereditary spinocerebellar ataxias 8, Emery-Dreifuss muscular dystrophy 4 and Senion-Loken syndrome 6 [Citation11–14]. Her mother contained the mutations of p.V3643L and p.M3376V, but not p.T2860A, while her sister did not contain any above mutation (). The examinations including electrocardiography did not reveal any abnormality in cardiac and renal functions.
Interviews with the patient and her family members denied disease family history (). The patient's father died of stroke and diabetes 15 years ago at age of 74. Her mother is still healthy with the age of 83. The patient has two elder sisters and one elder brother, without any detectable neurological abnormality. Her two children are also healthy. Retrospective review of the close relatives also did not find any neurological abnormality among her aunts and uncles from both paternal and maternal lines. The general situation of the patient became worse that she was unable to walk and was bedridden. Akinetic mutismwas noticed one month after discharged from hospital. Up to the end of Nov 2017 (21 months after onset), she is still alive.
Discussion
In this study, we have described a genetic-confirmed GSS case with a P102L mutation in PRNP, who also contains other three disease-associated missense point mutations in SYNE1. This patient seems to undergo the typical process of GSS, with relatively slow onset and disease progression [Citation15]. Cerebellar disorders manifested as unsteady gait, sleeping disturbance and lack of coordination in swallowing are the major problems at early stage. Obvious cognitive dysfunctions, pyramidal and extrapyramidal symptoms appear more than one year later after onset. Akinetic mutism appears even later. The predominant cerebellar symptoms and the obviously later appearances of other CJD-associated clinical manifestations make the diagnosis of ataxias and hereditary spinocerebellar ataxias at early period and of possible sCJD late.
This patient has received several times of brain MRI scanning. Coincidental with the appearance of CJD-associated symptoms, the sCJD-associated abnormalities on MRI are not reported in the earlier examinations, but noticed in the scanning 16 months later after onset. Review of other 9 Chinese cases of P102L GSS (unpublished data) illustrates that 8 patients show sCJD-associated abnormalities on MRI, highlighting the significance of MRI examination for P102L GSS, especially in the relative later stage. Periodic sharp wave complexes (PSWC) on EEG are not detected, but CSF 14-3-3 is positive in this patient. PSWC on EEG and CSF 14-3-3 positive are observed in 50% and 31% in Caucasian GSS patients, respectively [Citation16]. Approximately half of 9 Chinese patients with P102L GSS display PSWC on EEG and CSF 14-3-3 positive (unpublished data). Apparently, the diagnostic values of EEG and CSF 14-3-3 for P102L GSS are less than that of MRI.
With the help of gene testing, three missense heterologous mutations in SYNE1 have been found in this patient. Human SYNE1 encodes a very large protein (Syne1) that belongs to a new family of membrane proteins localized to the nuclear envelope [Citation17]. Syne1 distributes widely in various types of tissues and organs and participates in maintenance of nuclear organization and anchorage, especially at neuromuscular junctions [Citation17,Citation18]. Mutations in SYNE1 are associated with several hereditary diseases [Citation19]. One is Senior-Loken syndrome, which is an autosomal recessive disease characterized by the combination of nephronophthisis and Leber congenital amaurosis [Citation20]. The other one is Emery-Dreifuss muscular dystrophy 4, which is a condition that primarily affects skeletal muscles and cardiac muscle, usually with joint deformities called contractures as the earliest features[13]. The clinical features of the patient described in this study are completely not compliance with these two conditions. Another SYNE1 mutation associated disease is autosomal recessive hereditary spinocerebellar ataxias type 8, which is featured by cerebellar ataxia and lack of coordination, with the onset age of 18 to 65 year-old [Citation21]. Usually, the disease progresses extremely slowly and the patient has normal lifespan. Obviously, the clinical features of this patient are not consistent with hereditary spinocerebellar ataxias type 8. The pathogenic penetrations of those three mutations in SYNE1 are very limited for this patient.
The P102L GSS case in this study lacks of definite disease family history, though her mother contains also two mutations in her SYNE1. The patient's elder siblings and her close relatives of parent-generation are healthy during interview. Later, the PRNP sequencing assays for her mother and children propose that the mutation is probably inherited from her father. Out of 52 GSS cases in Western countries, 33 (69.7%) have had positive family history for prion diseases [Citation16]. However, up to 9 out of 11 Chinese P102L GSS patients recalled disease family history during the clinical interviews. In the later stage, the clinical manifestations of GSS sometimes are usually not distinguishable with that of sCJD. Hence, it proposes again the importance of PRNP gene testing for the diagnosis of genetic prion disease.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the referring patient and her family members for supplying the required information and donating specimens.
Additional information
Funding
References
- Chen C, Dong XP. Epidemiological characteristics of human prion diseases. Infect Dis Poverty. 2016 Jun 02;5(1):47. doi: 10.1186/s40249-016-0143-8
- Collins S, McLean CA, Masters CL. Gerstmann-Straussler-Scheinker syndrome,fatal familial insomnia, and kuru: a review of these less common human transmissible spongiform encephalopathies. J Clin Neurosci: official journal of the Neurosurgical Society of Australasia. 2001 Sep;8(5):387–97. doi: 10.1054/jocn.2001.0919
- Ironside JW, Ritchie DL, Head MW. Prion diseases. Handb Clin Neurol. 2017;145:393–403. doi: 10.1016/B978-0-12-802395-2.00028-6
- Schmitz M, Dittmar K, Llorens F, et al. Hereditary Human Prion Diseases: an Update. Mol Neurobiol. 2017 Aug;54(6):4138–4149. doi: 10.1007/s12035-016-9918-y
- Nonno R, Angelo Di Bari M, Agrimi U, et al. Transmissibility of Gerstmann-Straussler-Scheinker syndrome in rodent models: New insights into the molecular underpinnings of prion infectivity. Prion. 2016 Nov;10(6):421–433. doi: 10.1080/19336896.2016.1239686
- Chiesa R, Restelli E, Comerio L, et al. Transgenic mice recapitulate the phenotypic heterogeneity of genetic prion diseases without developing prion infectivity: Role of intracellular PrP retention in neurotoxicity. Prion. 2016 Mar 03;10(2):93–102. doi: 10.1080/19336896.2016.1139276
- Pirisinu L, Di Bari MA, D'Agostino C, et al. Gerstmann-Straussler-Scheinker disease subtypes efficiently transmit in bank voles as genuine prion diseases. Sci Rep. 2016 Feb 04;6:20443. doi: 10.1038/srep20443
- Shi Q, Zhou W, Chen C, et al. The Features of Genetic Prion Diseases Based on Chinese Surveillance Program. PloS one. 2015;10(10):e0139552. doi: 10.1371/journal.pone.0139552
- Shi Q, Zhou W, Chen C, et al. Quality evaluation for the surveillance system of human prion diseases in China based on the data from 2010 to 2016. Prion. 2016 Nov;10(6):484–491. doi: 10.1080/19336896.2016.1229731
- Gao C, Shi Q, Tian C, et al. The epidemiological, clinical, and laboratory features of sporadic Creutzfeldt-Jakob disease patients in China: surveillance data from 2006 to 2010. PloS one. 2011;6(8):e24231. doi: 10.1371/journal.pone.0024231
- Gros-Louis F, Dupre N, Dion P, et al. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat Genet. 2007 Jan;39(1):80–5. doi: 10.1038/ng1927
- Izumi Y, Miyamoto R, Morino H, et al. Cerebellar ataxia with SYNE1 mutation accompanying motor neuron disease. Neurol. 2013 Feb 05;80(6):600–1. doi: 10.1212/WNL.0b013e3182815529
- Puckelwartz M, McNally EM. Emery-Dreifuss muscular dystrophy. Handb Clin Neurol. 2011;101:155–66. doi: 10.1016/B978-0-08-045031-5.00012-8
- Ronquillo CC, Bernstein PS, Baehr W. Senior-Loken syndrome: a syndromic form of retinal dystrophy associated with nephronophthisis. Vision Res. 2012 Dec 15;75:88–97. doi: 10.1016/j.visres.2012.07.003
- Mastrianni JA. Genetic Prion Diseases. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews(R). Seattle (WA): University of Washington; 1993.
- Kovacs GG, Puopolo M, Ladogana A, et al. Genetic prion disease: the EUROCJD experience. Hum Genet. 2005 Nov;118(2):166–74. doi: 10.1007/s00439-005-0020-1
- Smith ER, Zhang XY, Capo-Chichi CD, et al. Increased expression of Syne1/nesprin-1 facilitates nuclear envelope structure changes in embryonic stem cell differentiation. Dev Dyn: an official publication of the American Association of Anatomists. 2011 Oct;240(10):2245–55. doi: 10.1002/dvdy.22717
- Crisp M, Liu Q, Roux K, et al. Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol. 2006 Jan 02;172(1):41–53. doi: 10.1083/jcb.200509124
- Chen CP. Prenatal diagnosis and genetic analysis of fetal akinesia deformation sequence and multiple pterygium syndrome associated with neuromuscular junction disorders: a review. Taiwan J Obstet Gynecol. 2012 Mar;51(1):12–7. doi: 10.1016/j.tjog.2012.01.004
- Kaur A, Dhir SK, Goyal G, et al. Senior Loken Syndrome. J Clin Diag Res: JCDR. 2016 Nov;10(11):SD03–SD04. doi: 10.7860/JCDR/2016/21832.8816
- Didonna A, Opal P. Advances in Sequencing Technologies for Understanding Hereditary Ataxias: A Review. JAMA Neurol. 2016 Dec 01;73(12):1485–1490. doi: 10.1001/jamaneurol.2016.3097